



Overview
Source: Perchet Thibaut1,2,3, Meunier Sylvain1,2,3, Sophie Novault4, Rachel Golub1,2,3
1 Unité de lymphopoiesis, Département d'immunologie, Institut Pasteur, Paris, France
2 INSERM U1223, Paris, France
3 Université Paris Diderot, Sorbonne Paris Cité, Cellule Pasteur, Paris, France
4 Flow Cytometry Platfrom, Cytometry and Biomarkers UtechS, Center for Translational Science, Institut Pasteur, Paris, France
La fonction globale du système immunitaire est de défendre le corps contre les organismes infectieux et autres envahisseurs. Les globules blancs, ou leucocytes, sont les principaux acteurs du système immunitaire. Lors de l'infection, ils sont activés et déclenchent une réponse immunitaire. Les leucocytes peuvent être divisés en diverses sous-populations (p. ex. cellules myéloïdes, lymphocytes, cellules dendritiques) en fonction de différents paramètres qui peuvent être biologiques, physiques et/ou fonctionnels (p. ex. taille, granularité et sécrétion). Une façon de caractériser les leucocytes est à travers leurs protéines de surface, qui sont principalement des récepteurs. Chaque population de leucocytes exprime une combinaison spécifique de récepteurs (p. ex. récepteurs cytotoxiques, activants, récepteurs de migration) qui peuvent définir des sous-ensembles entre les populations. Comme le système immunitaire englobe un large éventail de populations cellulaires, il est essentiel de les caractériser pour déchiffrer leur participation à la réponse immunitaire.
La cytométrie de flux (FC ou FCM) est une méthode largement utilisée pour analyser l'expression de la surface cellulaire et des molécules intracellulaires, caractérisant et définissant différents types de cellules dans un mélange hétérogène de cellules. Les cytomètres de débit sont composés de trois sous-systèmes principaux : la fluidique, l'optique et l'électronique. Le système fluidique transporte les cellules dans un flux de telle sorte qu'elles passent devant un laser une par une. Le système optique se compose de sources lumineuses (lasers) pour éclairer les particules, de filtres optiques pour diriger la lumière résultante et de signaux fluorescents vers des détecteurs appropriés. Enfin, le système électronique convertit les signaux lumineux détectés en signaux électroniques qui peuvent être traités par l'ordinateur. Comme une cellule individuelle passe devant le faisceau laser, il disperse la lumière. Un détecteur devant le faisceau mesure la diffusion vers l'avant (FS) et plusieurs détecteurs sur le côté mesurent la diffusion latérale (SC). Le FS est en corrélation avec la taille des cellules et le SC est proportionnel à la granularité des cellules. De cette manière, les populations cellulaires peuvent souvent être distinguées en fonction des différences dans leur taille et leur granularité seulement.
En plus d'analyser la taille, la forme et la complexité d'une cellule, la cytométrie du débit est largement utilisée pour détecter l'expression des récepteurs de surface cellulaire (1). Ceci est accompli en utilisant des anticorps monoclonaux fluorochrome-étiquetés qui se lient aux récepteurs cellulaires-spécifiques connus. Lors de l'excitation, ces fluorochromes liés émettent une lumière de longueur d'onde spécifique, appelée longueur d'onde d'émission, qui peut être détectée et notée. Les mesures de fluorescence fournissent des données quantitatives et qualitatives sur les récepteurs de surface cellulaire étiquetés fluorochrome. Les hématologues ont été les premiers à utiliser FC pour le suivi thérapeutique des populations de cellules immunitaires (2). Maintenant, il est employé pour un large éventail d'applications telles que l'immunophénotypage, la viabilité de cellules, l'expression de gène, le comptage de cellules, et l'analyse de GFP.
LE FACS (Fluorescent Activated Cell Sorter) est un type spécialisé de cytométrie d'écoulement, qui trie une population de cellules en sous-population à l'aide d'un étiquetage fluorescent. Tout comme la cytométrie de flux classique, les premières données FS, SC et fluorescentes sont collectées. Ensuite, la machine applique une charge (négative ou positive) et un système de déviation électrostatique (électroaimants) facilite la collecte des gouttelettes chargées contenant des cellules dans des tubes appropriés.
