



Overview
Source : Michael S. Lee1 et Tonya J. Webb1
1 Département de microbiologie et d'immunologie, University of Maryland School of Medicine et Marlene and Stewart Greenebaum Comprehensive Cancer Center, Baltimore, Maryland 21201
L'immunohistochimie (IHC) et l'immunocytochimie (ICC) sont des techniques utilisées pour visualiser l'expression et la localisation d'antigènes spécifiques à l'aide d'anticorps. La première utilisation publiée de l'IHC a été en 1941 quand Albert Coons a utilisé la technique pour visualiser la présence d'antigène pneumococcique dans les sections de tissus de souris infectées par Pneumococcus (1). Le nom, immunohistochimie, est dérivé des racines "immuno-," en référence aux anticorps, et "histo-," en référence aux sections de tissu utilisées dans IHC. La racine "cyto-" dans l'immunocytochimie met en évidence la différence clé entre l'ICC et IHC. Alors que le CiSE utilise des sections de tissus entiers, l'ICC utilise des cellules qui ont été isolées des tissus ou cultivées en culture. La différence dans les échantillons utilisés signifie que la préparation des échantillons diffère techniquement entre le CSI et l'ICC, mais sinon les protocoles de l'ICC et du CiSE sont identiques et on constatera que les termes sont fréquemment utilisés de manière interchangeable.
Au CSI et à l'ICC, des anticorps munis d'étiquettes chimiques ou fluorescentes, comme la peroxidase ou la rhodamine, respectivement, sont utilisés pour visualiser la distribution de tout antigène d'intérêt par une liaison spécifique de l'anticorps étiqueté à l'antigène. Dans le cas du CSI, de fines tranches de tissu sont immobilisées sur une glissière pour maintenir la structure du tissu avant d'être tachées, ce qui permet la visualisation d'antigènes dans le contexte de tissus entiers (figure 1). Dans le cas de l'ICC, les cellules sont réparties uniformément sur une diapositive avant d'être tachées, ce qui permet la visualisation de la distribution d'antigènes dans les cellules individuelles, mais pas dans la structure d'un tissu spécifique. En raison des similitudes entre les deux protocoles, ce protocole mettra l'accent sur le CSI afin d'aborder les complexités additionnelles de la préparation des échantillons impliquées dans le CSI.
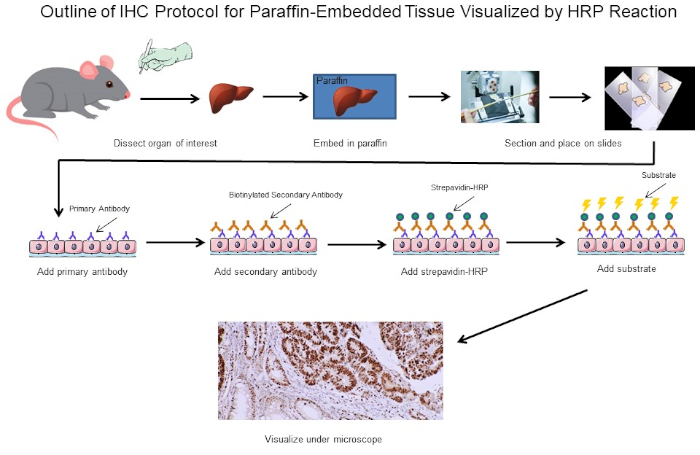
Figure 1 : Aperçu du Protocole du CSI. Contour visuel d'un protocole d'IHC pour le tissu paraffine-incorporé disséqué d'une souris. Ce protocole utilise un anticorps secondaire biotinylated et la strepavidtine-HRP pour visualiser l'emplacement de la liaison d'anticorps. D'autres options, telles que des anticorps étiquetés fluorescents, sont également possibles. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
La première décision importante lors de l'exécution du CSI est de savoir comment préparer les sections tissulaires afin de maintenir la structure du tissu tout au long du processus de coloration. Les deux principaux choix sont des sections fixées par formaline de tissu incorporé à la paraffine ou des sections fraîches de tissu congelé. Il n'y a pas de réponse simple quant à la méthode à utiliser car elle dépend de l'analyse en aval qui sera effectuée. On pense généralement que la fixation formaline des tissus incorporés à la paraffine est généralement mieux préservée de la morphologie tissulaire pour une imagerie optimale, tandis que la congélation des tissus frais peut préserver la fonction protéique pour les essais ultérieurs à l'extérieur du CSI. En outre, il a été démontré que les sections de tissus congelés frais conviennent mieux à l'analyse de l'expression génique (2). Une troisième considération est de savoir si oui ou non les anticorps pour votre antigène d'intérêt sont adaptés pour les sections de tissus fixes ou congelés, comme certains anticorps ont seulement été optimisés pour un type spécifique de section et peut ne pas fonctionner pour d'autres. Enfin, il faut aussi déterminer combien de temps ils ont besoin pour stocker les sections de tissus, car les échantillons congelés frais doivent être conservés à -80 oC et ne peuvent pas durer au-delà d'un an tandis que les sections fixes peuvent être stockées beaucoup plus longtemps à température ambiante. Ce sont quelques-unes des principales considérations pour déterminer s'il faut utiliser des sections fixées par formaline de tissu incorporé à la paraffine ou des sections fraîches de tissu congelé. En fin de compte, si l'on a assez de tissu, il peut être préférable juste d'avoir quelques-uns des deux.
