



Overview
Fuente: Laboratorio del Dr. Pablo Bower - Universidad de Purdue
El método de adiciones estándar es un método de análisis cuantitativo, que a menudo se utiliza cuando la muestra de interés tiene varios componentes que resultan en efectos de matriz, donde los componentes adicionales pueden reducir o aumentar la señal de absorbancia del analito. Resulta en errores significativos en los resultados del análisis.
Adiciones estándar se usan para eliminar efectos de matriz de una medición, puesto que se asume que la matriz afecta a todas las soluciones igual. Además, se utiliza para corregir la química fase separaciones realizadas en el proceso de extracción.
El método se realiza mediante la lectura de la intensidad (en este caso fluorescente) experimental de la solución desconocida y luego midiendo la intensidad de lo desconocido con cantidades variables de estándar conocido añadido. Los datos se trazan como fluorescencia intensidad vs. la cantidad del estándar añadido (lo desconocido, con ningún estándar añadido, se traza en el eje y). La línea de mínimos cuadrados se cruza con el eje x en el negativo de la concentración de lo desconocido, como se muestra en la figura 1.
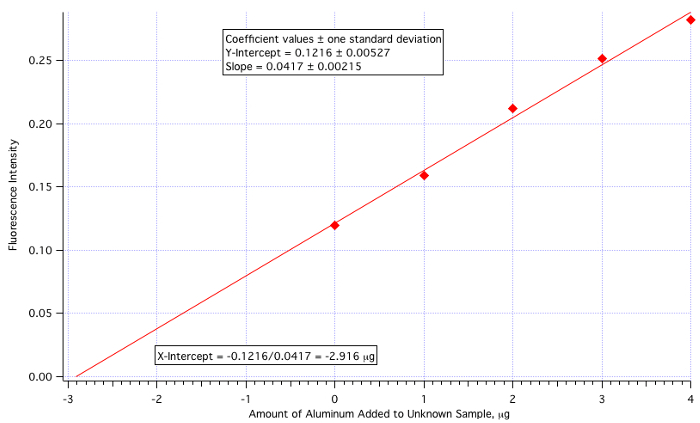
Figura 1. Representación gráfica del método de adición estándar de.
Principles
En este experimento, el método de adiciones estándar se demuestra como una herramienta analítica. El método es un procedimiento para el análisis cuantitativo de una especie sin la generación de una curva de calibración típica. Análisis de la adición estándar se logran midiendo intensidad espectroscópica antes y después de la adición de precisa alícuotas de una solución estándar conocida del analito.
Este experimento estudia las especies no fluorescentes por reaccionar de tal manera que forma un complejo fluorescente. Este enfoque se usa comúnmente en la investigación de los iones del metal. Iones de aluminio (Al3 +) se determinará mediante la formación de un complejo con 8-hidroxiquinoleína (8HQ). Al3 + se precipita por 8HQ de la solución acuosa y luego es extraído en cloroformo; la fluorescencia de la solución de cloroformo es medida y relacionado con la concentración de la solución3 + Al original. Sensibilidad en el rango de parte por millón (ppm o μg/mL) se espera que para este experimento.
La reacción es

La cantidad de aluminio en cada muestra durante este experimento se calcula como sigue:
| En blanco | 0 | ||
| Desconocido + 0 mL estándar | Vdesconocida(Cdesconocido) = 25 mL (Cdesconocido) | ||
| Desconocido + 1 mL estándar | Vdesconocida(Cdesconocido) + Vestándar(Cestándar) = 1 mL (1 μg/mL) 25 mL (Cdesconocido) | ||
| Desconocido + 2 mL estándar | V Desconocido (Cdesconocido) + V Estándar (Cestándar) = 25 mL (Cdesconocido) + 2 mL (1 μg/mL) | ||
| Desconocido + 3 mL estándar | V Desconocido (Cdesconocido) + V Estándar (Cestándar) = 25 mL (Cdesconocido) + 3 mL (1 μg/mL) | ||
| Desconocido + 4 mL estándar | V Desconocido (Cdesconocido) + V Estándar (Cestándar) = 25 mL (Cdesconocido) + 4 mL (1 μg/mL) | ||
Subscription Required. Please recommend JoVE to your librarian.