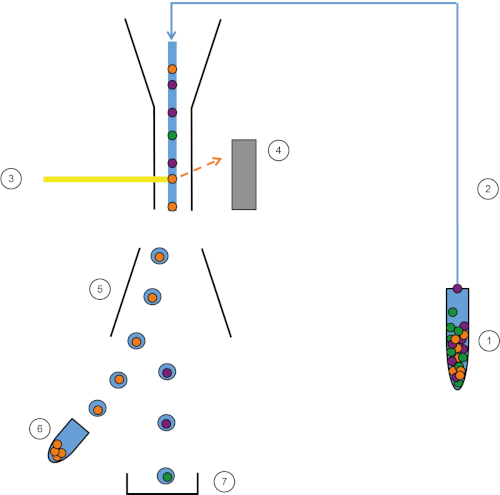
Figure 1 : Représentation schématique du FACS. L'échantillon (1) est aspiré dans le FACS (2) et passé devant le laser (3). La fluorescence cellulaire est détectere par les détecteurs de fluorescence (4). Enfin, les cellules sont incorporées dans des gouttelettes et les cellules d'intérêt sont déviées par des plaques de déviation (5) et collectées dans un tube de collecte (6). Les cellules restantes vont à la poubelle (7). Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
L'aspect tri du FACS présente de nombreux avantages. De nombreux tests peuvent aider à comprendre le rôle de cellules spécifiques dans le système immunitaire, telles que les analyses de l'expression des gènes comme RT-qPCR, cycle cellulaire, ou la sécrétion de cytokine. Cependant, les cellules doivent être purifiées en amont pour obtenir des résultats clairs et spécifiques. Ici, FACS est utile et les cellules désirées peuvent être triées avec une grande pureté, donnant des résultats très fiables et reproductibles. Les FACS peuvent également être utilisés pour trier les cellules en fonction de la coloration nucléaire ou d'autres taches intracellulaires et en fonction de la présence, de l'absence et de la densité des récepteurs de surface. FACS est maintenant une technique standard pour la purification des sous-populations de cellules et a la capacité de trier jusqu'à quatre populations simultanément.
Cet exercice de laboratoire démontre comment isoler les leucocytes spléniques et ensuite comment trier spécifiquement les cellules lymphoïdes B du mélange splénique de cellules de leucocyte utilisant FACS.
Procedure
1. Préparation
- Avant de commencer, enfilez des gants de laboratoire et les vêtements de protection appropriés.
- Stériliser tous les outils de dissection, d'abord avec un détergent, puis avec 70% d'éthanol, puis sécher à fond.
- Préparer 50mL de la solution de sel équilibrée de Hank (HBSS) contenant 2% de sérum foetal de veau (FCS).
2. Dissection
- À l'aide d'un système de distribution de dioxyde de carbone, euthanasiez la souris par hypoxie. Fixez la souris euthanasiée sur une plaque de dissection en position de supine et effectuez une laparotomie longitudinale à l'aide de ciseaux et de forceps.
- À l'aide de forceps, déplacez les intestins et l'estomac sur le côté droit de l'abdomen pour exposer l'estomac et la rate. La rate est attachée à l'estomac.
- À l'aide de forceps, détachez soigneusement la rate de l'estomac et placez-la dans le plat Petri contenant 5 mL de HBSS 2% FCS.
3. Isolement des cellules immunitaires
- Placer la rate dans une passoire cellulaire de 40 m sur le même plat Petri. Écraser la rate avec un piston pour la dissocier dans le même plat.
- Transférer la rate dissociée et le liquide dans un tube centrifugeuse de 15 ml.
- Centrifuger le tube à 370 x g pendant 7 min à 10 oC et jeter le supernatant en évitant le granule.
- Resuspendre la pastille dans 2mL d'acétate de potassium pour lyser les érythrocytes. Attendez 2 min, puis faites le volume jusqu'à 15mL en utilisant HBSS 2% FCS.
- Centrifuger le tube à nouveau à 370 x g pendant 7 min à 10oC. Jetez le supernatant et resuspendre la pastille dans 5mL de HBSS 2% FCS.
