



Overview
Source : Laboratoire du Dr Paul Bower - Purdue University
La méthode des ajouts dosés est une méthode d’analyse quantitative, qui est souvent utilisée lorsque l’échantillon comporte des éléments multiples qui donnent lieu à des effets de matrice, où les composants supplémentaires peuvent réduire ou augmenter le signal d’absorbance analyte. Qui se traduit par des erreurs significatives dans les résultats de l’analyse.
Ajouts dosés sont couramment utilisés pour éliminer les effets de matrice d’une mesure, car il est supposé que la matrice affecte toutes les solutions également. En outre, il sert à corriger pour la substance chimique séparations dans le processus d’extraction de phase.
La méthode est exécutée par la lecture de l’intensité (en l’occurrence fluorescente) expérimentale de la solution inconnue, puis en mesurant l’intensité de l’inconnu avec des quantités variables de norme connue ajouté. Les données sont tracées comme intensité de fluorescence vs. le montant de la norme ajouté (l’inconnu lui-même, avec aucune norme ajouté, est tracée sur l’axe des ordonnées). La ligne des moindres carrés entre en intersection avec l’axe des abscisses au négatif de la concentration de l’inconnu, comme illustré à la Figure 1.
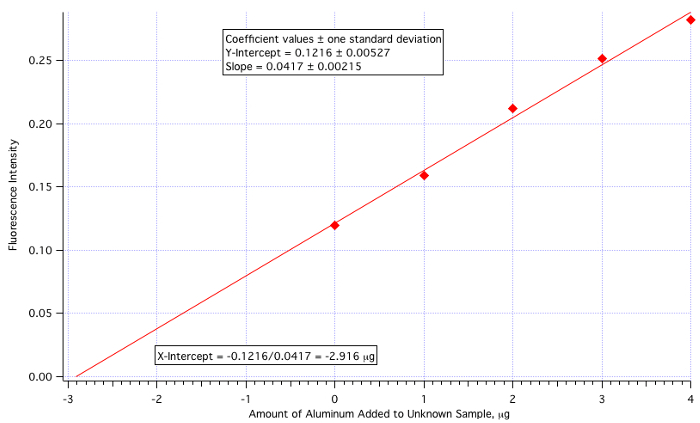
Figure 1. Une représentation graphique de la méthode des ajouts dosés.
Principles
Dans cette expérience, la méthode des ajouts dosés est démontrée comme outil d’analyse. La méthode est une procédure d’analyse quantitative d’une espèce sans la génération d’une courbe d’étalonnage typique. Ajout d’un étalon l’analyse est réalisée en mesurant l’intensité spectroscopique avant et après l’ajout de portions précises d’une solution étalon connue de l’analyte.
Cette expérience étudie les espèces fluorescentes en réagissant de manière à former un complexe fluorescent. Cette approche est couramment utilisée dans le cadre de l’enquête des ions métalliques. Les ions d’aluminium (Al)3 +seront déterminées en formant un complexe avec 8-Hydroxyquinoléine (8HQ). L’Al3 + est précipitée par 8HQ d’une solution aqueuse et est ensuite extraite dans le chloroforme ; la fluorescence de la solution de chloroforme est mesurée et liée à la concentration de la solution3 + Al d’origine. Sensibilité de l’ordre de parties par million (ppm ou μg/mL) est attendue pour cette expérience.
La réaction est

La quantité d’aluminium dans chaque échantillon lors de cette expérience est calculée comme suit :
| Vide | 0 | ||
| Standard mL inconnu + 0 | Vinconnu(Cinconnue) = 25 mL (Cinconnu) | ||
| Inconnu + 1 mL Standard | Vinconnu(Cinconnu) + VStandard(CStandard) = (Cinconnu) 25 mL + 1 mL (1 μg/mL) | ||
| Étalon inconnu + 2 mL | V Inconnu (Cinconnu) + V Standard (CStandard) = 25 mL (Cinconnu) + 2 mL (1 μg/mL) | ||
| Étalon inconnu + 3 mL | V Inconnu (Cinconnu) + V Standard (CStandard) = 25 mL (Cinconnu) + 3 mL (1 μg/mL) | ||
| Étalon inconnu + 4 mL | V Inconnu (Cinconnu) + V Standard (CStandard) = 25 mL (Cinconnu) + 4 mL (1 μg/mL) | ||
Subscription Required. Please recommend JoVE to your librarian.