Procedure
1. preparación de los reactivos
- Al3 + solución estándar de 100 ppm: disolver 0,9151 g de nitrato básico de aluminio (Al (NO3)3•9H2O) en un matraz aforado de 1 L con agua desionizada.
- Solución de 8HQ en 1 M de ácido acético (2% peso/vol): Añadir a un matraz aforado de 100 mL 2,0 g de 8-hidroxiquinoleína.
- Cuidadosamente añadir 5,74 mL ácido acético glacial al matraz de 100 mL y diluir hasta la marca con agua desionizada. Esto permite que la 8-hidroxiquinoleína disolver en la fase acuosa.
- 1 M NH4+/NH3 almacenador intermediario (pH ~ 8): Añadir 20 g de acetato de amonio (NH4OAc) a una botella de 100 mL.
- Agregar 7 mL de hidróxido de amonio 30% a esta botella de 100 mL y diluir hasta la marca con agua desionizada. Esto ayuda a neutralizar el ácido en la solución de 8HQ cuando se combinan.
- Otros reactivos incluyen sulfato de sodio anhidro (Na2SO4) y cloroformo (grado de especificaciones).
2. preparación de las muestras
- Preparar una 1,00 ppm solución patrón de Al3 + al añadir 1,0 mL del stock 100 ppm Al3 + solución con una pipeta a un matraz aforado de 100 mL.
- Coloque seis 125 mL embudos separatory en los anillos que están en un soporte de anillo grande situado en la campana. Debe etiquetarse como sigue: BL, 0, 1, 2, 3, 4. Asegúrese de que toda la cristalería esté escrupulosamente limpia, ya que es difícil obtener resultados cuantitativos si poco gotas de cloroformo se pegan a las paredes del vidrio.
- Agregar 25,00 mL de la solución de Al3 + desconocida a los cinco embudos separatory etiquetado como 0, 1, 2, 3, & 4. En este ejemplo, la concentración desconocida es 0,110 ppm.
- Añadir 0, 1.00, 2.00, 3.00 y 4,00 mL de la 1,00 ppm Al3 + solución estándar, respectivamente, a los 5 embudos con una pipeta de 1 mL.
- Preparar un espacio en blanco añadiendo 25,00 mL de agua destilada en el matraz etiquetado BL.
- Añadir 1,0 mL de la solución de 8-hidroxiquinoleína con una pipeta a cada uno de las seis soluciones.
- Añadir 3,0 mL de solución tampón con una pipeta a cada uno de las seis soluciones.
- Extraiga cada solución dos veces con 10 mL de cloroformo, agitando vigorosamente durante 1 minuto cada vez. No olvide ventilar de vez en cuando el embudo para liberar la acumulación de presión. (Nota: una buena extracción sólo ocurre cuando hay mucho líquido-líquido de contacto entre las fases).
- Recoger el cloroformo en un matraz 100 mL etiquetado de limpio y seco. Cloroformo tiene una densidad de cerca de 1.5 g/cm3, por lo que es la capa más baja. No debería haber ningún rastro de izquierda de color amarillo en la fase acuosa después de una extracción completa.
- Transferir el extracto cloroformo combinada de cada vaso en su respectivo matraz aforado de 25 mL y diluir hasta la marca con cloroformo. Asegúrese de colocar tapones en cada Fiola para evitar cualquier cloroformo se evapora.
- Añadir ~ 1 g de sulfato de sodio anhidro (Na2SO4) a cada uno de los seis vasos de 100 mL en el paso 2.9. El sulfato de sodio ayuda a eliminar cualquier rastro de agua que puede estar presente en el extracto de cloroformo.
- Transferir las soluciones a sus vasos respetados. Agitar cuidadosamente para facilitar la deshidratación de agua en la muestra.
- Decantar los extractos de cloroformo en una celda de cuarzo Fluorímetro (Nota: cloroformo disolverá un plástico poliestireno celular).