- Comptez les cellules à l'aide d'un test de coloration bleu trypan et ajuster la concentration cellulaire finale à 107 cellules/mL en utilisant le volume approprié de HBSS 2% FCS.
4. Staining cellulaire
- Transférer 200 'L de la suspension cellulaire dans six tubes FACS, étiquetés 1 à 6.
- Centrifuger les tubes à 370 x g pendant 7 min à 10oC et jeter le supernatant en évitant la pastille.
- Ensuite, préparez six nouveaux mélanges d'anticorps en ajoutant la quantité appropriée d'anticorps à 200 L HBSS 2% FCS selon le tableau 1.
- Ensuite, transférez ces mélanges d'anticorps dans les tubes FACS numérotés correspondants.
- Incuber les suspensions cellulaires mélangées aux anticorps pendant 20 minutes sur la glace dans l'obscurité.
- Ajouter 1mL de HBSS 2% FCS à chaque tube, puis centrifugeuse à nouveau à 370 x g pendant 3 min à 10oC.
- Jetez le supernatant et suspendez la pastille dans 200 'L de HBSS 2% FCS.
- Transférer les granulés resuspendus sur de nouveaux tubes FACS.

Tableau 1 : Composition du mélange d'anticorps. Six mélanges de 200 oL d'anticorps HBSS ont été préparés pour l'expérience. Mix 1 est pour le réglage PMT, mélanges 2 à 5 sont pour les paramètres de compensation, et le mélange 6 est pour le tri cellulaire.
5. Calibration FACS
- Tout d'abord, allumez le trieur et effectuez «Fluidics Startup».
- Allumez le ruisseau, puis attendez 15 min pour que le flux se stabilise.
- Ajuster l'amplitude du cours d'eau pour obtenir la formation de largage détaché. Ensuite, cliquez sur le "Sweet Spot" pour compléter l'ajustement de l'amplitude.
- Mettez le filtre de densité neutre (N.D.) - 1.0 et ouvrez l'interface «CST» qui signifie configuration et suivi des cytomètres.
- Pour effectuer un contrôle quotidien de la qualité, diluer d'abord les perles CST avec le milieu FACS suivant les instructions du fabricant, puis effectuer le contrôle CST.
- Une fois le contrôle CST terminé, remplacez le filtre N.D. 1.0 par le filtre N.D. 2.0 sur le cytomètre.
- Ensuite, diluer les perles de retard de chute dans le milieu FACS suivant les instructions du fabricant, puis charger dans le FACS.
- Pour s'assurer que le tri approprié effectue Drop Delay-
- Premier clic "Voltage" puis "Optical Filter". Le quadrant droit doit être égal à 100%. Si nécessaire, ajustez la vis laser rouge sur le cytomètre gauche ou droite pour obtenir 100% dans le quadrant droit.
- Ensuite, effectuez un test pour vous assurer que le flux tombe dans le tube de collecte. Pour ce faire, cliquez sur "Waste Drawer" et commencez "Test Sort". Vérifiez que les flux latéraux tombent dans les tubes de collecte. S'ils ne le font pas, réglez la tension jusqu'à ce qu'ils le fassent.
- Naviguez vers le modèle expérimental. Ensuite, ouvrez l'expérience "Accudrop Drop Delay" et cliquez sur "Trier la mise en page".
- Modifier le débit pour obtenir 1000 à 3000 événements par seconde.
- Cliquez sur "Voltage" puis "Filtre optique". Le quadrant gauche doit être égal à 0 et le quadrant droit à 100.
- Dans le "Trier Layout" fenêtre cliquez sur "Trier" et puis cliquez sur "Annuler". Le quadrant gauche doit être égal à 100 et le quadrant droit à 0. Si le quadrant gauche est inférieur à 95 cliquez sur "Auto Delay" pour l'ajuster automatiquement.
6. Cytométrie de flux et contrôle de pureté
- Commencer la cytométrie d'écoulement en commençant par le tube 1 (cellules non tachées) pour définir la morphologie cellulaire et les pics négatifs des fluorochromes. Configurez la diffusion vers l'avant et la partie et définissez les tensions de chaque paramètre fluorescent. Placez la population négative dans la première décennie en utilisant les grilles sur chaque parcelle de point.