Dans cette expérience, nous avons entrepris de déterminer si l'expression de cyclin D1 a été augmentée dans les rates agrandies d'un modèle spontané de souris du développement de lymphome. Des échantillons de tissu splénique ont d'abord été isolés de souris de type sauvage, de souris transgéniques qui n'ont pas de lymphome, ou de souris transgéniques qui ont développé spontanément un lymphome. Les échantillons de tissu de rate ont été fixés dans le paraformaldéhyde, incorporés dans le paraffine, sectionné, souillé utilisant un anticorps primaire anti-cyclin d'anticyclin de souris suivi d'un anticorps secondaire d'anti-souris de cheval, et développé utilisant 3,3-diaminobenzidine (DAB). Les sections ont ensuite été contre-tachées dans Harris Hematoxylin Solution, puis les sections ont été imaged à 20X grossissement.
Réactifs
Sections paraffines
- 4% Paraformaldéhyde (PFA)
- Éthanol (anhydrous dénaturé, grade histologique 100%, 95%, 80%, 75% et 50%). Peut être dilué à partir de 100% de stock à l'aide d'eau double distillée (ddH2O)
- Xylène
- Lamlede de verre compatible iHC pour s'assurer que la section tissulaire reste attachée tout au long de la procédure. Les lames de verre compatibles IHC ont un revêtement spécialisé et sont facilement disponibles auprès de plusieurs détaillants. Si vous effectuez icc, utilisez une diapositive chambrée. Les glissières chambrées permettent d'ensemencées dans les chambres et placées dans l'incubateur jusqu'à ce que les cellules se fixent à la glissière et atteignent la confluence appropriée, à ce moment-là les chambres peuvent être enlevées et la coloration peut se dérouler de la même manière que le CSI.
- pétrole
- 0,3 % Peroxyde d'hydrogène (H2O2)/méthanol : Pour préparer, ajouter 1 ml 30 % H2O2 à 99 ml de méthanol. Magasinez à -20oC
- Tampon de récupération d'antigène : IHC citrate buffer pH 6.0
Sections fraîches congelées
- Composé optimal de température de coupe (OCT)
- Fixation optimale : 4 % de PFA ou d'acétone refroidi à -20 oC
Coloration
- Tampon de blocage : doit être déterminé par l'utilisateur. Un exemple est le sérum de cheval dilué dans 1X PBS
- Anticorps primaires dilués : voir les spécifications du fabricant
- Anticorps secondaires biotinylated dilués : voir les spécifications du fabricant
- Peroxidase avide-raifort dilué (HRP) : Seulement pour la visualisation de peroxidase. Voir les spécifications du fabricant.
- DAB ou un autre substrat compatible
- Counterstain (facultatif)
- Éthanol (anhydre dénaturée, grade histologique 100% et 95%)
- Xylène
- Mont Organo/Limonène
Procedure
1. Préparation des cellules pour l'immunocytochimie
- Cellules de semences d'intérêt sur des toboggans chambrés ou des plaques de couverture chambrées en ajoutant 0,5 ml de suspension cellulaire aux puits d'une plaque de culture de 24 puits.
Note: Certaines cellules peuvent avoir besoin de croissance sur les couvertures traitées, comme les couvertures traitées à la polylysine. Les conditions de traitement optimales doivent être déterminées par l'utilisateur en fonction du type de cellule utilisé. - Placer la plaque dans un incubateur humidifié de CO2 et permettre aux cellules de se développer à 37 oC jusqu'à 50-70% de confluents.