Procedure
1. préparation des réactifs
- Al3 + solution étalon de 100 ppm : dissoudre 0,9151 g de nitrate basique d’aluminium (Al (NO3)3•9H2O) dans une fiole jaugée de 1 L avec l’eau distillée.
- 8HQ solution dans l’acide acétique 1 M (2 % wt/vol) : ajouter 2,0 g de 8-Hydroxyquinoléine dans une fiole jaugée de 100 mL.
- Soigneusement ajouter 5,74 mL d’acide acétique glacial à la fiole de 100 mL, puis compléter au trait avec l’eau distillée. Cela permet à la 8-Hydroxyquinoléine de dissoudre en phase aqueuse.
- 1 M NH4+/NH3 tampon (pH ~ 8) : ajouter 20 g d’acétate d’ammonium (NH4OAc) dans une bouteille de 100 mL.
- Ajouter 7 mL d’hydroxyde d’ammonium 30 % à cette bouteille de 100 mL et compléter au trait avec l’eau distillée. Cela aide à neutraliser l’acide dans la solution 8HQ lorsqu’il est combiné.
- Les autres réactifs comprennent sulfate de sodium anhydre (Na2SO4) et le chloroforme (qualité technique).
2. préparation des échantillons
- Préparer une Al3 + solution titrée de 1,00 ppm en ajoutant 1,0 mL de l’étalon de 100 ppm Al3 + solution avec une pipette dans une fiole jaugée de 100 mL.
- Placez les six 125 mL ampoule à entonnoirs dans les anneaux qui se trouvent sur un stand de grand anneau situé dans la hotte. Ils doivent être étiquetés comme suit : BL, 0, 1, 2, 3, 4. Veillez à ce que toute la verrerie est rigoureusement propre, car il est difficile d’obtenir des résultats quantitatifs si petites perles du chloroforme collent aux parois de la verrerie.
- Ajouter 25,00 mL de la solution3 + Al inconnue à cinq cheminées ampoule à marqué 0, 1, 2, 3, & 4. Dans cet exemple, la concentration inconnue est 0,110 ppm.
- Ajouter 0, 1.00, 2.00, 3.00 et 4,00 mL de la solution3 + 1,00 ppm étalon Al, respectivement, pour les 5 cheminées avec une pipette 1 mL.
- Préparer un blanc en ajoutant 25,00 mL d’eau distillée à l’ampoule à décanter étiquetée BL.
- Ajouter 1,0 mL de la solution de 8-Hydroxyquinoléine avec une pipette à chacune des six solutions.
- Ajouter 3,0 mL de solution tampon avec une pipette à chacune des six solutions.
- Extraire chaque solution deux fois avec 10 mL de chloroforme, secouer vigoureusement pendant 1 min à chaque fois. N’oubliez pas de ventiler occasionnellement l’ampoule à décanter pour libérer l’accumulation de pression. (Remarque : une bonne extraction n’a lieu que lorsqu’il y a beaucoup de liquide-liquide de contact entre les phases).
- Recueillir le chloroforme dans un bécher étiqueté de 100 mL propre et sec. Chloroforme a une densité de près de 1,5 g/cm3, donc c’est la couche inférieure. Il ne devrait y avoir aucune trace de gauche de couleur jaune dans la phase aqueuse après une extraction complète.
- Transférer le combinés de chloroforme extrait chaque bécher dans leur respectif fiole jaugée de 25 mL et compléter au trait avec du chloroforme. N’oubliez pas de placer des bouchons dans chaque fiole pour garder toute chloroforme de s’évaporer.
- Ajouter environ 1 g de sulfate de sodium anhydre (Na2SO4) à chacun des six béchers de 100 mL de l’étape 2,9. Le sulfate de sodium aide à éliminer toute trace d’eau qui peut-être être présent dans l’extrait de chloroforme.
- Transférer les solutions à leurs gobelets respectés. Agiter soigneusement pour faciliter la déshydratation de l’eau dans l’échantillon.
- Décanter les extraits de chloroforme dans une cellule de fluorimètre quartz (Note : chloroforme se dissoudra une cellule en plastique polystyrène).
3. sélectionner la longueur d’onde d’Excitation
Déterminer les longueurs d’onde d’excitation et d’émission en exécutant des analyses, puis simplement lire et enregistrer l’intensité de la fluorescence de tous les échantillons à ces valeurs. Les bandes passantes d’excitation et d’émission sont préréglées à 5 nm. Le complexe absorbe dans l’UV proche, donc la longueur d’onde d’excitation doit être environ 385 nm. Au début, surveiller la fluorescence à 500 nm de la branche des émissions.