- Ensuite, charger le tube 2 (contrôle de couleur unique) dans le cytomètre. Ajustez le chevauchement spectral jusqu'à ce que les médianes de population négatives et positives soient alignées ou utilisez le logiciel de calcul automatique. Il est important de maintenir le signal à l'échelle. Les contrôles de compensation doivent correspondre aux paramètres expérimentaux des fluorochromes et des détecteurs. Enregistrez 10 000 événements.
- Répétez ces étapes avec le tube 3, 4 et 5 (autres commandes de couleur unique).
- Ensuite, chargez le tube 6 (cellules multi-tachées) et définissez les populations cellulaires d'intérêt en utilisant une stratégie de gating spécifique.
- Dans la fenêtre De tri, sélectionnez la population cellulaire d'intérêt. Sélectionnez le seuil cellulaire dans les «événements cibles» et le niveau de précision dans la «Précision». Ici, une seule population est triée, cependant, quatre populations différentes peuvent être triées en même temps.
- Une fois prêt cliquez sur "Sort" et "OK", puis attendez le tri cellulaire.
- Une fois le tri cellulaire terminé, effectuez un contrôle de pureté en proutant d'abord 10 ll de cellules triées dans un nouveau tube FACS avec 90 microlitres de HBSS 2% FCS.
- Ensuite, chargez le tube du cytomètre. Enregistrez et analysez les phénotypes des cellules pour vérifier que la stratégie de gating a fonctionné comme prévu.
7. Analyse des données
- Ouvrez le logiciel 'FlowJo' et faites glisser les fichiers pour chaque tube dans la fenêtre "All sample".
- Double cliquez sur un fichier pour l'ouvrir dans une nouvelle fenêtre.
- Cliquez sur le "Polygone" et recréez la stratégie de gating utilisée précédemment.
- Répétez les étapes avec tous les autres fichiers.
- Pour visualiser les parcelles de points, cliquez sur "Layout editor" et faites glisser les populations d'intérêt du tube 6 et le contrôle de la pureté dans l'onglet éditeur de mise en page. Les cellules ne doivent apparaître que dans la population d'intérêt pour le contrôle de la pureté (voir Figure 2).
- Pour vérifier la pureté des lymphocytes B dans les cellules triées, cliquez sur "Table editor". Population de lymphocytes De drag B du tube 6 et contrôle de pureté dans le tableau.
- Sur "Statistique" menu sélectionnez la fréquence descellules CD45 pour tester la pureté de cette population cellulaire, puis cliquez sur " Createtable".
- Les valeurs de paramètres apparaissent dans une nouvelle table. Dans la fenêtre de contrôle de la pureté, vérifiez la fréquence des lymphocytes B dans les cellules CD45, qui devraient être supérieures à 98 % (voir figure 2, panneau inférieur).
Le système immunitaire protège le corps contre les agents pathogènes envahissants en générant des leucocytes, également appelés globules blancs. Quand un agent pathogène infecte avec succès un organisme, une grande variété de leucocytes sont activés et cette réaction coordonnée est appelée une réponse immunitaire.
Fréquemment, il est utile pour les chercheurs d'être en mesure d'identifier le type spécifique et le nombre de cellules immunitaires qui ont été activées en réponse à un agent pathogène. La cytométrie de flux est une technique qui permet aux chercheurs de séparer les cellules en fonction d'épitopes spécifiques exprimés sur leurs surfaces. Ceci est accompli à l'aide d'anticorps monoclonaux marqués fluorochrome qui se lient à des épitopes spécifiques connus des cellules immunitaires, et lors de l'excitation, ces fluorochromes liés émettent une longueur d'onde de lumière qui peut être détectée et marquée par un cytomètre de flux.
Les cytomètres de flux sont composés de trois systèmes. Le système fluide transporte les cellules dans un flux de telle sorte qu'elles passent devant un laser une par une. Le système optique est composé de lasers et de détecteurs qui reconnaissent la présence ou l'absence des fluorophores. Enfin, le système électronique convertit les données optiques collectées en fichiers électroniques pour analyse.