3. seleccionar la longitud de onda de excitación
Determinar las longitudes de onda de excitación y emisión ejecutando exploraciones, entonces simplemente leer y registrar la intensidad de fluorescencia de las muestras en los valores. Los anchos de banda de excitación y emisión preajustados de 5 nm. El complejo absorbe en el UV cercano, así que la longitud de onda de excitación debe ser alrededor de 385 nm. Al principio, controlar la fluorescencia de 500 nm en la rama de la emisión.
- En el Fluorímetro, verifique que ambos el interiores y exteriores los ventiladores en el Fluorímetro son encendidos antes de encender la lámpara de xenón. La lámpara de xenón se calienta mucho y requiere refrigeración continua.
- Encenderá el alto voltaje (HV) para el detector PMT a 400 V.
- Abra ambos de las persianas.
- Abrir el programa de adquisición de datos en la computadora, aquí es "100nmFluorScan".
- Coloque el "muestra + 2 mL agregado" (2) solución en la celda de cuarzo para determinar las longitudes de onda de excitación y emisión mejor.
- Con la longitud de onda de emisión establecido inicialmente a 500 nm, ejecutar un análisis de excitación de 335-435 nm con una velocidad de exploración de 2 nm/s.
- De la parcela resultante de la fluorescencia, determinar la máxima longitud de onda de excitación de la fluorescencia (EXλmax) y ajustar el instrumento a ese valor.
4. seleccionar la longitud de onda de emisión
- Seleccionar la longitud de onda de Fluorímetro emisión a 450 nm.
- Establecer el rango de longitud de onda de emisión para ejecutar una exploración de nm 100 de 450-550 nm.
- Para iniciar la exploración, haga clic en el botón "iniciar prueba" en el programa al mismo tiempo de pulsar el botón "START" en el panel frontal de la Fluorímetro.
- De la parcela resultante de la fluorescencia, determinar la fluorescencia máxima emisión de longitud de onda (EMλmax) y ajustar el instrumento a ese valor (figura 2).

Figura 2. Determinar el óptimo EXλmáximo y EMλmax longitudes de onda.
5. medición de la fluorescencia de las muestras
- Todas las muestras van amax EMλ ymaxEXλ. Las exploraciones no son necesarios para cada muestra, pero sólo el valor de la fluorescencia en estas condiciones. A partir de la muestra más diluida (en blanco), coloque en una celda de cuarzo y luego en el instrumento. Registrar la intensidad fluorescente en el cuaderno de laboratorio.
- Repita para el resto de las muestras.
- Recuerde que la intensidad relativa en blanco debe restarse de las intensidades relativas de cada solución antes de crear el cuadro de calibración.
6. creación de la trama de la adición estándar
- Parcela la intensidad de fluorescencia vs μg de Al3 + añadido.
- Determinar el valor de mínimos cuadrados de la parcela resultante y registrar la pendiente y el intercepto.
- Determinar los μg de Al3 + en la muestra desconocida de la ecuación μg de Al3 + = -b/m
- Sabiendo que el aluminio desconocido tenía un volumen de 25,0 mL agrega a cada muestra, determinar la concentración de aluminio en lo desconocido.
El método de adición estándar es una técnica de análisis cuantitativo utilizada para minimizar los efectos de matriz que interfieran con las señales de medición de analitos.
Las concentraciones del componente desconocido son a menudo dilucidar a través de una gama de técnicas analíticas, tales como luz espectroscopia y espectrometría de masas electroquímica. Sin embargo, la medición puede verse afectado por otros componentes de la muestra, llamada la matriz y causar la reducción inadvertida o realce de la señal, llamado efectos de matriz. Estos efectos pueden sesgar los resultados y causar errores significativos en el análisis.
El método de adición estándar puede utilizarse para minimizar efectos de matriz en las señales de medición. Esto se realiza mediante la adición de volúmenes precisa de una solución conocida de analito a la muestra.
Este video se introduce los conceptos básicos del método de adición de estándares y demostrar cómo realizar la técnica en el laboratorio mediante la medición de la fluorescencia.