- Sur le fluorimètre, vérifiez que les deux les internes et externes ventilateurs de refroidissement dans le fluorimètre sont allumés avant d’allumer la Lampe xénon. La Lampe xénon devient très chaude et nécessite un refroidissement continu.
- Activez la haute tension (HT) pour le détecteur PMT à 400 V.
- Ouvrez les deux des volets.
- Ouvrez le programme d’acquisition de données sur l’ordinateur, c’est ici « 100nmFluorScan ».
- Placez le « Sample + 2 mL ajouté » solution (2) dans la cellule de quartz à utiliser pour déterminer les longueurs d’onde d’excitation et d’émission exemplaires.
- Avec la longueur d’onde d’émission initialement fixé à 500 nm, exécuter une analyse de l’excitation de 335-435 nm avec une vitesse de balayage de 2 nm/s.
- De la parcelle résultant de la fluorescence, déterminer maximum longueur d’onde de fluorescence excitation (EXλmax) et la valeur de l’instrument à cette valeur.
4. sélection de la longueur d’onde d’émission
- Définissez la longueur d’onde d’émission de fluorimètre à 450 nm.
- Définissez la plage de longueur d’onde d’émission à scanner 100 nm de 450 à 550 nm.
- Pour lancer la numérisation, cliquez sur le bouton de « début du procès » sur le programme en même temps après avoir appuyé sur le bouton « Démarrer » sur le panneau avant du fluorimètre.
- De la parcelle résultant de la fluorescence, déterminer la longueur d’onde des émissions maximales de fluorescence (EMλmax) et la valeur de l’instrument à cette valeur (Figure 2).

La figure 2. Détermination optimale EXλmax et EMλmax longueurs d’onde.
5. mesure de la Fluorescence des échantillons
- Tous les échantillons sont exécutés à EMλmax et EXλmax. Les analyses ne sont pas nécessaires pour chaque échantillon, mais seulement la valeur de fluorescence à ces conditions. À partir de l’échantillon plus diluée (vide), placer dans une cellule de quartz, puis dans l’instrument. Enregistrer l’intensité de fluorescence dans le cahier de laboratoire.
- Répétez pour tous les autres échantillons.
- N’oubliez pas que l’intensité Relative de vide doit être soustraite de l’intensité Relative de chacune des solutions avant de créer le tableau de calibrage.
6. créer l’intrigue des ajouts dosés
- Tracer l’intensité de fluorescence vs µg de Al3 + ajouté.
- Déterminer la valeur de la méthode des moindres carrés de l’intrigue qui en résulte et enregistrer la pente et l’intersection.
- Déterminer les µg de Al3 + dans l’échantillon inconnu de l’équation µg Al3 + = -b/m
- Sachant que l’aluminium inconnu avait un volume de 25,0 mL ajouté à chaque échantillon, déterminer la concentration de l’aluminium dans l’inconnu.
La méthode des ajouts dosés est une technique d’analyse quantitative permettant de minimiser les effets de matrice qui interfèrent avec les signaux de mesure d’analyte.
Les concentrations inconnues composant sont souvent élucidées grâce à un éventail de techniques analytiques, telles que la spectroscopie lumineuse, spectrométrie de masse et l’électrochimie. Toutefois, la mesure peut être affectée par d’autres composants de l’échantillon, appelée la matrice et provoquer la réduction involontaire ou l’amélioration du signal, appelé les effets de matrice. Ces effets peuvent biaiser les résultats et entraîner d’importantes erreurs dans l’analyse.
La méthode des ajouts dosés peut être utilisée pour minimiser les effets de matrice sur les signaux de mesure. Ceci est effectué en ajoutant des volumes précis d’une solution d’analyte connues à l’échantillon.
Cette vidéo va introduire les bases de la méthode d’ajout de normes et montrent comment effectuer la technique en laboratoire au moyen d’une mesure de fluorescence.
Les effets de matrice peuvent survenir dans des échantillons complexes où un certain nombre d’autres molécules interagissent avec l’analyte. Par exemple, cela peut se produire lorsque les molécules se lient ou s’agglomérer avec l’analyte, changeant ainsi sa capacité à fluorescence. Ou de la matrice peut modifier la force ionique de la solution globale, en modifiant les propriétés spécifiques de l’analyte.