Une extension de la cytométrie de flux est le Trieur de cellules activé par fluorescence, ou FACS, qui permet l'enrichissement de populations cellulaires spécifiques afin qu'elles puissent être étudiées indépendamment. Le tri cellulaire est effectué à l'aide d'une buse vibrante dans le flux fluidique qui forme des micro gouttelettes, chacune contenant une seule cellule. Ensuite, un détecteur détermine si oui ou non la lumière fluorescente est émise par chaque gouttelette, et sur la base de cette information, un électroaimant donne à chaque cellule une charge négative ou positive. Ensuite, un champ électrique solide trie les gouttelettes chargées différemment dans des récipients séparés. En fin de compte, l'un des conteneurs contiendra une population homogène de cellules basées sur l'expression d'une molécule spécifique de surface cellulaire.
Dans cette vidéo, vous apprendrez à utiliser la cytométrie du débit pour isoler les leucocytes du tissu de la rate de souris et du FACS pour sélectionner les lymphocytes B.
Pour commencer, enfilez des gants de laboratoire et les vêtements de protection appropriés. Ensuite, lavez une paire de ciseaux disséquants et de forceps d'abord avec du détergent, puis avec 70% d'éthanol, puis séchez-les avec un essuie-tout propre.
Ensuite, ajoutez 49 millilitres de la solution de sel équilibré de Hank, ou HBSS, à un tube de 50 millilitres. Ajoutez un millilitre de sérum de veau fœtal, ou FCS, pour créer une solution HBSS 2% FCS et mélangez en pipetting doucement vers le haut et vers le bas environ 10 fois.
Ensuite, placez une souris euthanasiée dans la position de supine sur une plaque de dissection. Avec les ciseaux et les forceps, effectuer une laparotomie longitudinale pour accéder à la cavité abdominale. Utilisez les forceps pour déplacer les intestins sur le côté droit de l'abdomen d'un côté pour exposer l'estomac et la rate. La rate est attachée à l'estomac. Ensuite, avec une pipette, placez cinq millilitres du HBSS 2% FCS dans un plat Petri. À l'aide de forceps, détachez soigneusement la rate de l'estomac et placez la rate dans le plat Petri.
Pour isoler les cellules immunitaires, placez d'abord la rate sur une passoire à cellules de 40 microns dans un plat Petri. Écraser la rate avec un piston pour la dissocier dans le plat. Ensuite, pipette la rate dissociée et le liquide du plat Petri dans un tube de centrifugeuse de 15 millilitres. Centrifuger le tube à 370 fois g pendant sept minutes à 10 degrés Celsius, puis récupérer le tube avec soin afin de ne pas déranger la pastille.
Maintenant, retirez le supernatant, en évitant le granule, et jetez le liquide dans un récipient de déchets approprié. Ensuite, ajoutez deux millilitres de tampon de lysing ACK dans le tube de centrifugeuse pour resuspendre et lyser les érythrocytes. Attendez deux minutes, puis ajoutez HBSS 2% FCS pour obtenir un volume total de 15 millilitres. Répétez la centrifugation. Récupérer le tube soigneusement et jeter le supernatant. Resuspendre la pastille à nouveau en cinq millilitres de HBSS 2% FCS.
Pour compter les cellules suspendues, diluer cinq microlitres de la suspension cellulaire avec cinq microlitres de Trypan Blue. Ensuite, déposez délicatement une goutte de cinq microlitres de cette suspension cellulaire diluée entre le couvre-verre et la glissière Malassez. Maintenant, sous un microscope à 40X grossissement, compter le nombre de cellules présentes. Ensuite, ajuster la concentration cellulaire à 10 à la septième cellule par millilitre en ajoutant le volume approprié de HBSS 2% FCS.
Pour tacher les cellules immunitaires, commencez par étiqueter six tubes FACS de un à six. Ensuite, transférer 200 microlitres de la solution cellulaire dans chacun des six tubes. Centrifuger ces tubes à 370 fois g pendant sept minutes à 10 degrés Celsius et enlever le supernatant.