Efectos de matriz pueden presentarse en muestras complejas donde un número de otras moléculas interactúa con el analito. Por ejemplo, esto puede ocurrir cuando las moléculas se unen o aglomeración con el analito, cambiando así su capacidad de emitir fluorescencia. O la matriz puede cambiar la fuerza iónica de la solución general, cambio de las propiedades específicas del analito.
Para mitigar estos efectos usando el método de adición estándar, se agrega una amplia gama de volúmenes de una solución estándar del analito a volúmenes iguales de la muestra. Los volúmenes de solución son después hace igual usando solvente.
Luego, se mide la señal de las muestras con y sin la adición estándar. Los datos se trazan como intensidad versus la cantidad del estándar añadido a la muestra, en lugar de una curva de calibración clásico. La concentración real del analito en cualquier frasco de dado se define por la siguiente ecuación. La respuesta instrumental será igual algunas veces constante la concentración de analito. La ecuación resultante toma la forma lineal y = mx + b. Así, cuando la trama se extrapola a cero de absorbancia, la ordenada al origen es igual a la concentración desconocida de la muestra.
La trama de la señal debe ser lineal en el intervalo de concentraciones de interés. Además, la interferencia no debe variar la relación del analito a los cambios de la matriz de muestra. Finalmente, la matriz sí mismo no debe generar las señales de medición por sí sola.
El siguiente experimento aluminio de estudios, una especie no fluorescente, por reaccionar con 8-hidroxiquinoleína o 8HQ, para formar el ALQ fluorescente3 complejos.
La fluorescencia del aluminio complejo en un solvente orgánico es medida y luego relaciona la concentración de la solución original de aluminio. Este enfoque es común en el análisis de los iones del metal.
Ahora que se han expuesto los conceptos básicos del método de adición estándar, y explicaron los fundamentos del experimento, vamos a realizar la técnica en el laboratorio.
En primer lugar, preparar la solución madre de 100 ppm de aluminio en el agua y entonces utilizarlo para preparar una solución estándar de 1 ppm.
A continuación, añadir 2 g de 8-hidroxiquinoleína o 8HQ, a un matraz aforado de 100 mL.
Cuidadosamente añadir 5,74 mL ácido acético glacial y diluir a la marca de 100 mL con agua desionizada. Este paso permite la 8HQ disolver en la fase acuosa.
A continuación, preparar el tampón añadiendo 20 g de acetato de amonio y 7 mL de hidróxido de amonio 30% a una botella marcada de 100 mL y diluir. Verificar el pH con un palo de indicador de pH. Este buffer ayuda a neutralizar el ácido en la solución de 8HQ cuando se combinan.
Otros reactivos necesitan incluyen sulfato de sodio anhidro y cloroformo grado espectrofotométrico.
Ahora preparamos las muestras, en este caso mediante la extracción de la muestra acuosa en la fase orgánica utilizando extracción líquido-líquido. Coloque seis 125 mL embudos separatory en anillos de soporte de anillo interior de la campana. Asegúrese de que toda la cristalería está escrupulosamente limpia, como cristalería sucia sesgar resultados. Secuencialmente de la etiqueta los embudos "en blanco", "0", "1", "2", "3" y "4".
Utilizando una pipeta, añadir 25 mL de la solución de aluminio desconocido a cada uno de los cinco embudos separatory con la etiqueta "0" a "4". Preparar el espacio en blanco añadiendo 25 mL de agua desionizada en el embudo con la etiqueta "en blanco".
A continuación, añadir 1, 2, 3 y 4 mL de la solución estándar de 1 ppm a los embudos numerados correspondientes. No añadir ninguna solución estándar a los embudos en blanco o 0.
Añadir 1 mL de la solución de 8HQ y 3 mL de solución buffer a cada uno de los embudos de 6.
Realizar una extracción líquido-líquido añadiendo 10 mL de cloroformo a cada matraz. Agite vigorosamente el embudo y ventilar de vez en cuando el embudo para liberar la acumulación de presión. Coloque el embudo en el anillo y deje que las capas líquidas separar.