Pour atténuer ces effets à l’aide de la méthode des ajouts dosés, une gamme de volumes d’une solution étalon de l’analyte est ajoutée à volumes égaux de l’échantillon. Les volumes de la solution sont alors apportées égale à l’aide de solvants.
Ensuite, le signal est mesuré pour les échantillons avec et sans l’ajout d’un étalon. Les données sont tracées en intensité par rapport à la quantité de la norme a ajouté à l’échantillon, plutôt qu’une courbe d’étalonnage classique. La concentration réelle de l’analyte dans n’importe quel ballon donné est définie par l’équation suivante. La réponse instrumentale est égale à certains moments constantes la concentration de l’analyte. L’équation résultante prend la forme linéaire y = mx + b. Ainsi, quand l’intrigue est extrapolé à zéro d’absorbance, le point d’intersection est égale à la concentration inconnue de l’échantillon.
L’intrigue de signal doit être linéaire dans l’intervalle de concentration de préoccupation. En outre, l’interférence ne devrait pas varier comme le ratio de l’analyte aux changements de matrice d’échantillon. Enfin, la matrice elle-même ne doit pas générer des signaux de mesure sur ses propres.
Ce qui suit expérimenter des études en aluminium, une espèce non fluorescent, par il réagissant avec 8-hydroxyquinoline ou 8HQ, pour former l' ALQ fluorescent3 complexes.
La fluorescence de l’aluminium complexe dans un solvant organique est mesurée et ensuite liée à la concentration de la solution d’origine en aluminium. Cette approche est commune dans l’analyse des ions métalliques.
Maintenant que les bases de la méthode des ajouts dosés ont été soulignées, et a expliqué les bases de l’expérience, nous allons effectuer la technique en laboratoire.
Tout d’abord, préparer la solution mère de 100 ppm d’aluminium dans l’eau et puis l’utiliser pour préparer une solution titrée de 1 ppm.
Ensuite, ajouter 2 g de 8-Hydroxyquinoléine ou 8HQ, dans une fiole jaugée de 100 mL.
Soigneusement ajouter 5,74 mL d’acide acétique cristallisable, puis diluer à la marque de 100 mL avec de l’eau désionisée. Cette étape permet la 8HQ se dissoudre dans la phase aqueuse.
Ensuite, préparer le tampon en ajoutant 20 g d’acétate d’ammonium et 7 mL d’hydroxyde d’ammonium 30 % sur une bouteille marquée à 100 mL et diluer. Vérifier le pH avec un bâton d’indicateur de pH. Ce tampon aide à neutraliser l’acide dans la solution 8HQ lorsqu’il est combiné.
Les autres réactifs nécessaires comprennent sulfate de sodium anhydre et le chloroforme de qualité spectrophotométrique.
Maintenant préparer les échantillons, en l’occurrence par l’extraction de l’échantillon aqueux dans la phase organique à l’aide d’extraction liquide-liquide. Placez les six 125 mL ampoule à entonnoirs anneau-stand anneaux situés à l’intérieur de la hotte. Assurez-vous que toute la verrerie est rigoureusement propre, comme la verrerie sale va fausser les résultats. L’étiquette séquentiellement les cheminées « en blanc », « 0 », « 1 », « 2 », « 3 » et « 4 ».
À l’aide d’une pipette, ajouter 25 mL de la solution inconnue en aluminium à chacun des cinq cheminées ampoule à marquée « 0 » à « 4 ». Préparer le flan en ajoutant 25 mL d’eau désionisée à l’entonnoir étiqueté « en blanc ».
Ensuite, ajouter 1, 2, 3 et 4 mL de la solution étalon de 1 ppm pour les cheminées numérotées correspondantes. N’ajouter aucune solution standard pour les cheminées vierges ou 0.
Ajouter 1 mL de la solution 8HQ et 3 mL de solution tampon dans chacune des 6 cheminées.
Effectuer une extraction liquide-liquide en ajoutant 10 mL de chloroforme dans chaque fiole. Vigoureusement l’entonnoir et occasionnellement désaérer l’entonnoir pour libérer l’accumulation de pression. Placez l’entonnoir dans le ring et laisser les couches liquides à séparer.
Ensuite, recueillir la phase chloroformique dans un bécher de 100 mL propre, sec et étiqueté. Étant donné que le chloroforme a une densité plus élevée que l’eau, c’est la couche inférieure dans l’entonnoir.