Ensuite, étiquetez six nouveaux tubes FACS comme un à six et la pipette 200 microlitres de HBSS 2% FCS dans chacun. Préparer les six nouveaux mélanges d'anticorps en ajoutant la quantité appropriée d'anticorps à chaque tube selon la table un. Mélanger un est pour les cellules non tachées sans ajout d'anticorps. Les mélanges de deux à cinq contiennent chacun un anticorps unique différent pour les paramètres de compensation. Mix six contient les quatre anticorps pour les cellules multi-tachées à utiliser pour le tri.
Ensuite, transférez ces mélanges d'anticorps dans les tubes FACS numérotés correspondants. Incuber ces solutions pendant 20 minutes à quatre degrés Celsius ou sur la glace dans l'obscurité. Ensuite, ajouter un millilitre de HBSS 2% FCS à chaque tube, puis centrifugeuse à nouveau. Jeter le supernatant, puis resuspendre les granulés dans 200 microlitres de HBSS 2% FCS. Enfin, transférez les granulés suspendus sur de nouveaux tubes FACS étiquetés.
Pour effectuer FACS, tournez d'abord sur le trieur. Ensuite, sélectionnez le menu cytomètre et cliquez sur fluidics startup. Suivez les instructions à l'écran.
Sur l'onglet flux, cliquez sur la croix rouge pour allumer le flux, puis attendez 15 minutes pour que le flux se stabilise. Ajustez l'amplitude du flux jusqu'à ce que vous voyez une goutte claire détachée apparaître sur l'onglet du flux. Ensuite, cliquez sur sweet spot pour compléter l'ajustement de l'amplitude. Insérez le filtre Neutral Density, ou ND, 1.0 devant le laser.
Ouvrez le menu cytomètre en haut de l'écran et sélectionnez CST, qui signifie configuration cytomètre et suivi. Pour effectuer un contrôle quotidien de la qualité, diluer d'abord les perles CST avec le milieu FACS dans un tube FACS suivant les instructions du fabricant. Ensuite, chargez le tube dans la machine et effectuez le contrôle CST en cliquant sur l'onglet CST.
Lorsque le contrôle CST est terminé, remplacez le filtre ND 1.0 par le filtre ND 2.0 sur le cytomètre. Ensuite, diluer les perles de retard de chute dans le milieu FACS suivant les instructions du fabricant, puis charger le tube dans le FACS. Pour assurer un tri approprié, effectuer le retard de chute en cliquant d'abord sur la tension, puis le filtre optique. Le quadrant droit du filtre optique doit être égal à 100%, ce qui indique que 100% des gouttes sont enregistrées par la machine. Si nécessaire, ajustez la vis laser rouge sur le cytomètre gauche ou droite pour obtenir 100% dans le quadrant droit. Il est important de s'assurer que le cours d'eau tombe dans le tube de collecte. Pour ce faire, effectuer une sorte de test en cliquant sur le tiroir à déchets, puis le tri de test. Vérifiez que les flux latéraux tombent dans les tubes de collecte. S'ils ne le font pas, réglez la tension sous l'onglet de tri jusqu'à ce qu'ils le fassent.
Naviguez vers le modèle expérimental en sélectionnant l'onglet navigateur et en cliquant sur la vue partagée. Ensuite, ouvrez l'expérience Accudrop-DROP DELAY et cliquez sur le bouton de mise en page du tri. Maintenant, modifiez le taux de seuil sur le tableau de bord d'acquisition en manipulant le débit jusqu'à ce qu'il atteigne 3 000 événements par seconde. Cliquez sur la tension, puis cliquez sur filtre optique. Le quadrant gauche doit être égal à zéro et le quadrant droit à 100.
Enfin, dans la fenêtre de mise en page de tri, cliquez sur trier, puis cliquez sur annuler. Le quadrant gauche doit être égal à 100 et le quadrant droit à zéro. Si le quadrant gauche est inférieur à 95, cliquez sur le délai automatique pour demander au logiciel d'augmenter automatiquement la tension pour obtenir 100% des gouttes dans le quadrant gauche.