A continuación, recoger la fase de cloroformo en un matraz de 100 mL limpio, seco y rotulado. Cloroformo tiene una mayor densidad que el agua, es la capa más baja en el embudo.
Transferir el extracto de cloroformo en un matraz aforado de 25 mL y la tapa de cada frasco para evitar la evaporación.
Llevar a cabo una segunda extracción líquido-líquido en la solución acuosa restante, agregar 10 mL de cloroformo a cada embudo. Agitar el embudo, como antes, para transferir cualquier analito restante a la fase de cloroformo. No debe haber ningún color amarillo en la fase acuosa superior.
Repetir la segunda extracción para cada embudo y recoge las fases de cloroformo en la correspondiente etiqueta vasos. Vierta el cloroformo recogido en sus respectivos matraces aforados y diluir hasta la marca con cloroformo fresco.
Para quitar el agua del rastro, agregar aproximadamente 1 g de sulfato de sodio anhidro a cada uno de los seis vasos de precipitado de 100 mL. Transferir las soluciones en sus respectivos vasos de precipitados y agitar para facilitar la deshidratación de la muestra.
Decantar el extracto de cloroformo en una celda de cuarzo Fluorímetro.
Configurar el Fluorímetro según las instrucciones del fabricante y ajustar la tensión a 400 V. A continuación, abra el programa de adquisición de datos en el ordenador.
Para determinar las longitudes de onda de excitación y emisión mejor utilizar la muestra 2. Ajuste la longitud de onda de emisión a 500 nm y ejecutar una excitación escanear desde 335-435 nm, con una velocidad de exploración de 2 nm/s.
De la trama de la fluorescencia, determinar la máxima longitud de onda de excitación. Ajuste el instrumento a valor de longitud de onda de excitación, en este caso 399 nm.
A continuación, determinar la longitud de onda de emisión mediante la realización de un análisis de 450-550 nm. De la parcela resultante de la fluorescencia, determinar la máxima longitud de onda y establecer la longitud de onda de emisión, en este caso 520 nm.
Medir cada muestra, incluyendo el espacio en blanco en la longitud de onda de excitación y de emisión seleccionado. Grabar cada lectura de intensidad de fluorescencia.
Reste la medida fluorescencia de la muestra en blanco de cada una de las otras 5 muestras.
Parcela la intensidad de fluorescencia de cada una de las cinco muestras versus la cantidad de aluminio que se añade a la muestra. Determinar el valor de mínimos cuadrados de la parcela resultante y registrar la pendiente y la intercepción.
La representación gráfica de la intensidad de fluorescencia vs cantidad de aluminio añadido produjo una línea de mínimos cuadrados como se muestra. La cantidad de aluminio en la muestra puede calcularse entonces con esta línea. Puesto que la cantidad de desconocido añadido fue de 25 mL, el valor determinado, 2.916 μg se divide por 25 mL. Esto da un resultado final de 0.117 μg/mL o de 0,117 ppm. Esto es bastante cercano al valor conocido de 0,110 ppm.
Ahora, echemos un vistazo a algunas otras técnicas analíticas que pueden haber sesgado resultados debido a los efectos de matriz.
Espectroscopia de absorción atómica es un método analítico que mide la absorbancia de luz por un analito blanco en la fase gaseosa. Para la mayoría de las muestras, una curva de calibración simple relacionadas con absorción a la concentración de la muestra, puede servir como un método fiable para cuantificar una concentración desconocida.
Sin embargo, esta técnica puede perder precisión si otros componentes de la mezcla interactúan con el analito blanco y suprimen o mejoran la absorción. El método de adición estándar puede utilizarse en este caso para tener en cuenta los efectos de estas interacciones, especialmente en las muestras donde no se puede quitar la matriz antes del análisis.
Calibración del instrumento desempeña un papel crucial en la precisión de una medición. El método de adición estándar se utiliza a menudo para ayudar en la calibración de instrumentos tales como ICP-MS. ICP-MS es un método comparativo, lo que significa que la medición de una muestra desconocida se basa en la medición de un producto químico estándar.