Transvaser l’extrait de chloroforme dans une fiole jaugée de 25 mL et cap chaque fiole pour empêcher l’évaporation.
Effectuer une deuxième extraction liquide-liquide sur le reste de la solution aqueuse, en ajoutant 10 mL de chloroforme dans chaque entonnoir. Secouez l’entonnoir, comme avant, pour transférer un analyte restant à la phase chloroformique. Il ne devrait y avoir aucune couleur jaune à gauche dans la phase aqueuse supérieure.
Répétez la deuxième extraction pour chaque entonnoir, puis recueillir les phases de chloroforme dans correspondant étiqueté béchers. Versez le chloroforme recueilli dans leurs fioles jaugées respectifs et compléter au trait avec du chloroforme fraîche.
Pour enlever la trace de l’eau, ajouter environ 1 g de sulfate de sodium anhydre à chacun des six béchers de 100 mL. Transfert des solutions dans leurs gobelets respectifs et agiter pour faciliter la déshydratation de l’échantillon.
Décanter l’extrait de chloroforme dans une cellule de fluorimètre de quartz.
Mettre en place le fluorimètre selon les instructions du fabricant et régler la tension de 400 V. Ensuite, ouvrez le programme d’acquisition de données sur l’ordinateur.
Échantillon 2 permet de déterminer les longueurs d’onde d’excitation et d’émission exemplaires. Définissez la longueur d’onde d’émission à 500 nm et exécuter une excitation numériser à partir de 335-435 nm, avec une vitesse de balayage de 2 nm/s.
À partir de l’intrigue de fluorescence, déterminer la longueur d’onde maximale pour l’excitation. La valeur de l’instrument à cette valeur de longueur d’onde d’excitation, dans ce cas 399 nm.
Ensuite, déterminez la longueur d’onde d’émission en effectuant une analyse de 450 à 550 nm. De la parcelle résultant de la fluorescence, déterminer la longueur d’onde maximale et la valeur de la longueur d’onde d’émission, dans ce cas 520 nm.
Mesurer chaque échantillon, y compris le blanc à la longueur d’onde d’excitation et d’émission sélectionnée. Enregistrer chaque lecture d’intensité de fluorescence.
Soustraire la fluorescence mesurée de l’échantillon témoin de chacune des 5 autres échantillons.
Tracer l’intensité de la fluorescence de chacun des cinq échantillons par rapport à la quantité d’aluminium ajoutée à l’échantillon. Déterminer la valeur de la méthode des moindres carrés de l’intrigue qui en résulte et consigner la pente et l’ordonnée à l’origine.
L’intrigue de l’intensité de fluorescence vs quantité d’aluminium ajoutée a donné une ligne de moindres carrés tel qu’illustré. La quantité d’aluminium dans l’échantillon peut alors être calculée à l’aide de cette ligne. Puisque la quantité d’inconnu ajouté était 25 mL, la valeur déterminée, 2.916 μg est divisé par 25 mL. Cela donne un résultat final de 0,117 µg/mL, soit 0,117 ppm. C’est assez proche de la valeur connue de 0,110 ppm.
Maintenant, regardons quelques autres techniques d’analyse qui peuvent avoir biaisé les résultats en raison des effets de matrice.
Spectroscopie d’absorption atomique est une méthode d’analyse qui mesure l’absorbance de la lumière par un analyte cible en phase gazeuse. Pour la plupart des échantillons, une courbe d’étalonnage simple absorption à la concentration de l’échantillon, peut servir d’une méthode fiable pour quantifier une concentration inconnue.
Cependant, cette technique peut perdre de précision si les autres composants du mélange interagissent avec l’analyte cible et suppriment ou renforcer l’absorption. La méthode des ajouts dosés peut être utilisée dans ce cas pour expliquer les effets de ces interactions, en particulier dans les échantillons où la matrice ne peut pas être retirée avant l’analyse.
Étalonnage de l’instrument joue un rôle crucial dans l’exactitude de la mesure. La méthode des ajouts dosés est souvent utilisée pour aider à l’étalonnage des instruments tels que PIC-Mme ICP-MS est une méthode comparative, ce qui signifie que la mesure d’un échantillon inconnu est basée sur la mesure d’une substance chimique standard.