Pour commencer la cytométrie d'écoulement, nous allons d'abord utiliser des cellules non tachées pour définir la morphologie cellulaire et les pics négatifs des fluorochromes. Pour ce faire, placez le tube un contenant des cellules non tachées dans la machine et sous la charge de clic d'onglet de tableau de bord d'acquisition. Dans l'onglet cytomètre, ajustez les tensions de diffusion vers l'avant et latérales jusqu'à ce que vous voyiez votre population cellulaire comme une concentration dense de points sur l'écran. Les lymphocytes sont de petites cellules, de sorte qu'ils auront une faible diffusion vers l'avant et faible dispersion latérale.
Ensuite, supprimez la fluorescence de fond en ajustant la tension pour les fluorochromes dans l'onglet cytomètre jusqu'à ce que les populations cellulaires à un niveau négatif sont dans la première décennie dans l'onglet feuille de travail globale. Dans le menu cytomètre, cliquez sur la configuration de la vue et vérifiez que tous les fluorochromes sont présents. Ensuite, placez le tube deux dans le cytomètre et cliquez sur la charge. Ajustez le chevauchement spectral dans l'onglet cytomètre jusqu'à ce que les médianes de population négatives et positives soient alignées dans l'onglet feuille de travail globale. Sur l'onglet acquisition, définiz les événements pour enregistrer le paramètre à 10 000 et cliquez sur l'enregistrement. Répétez ces étapes avec des tubes trois, quatre et cinq.
Ensuite, charger le tube six qui contient les cellules multi-tachées. Pour isoler les lymphocytes B, d'abord mettre en place les paramètres pour trier les cellules en fonction de leur morphologie. Dans la première fenêtre, tracez la zone de dispersion vers l'avant FSC-A sur l'axe y et la zone de dispersion latérale SSC-A sur l'axe X. Dans l'intrigue de dispersion, chaque point représente une cellule. Cliquez sur la porte en polygone sur la feuille de travail globale, puis sélectionnez la population avec une faible diffusion vers l'avant et une dispersion latérale intermédiaire. Sur une nouvelle fenêtre d'intrigue point, cliquez à droite sur la fenêtre et sélectionnez afficher les populations du menu et cliquez sur P1.
Puis, dans la nouvelle fenêtre, portez les cellules positives CD45 viables en traçant la viabilité sur l'axe y et CD45 sur l'axe X. Utilisez la porte en polygone pour encercler les cellules avec une faible viabilité et un signal CD45 élevé et sélectionnez P2 pour afficher les cellules sélectionnées dans une nouvelle fenêtre. Dans la fenêtre suivante, porte pour les leucocytes positifs CD45, à l'exclusion des lymphocytes T. Avec CD45 sur l'axe x et CD3 sur l'axe y, encerclez la population avec un signal CD45 élevé et un signal CD3 négatif faible et sélectionnez P3. Enfin, porte pour les cellules positives CD19 qui identifient les lymphocytes B. Avec CD19 sur l'axe y et CD3 sur l'axe x, encerclez la population avec un signal CD19 élevé et un signal CD3 négatif faible et sélectionnez P4.
Tous les paramètres de tri sont maintenant défini. Ensuite, dans la fenêtre de mise en page de tri, sélectionnez votre population cellulaire d'intérêt- P4, qui est la quatrième population qui a été fermée, et dit à la machine de trier uniquement les lymphocytes B. Définir les événements cibles à 10 000 cellules et définir la précision à la pureté. Nous ne trions qu'une seule population. Cependant, jusqu'à quatre populations différentes peuvent être triées en même temps. Une fois prêt, cliquez sur trier et OK. Ensuite, attendez le tri cellulaire.
Une fois le tri cellulaire terminé, effectuez un contrôle de pureté en canalisant 10 microlitres des cellules triées dans un nouveau tube FACS avec 90 microlitres de HBSS 2% FCS. Placez le tube dans le cytomètre, cliquez sur la charge, puis cliquez sur l'enregistrement pour analyser les phénotypes des cellules pour vérifier que la stratégie de gating a fonctionné comme prévu.
Maintenant, nous allons analyser les cellules triées pour déterminer le pourcentage de lymphocytes B parmi les leucocytes qui ont été isolés de la rate de la souris. Pour commencer, cliquez deux fois sur l'icône FlowJo et faites glisser les fichiers de chaque tube dans la fenêtre de l'échantillon.