Así, la incertidumbre de la medición de un desconocido no puede ser mejor que la incertidumbre de la calibración. Por lo tanto, el método de adición estándar puede utilizarse para crear una curva de calibración que es más preciso que el método estándar y es responsable de las interacciones de la matriz de la muestra.
Muchas moléculas biológicas se analizan mediante cromatografía líquida de alto rendimiento, o HPLC. HPLC es una técnica que separa y analiza mezclas complejas basados en las propiedades de la molécula como polaridad, la carga y el tamaño. El tiempo en que el analito deja la columna permite identificar cada componente en la mezcla.
Moléculas biológicas pueden interactuar a menudo en una mezcla y se ve muy afectadas por la matriz que se suspenden en. A menudo, el método de adición estándar se utiliza para crear una curva de calibración que explica estos efectos.
Sólo ha visto la introducción de Zeus para el método de adición estándar. Ahora debería entender cómo realizar la técnica para tener en cuenta para efectos de la matriz de análisis de la muestra.
¡Gracias por ver!
Subscription Required. Please recommend JoVE to your librarian.
Results
Una exploración de la longitud de onda de excitación de 335-435 demostró la absorción más alta en 399 nm, por lo que el monocromador de excitación fue fijado para ese valor. Luego se realizó la exploración de la emisión de 450-550 nm, y la señal más fuerte fue encontrada para ser a 520 nm. Estas son las longitudes de onda que se utilizan para todas las muestras.
| Muestra | Intensidad de fluorescencia | Intensidad de fluorescencia corregida |
| En blanco | 0.008 | 0.000 |
| Muestra | 0.128 | 0.120 |
| Muestra + 1 mL | 0,167 | 0.159 |
| Muestra + 2 mL | 0.220 | 0.212 |
| Muestra + 3 mL | 0.260 | 0.252 |
| Muestra + 4 mL | 0.290 | 0.282 |
Una parcela de fluorescencia (figura 3) vs μg de Al3 + añadido (figura 4) produjo una línea de mínimos cuadrados de:
Intensidad de fluorescencia = 0.0417 x (μg Al3 + añadido) + 0.1216
Cantidad de Al3 + =-(Y-Int)/pendiente =-0.1216/0.0417 =-2.916 μg/mL
Puesto que la cantidad de desconocido añadido fue de 25 mL, entonces el valor de 2.916 μg/mL debe ser dividido por 25.
Concentración de aluminio desconocido = 2.916 μg/mL / mL = 0.117 25,0 μg/mL = 0,117 ppm
que es bastante cercano al valor real de 0,110 ppm (6.4% error).

Figura 3. Fluorescencia de las muestras.

Figura 4. El diagrama de calibración de adición estándar.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
El método de adiciones estándar es a menudo la técnica utilizada cuando se desean resultados cuantitativos precisos, utilizado en Análisis analítico como absorción atómica, espectroscopia de fluorescencia, ICP-OES y cromatografía de gases. Esto se suele usar cuando hay otros componentes en la muestra del interés que provoca una reducción o aumento de la absorbancia para resultados cuantitativos. Cuando esto sucede, simplemente uno no puede comparar la señal de analitos usando el enfoque de la curva de calibración tradicional. De hecho, evaluación del efecto de matriz debe ser una parte obligatoria del procedimiento de validación.
Otro ejemplo donde pueden utilizarse adiciones estándar es cuando la extracción de plata de residuos fotográficos antiguos. La basura contiene Haluros de plata y puede ser extraída para que la plata puede ser reclamada. Por clavar la desconocida "los residuos" con cantidades conocidas de plata, este método puede predecir la cantidad de plata obtenida de la película fotográfica.
Los trabajadores expuestos a benceno fabricación plantas a menudo son probados para verificar con seguridad debajo de los niveles aceptados de benceno. La orina es la prueba de la química, y es la matriz biológica. También, la cantidad de analito supresión varía para diferentes personas, por lo que un kit de calibración único no funcionará. Con el método de adición estándar, cada empleado puede ser probado y evaluado con precisión.
Subscription Required. Please recommend JoVE to your librarian.