Ainsi, l’incertitude d’une mesure d’un inconnu ne peut pas être mieux que l’incertitude d’étalonnage. La méthode des ajouts dosés peut donc être utilisée pour créer une courbe d’étalonnage qui est plus précis que la méthode standard et tient compte des interactions entre la matrice de l’échantillon.
De nombreuses molécules biologiques sont analysées à l’aide de la chromatographie liquide à haute performance, ou HPLC. HPLC est une technique qui sépare et analyse de mélanges complexes basées sur les propriétés de la molécule comme la polarité, charge et taille. L’heure à laquelle l’analyte quitte la colonne permet à l’utilisateur identifier chaque composant dans le mélange.
Molécules biologiques peuvent souvent interagir dans un mélange et sont grandement affectés par la matrice dans qu'ils sont suspendus. Souvent, la méthode des ajouts dosés est utilisée pour créer une courbe d’étalonnage qui explique ces effets.
Vous avez juste regardé introduction de Jupiter à la méthode des ajouts dosés. Vous devez maintenant comprendre comment effectuer la technique pour tenir compte des effets de matrice dans l’analyse de l’échantillon.
Merci de regarder !
Subscription Required. Please recommend JoVE to your librarian.
Results
Une analyse de la longueur d’onde d’excitation de 335-435 a montré l’absorption plus élevée à 399 nm, donc le monochromateur d’excitation a été fixé pour cette valeur. Puis l’analyse des émissions a été réalisée de 450 – 550 nm, et le signal le plus fort s’est avéré pour être à 520 nm. Ce sont les longueurs d’onde qui sont utilisés pour tous les échantillons.
| Échantillon | Intensité de la fluorescence | Intensité de la Fluorescence a été corrigé |
| Vide | 0,008 | 0,000 |
| Échantillon | 0,128 | 0,120 |
| Échantillon + 1 mL | 0,167 | 0.159 |
| Échantillon + 2 mL | 0,220 | 0,212 |
| Échantillon + 3 mL | 0.260 | 0,252 |
| Échantillon + 4 mL | 0,290 | 0,282 |
Une parcelle de fluorescence (Figure 3) vs µg de Al3 + ajouté (Figure 4) a donné une ligne de carrés de :
Intensité de la fluorescence = 0,0417 x (µg de Al3 + ajouté) + 0.1216
Quantité de Al3 + =-(Y-Int)/pente =-0.1216/0.0417 =-2.916 µg/mL
La quantité d’inconnu ajouté étant 25 mL, puis la valeur de 2.916 µg/mL doit être divisé par 25.
Concentration d’aluminium inconnu = 2.916 µg/mL / 25,0 mL = 0,117 µg/mL = 0,117 ppm
qui est assez proche de la valeur réelle de 0,110 ppm (6,4 % d’erreur).

La figure 3. Fluorescence des échantillons.

La figure 4. L’intrigue de calibration des ajouts dosés.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
La méthode des ajouts dosés est souvent la technique utilisée quand des résultats quantitatifs précis sont souhaitées, utilisées dans l’analyse analytique comme d’absorption atomique, la spectroscopie de fluorescence, ICP-OES et chromatographie en phase gazeuse. Ceci est souvent utilisé lorsqu’il existe des autres composants dans l’échantillon qui provoque soit une réduction ou une augmentation de l’absorbance souhaité pour les résultats quantitatifs. Lorsque c’est le cas, on ne peut pas simplement comparer le signal d’analytes aux normes à l’aide de l’approche traditionnelle d’étalonnage de la courbe. En fait, l’évaluation effet matrice devrait être une partie obligatoire de la procédure de validation.
Lors de l’extraction d’argent de vieux déchets photographiques est un autre exemple où des ajouts dosés peuvent être utilisés. Les déchets contient des halogénures d’argent et peuvent être extraites afin que l’argent peut être récupérée. Par dopage l’inconnu « déchets » avec des quantités connues d’argent, cette méthode peut prévoir le montant de l’argent provenant de la pellicule photographique.
Les travailleurs qui sont exposés au benzène, usines de fabrication sont souvent testés pour vérifier qu’ils sont sans danger en dessous des niveaux acceptés du benzène. Leur urine est testé pour la substance chimique, et c’est la matrice biologique. En outre, la quantité d’analyte répression varie pour différentes personnes, donc un kit de calibrage simple ne fonctionnera pas. Avec la méthode des ajouts dosés, chaque employé peut être testé et évalué avec précision.
Subscription Required. Please recommend JoVE to your librarian.