Cliquez sur le polygone et recréez les stratégies de gating qui ont été utilisées dans la section précédente. Ensuite, cliquez sur l'éditeur de mise en page et faites glisser les populations de lymphocytes B d'intérêt du tube six et le contrôle de la pureté dans l'onglet éditeur de mise en page. Les parcelles de points représentant les lymphocytes B apparaîtront. Dans cet exemple, l'intrigue en haut à droite représente les lymphocytes B triés de la suspension totale des cellules de la rate et la parcelle en bas à droite est le contrôle de pureté. Les cellules ne doivent apparaître que dans la population d'intérêt dans le contrôle de la pureté.
Pour vérifier la pureté des lymphocytes B dans les cellules triées, cliquez sur l'éditeur de table. Faites glisser la population de lymphocytes B du tube six et contrôledez la pureté dans la table. Sur le menu statistique, sélectionnez la fréquence des cellules positives CD45 pour tester la pureté de cette population cellulaire. Ensuite, cliquez sur créer la table. Les valeurs de paramètres apparaissent dans une nouvelle table. Dans la fenêtre de contrôle de la pureté, vérifiez la fréquence des lymphocytes B dans les cellules positives CD45, qui devraient être supérieures à 98%.
Subscription Required. Please recommend JoVE to your librarian.
Results
Dans ce protocole, nous avons purifié les lymphocytes B spléniques à l'aide de la technologie FACS. Nous avons d'abord isolé les leucocytes de la rate et les avons tachés. À l'aide d'une combinaison de marqueurs de surface des cellules B, nous avons créé une stratégie de gating pour les trier (figure 2, panneau supérieur). À la fin de l'expérience, nous avons vérifié si les cellules du tube de collecte étaient des cellules B par le biais d'un « test de pureté ». Nous avons maintenu la même stratégie de gating et observé que plus de 98% des cellules étaient en effet des cellules B (figure 2, panneau inférieur). Ainsi, FACS est un protocole efficace pour isoler les populations de cellules immunitaires avec un haut degré de pureté. Les cellules collectées peuvent ensuite être utilisées pour des expériences en aval telles que la culture cellulaire, RT-qPCR, et les essais de cytotoxicité.

Figure 2 : Stratégie de gating et test de pureté post-tri. (A) Les cellules ont d'abord été fermées en fonction de leur morphologie (à gauche : FSC-A, SSC-A), puis seulement vivantes (au milieu à gauche : viabilité, CD45), CD45et cellules (CD45, CD3) ont été tracées contre CD19 et CD3. Seules les cellules CD19ont été triées. (B) Résultats des tests de pureté d'une fraction des cellules obtenues après le tri cellulaire. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
La cytométrie de flux est une technique de première main pour caractériser et trier les populations de cellules immunitaires avec un haut degré de pureté. C'est un outil primordial dans le domaine de la recherche car il permet l'enrichissement de populations cellulaires spécifiques et de déchiffrer la réponse immunitaire aux agents pathogènes. Avec l'augmentation du nombre de fluorochromes et de cytomètres disponibles, le nombre de paramètres détectables est fortement augmenté. Par conséquent, l'analyse bioinformatique des données du FACS a commencé à émerger et a ouvert de nouveaux horizons à la cytométrie des flux (3). La cytométrie de flux offre d'autres applications en hématologie et en oncologie (4) où elle est utilisée pour développer des outils de diagnostic.
Subscription Required. Please recommend JoVE to your librarian.
References
- Lanier, L. L. Just the FACS. The Journal of Immunology, 193 (5), 2043-2044 (2014).
- Walker, J. M. Epiblast Stem Cells IN Series Editor.
- Tung, J. W., Heydari, K., Tirouvanziam, R., Sahaf, B., Parks. D. R., Herzenberg, L. A., and Herzenberg. L. A. Modern Flow Cytometry: A Practical Approach. Clinics in Laboratory Medicine. 27 (3), 453-468 (2007).
- Walker, J. M. Tumor Angiogenesis Assays IN Series Editor.