- Une fois que les cellules atteignent la confluence optimale, retirez le support de culture de chaque puits et puis fixez les cellules en les incuber dans 0.5 ml de 4% PFA (dilué dans 1X PBS) et incubependant pendant 20 min à la température ambiante.
- Retirer le fixatif et laver les puits trois fois avec 1 ml de 1X PBS.
- Ensuite, perméabilisez les cellules en ajoutant 0,5 ml de 0,1 % de Triton-X-100 en 1X PBS à chaque puits et incuber pendant 15 min à température ambiante.
- Aspirer le tampon de perméabilisation et laver les puits trois fois avec 1 ml de 1X PBS.
- Les cellules sur les couvertures sont maintenant fixes et perméabilisées. Procéder à la procédure de coloration démontrée pour l'exemple d'immunohistochimie suivant- à l'exception que les incubations devraient être effectuées dans les puits de la plaque de puits 24 plutôt que directement sur une diapositive de section de tissu.
2. Préparation de sections formalines, paraffines pour la coloration
- Obtenir des sections de tissus incorporées à la formaline fixée s'y prêtent à la paraffine.
Déparaffinisation
- Immerger les lames dans 100% xylène 2 fois pendant 5 min chacun.
Réhydratation
- Immerger les lames dans 100% d'éthanol 2 fois pendant 3 min chacun.
- Immerger les lames dans 95 % d'éthanol pendant 3 min.
- Immerger les lames dans 70 % d'éthanol pendant 3 min.
- Immerger les lames dans 50 % d'éthanol pendant 3 min.
Bloquer l'activité endogène de peroxidase
- Incuber les toboggans en 100 ml de 0,3 % H2O2 pendant 30 min à température ambiante.
- Laver les diapositives avec 1X PBS 2 fois pendant 5 min chacun.
Récupération d'antigène
- Immerger les diapositives dans le tampon de citrate IHC (pH 6) et les faire bouillir pendant 20 min.
- Les glissières de tissu sont maintenant prêtes pour la coloration.
3. Préparation de sections fraîchement congelées et intégrées à l'OTC pour la coloration
- Placer 5 mm de tissu isolé frais dans un moule et ajouter OCT jusqu'à ce que la section soit complètement recouverte.
- Immerger lentement le bloc tissulaire dans l'azote liquide jusqu'à ce qu'il soit complètement congelé. L'échantillon peut maintenant être stocké à -80 oC jusqu'à 1 an.
- Une fois prêt pour la section, transférer le bloc de tissu congelé à un cryostat et permettre à l'ensemble de la configuration de venir à -20 oC.
- Couper des sections de tissu de 5 à 10 m d'épaisseur à l'aide d'un cryostat et utiliser un pinceau pour placer les sections directement sur des lames de verre compatibles au CSI.
- Laisser sécher les glissières toute la nuit à température ambiante. Les diapositives peuvent également être stockées à -80 oC.
- Immerger les glissières dans 250 ml de 4 % de PFA pendant 15 min à température ambiante pour fixer les lames avant la coloration. La méthode de fixation optimale doit être déterminée par l'utilisateur.
- Immerger les toboggans dans 250 ml de 1X PBS 2 fois pendant 5 min chacun.
- Immerger les toboggans dans 250 ml de 0,3 % H2O2 pendant 30 min à température ambiante afin de bloquer toute activité endogène de peroxidase.
- Immerger les toboggans dans 250 ml de 1X PBS 2 fois pendant 5 min chacun.
- Les diapositives sont maintenant prêtes pour la coloration.
4. Staining
- Encerclez le tissu à l'aide d'une barrière hydrophobe à l'aide d'un enclos à barrière.
Blocage
- À l'aide d'une pipette, placer 100 l de tampon de blocage (sérum de cheval dilué en 1X PBS) - sur la section pendant 1 heure à température ambiante.
- Supprimer le tampon de blocage à l'aide d'une pipette.
Incubation primaire d'anticorps
- Incuber le tissu encerclé avec une solution d'anticorps primaire diluée de 100 l (la cycline anti-humaine D1 diluée 1:100 dans le tampon de blocage) pendant 30 min à température ambiante.
- Égoutter l'anticorps primaire de chaque diapositive et laver les diapositives avec 1X PBS 2 fois pendant 5 min chacun.
Incubation secondaire d'anticorps
- Incuber l'échantillon avec 100 l d'anticorps secondaires biotinylated dilués (biotinylated cheval anti-souris IgG dilué 1:200) pendant 30 min à température ambiante.
- Retirez l'anticorps secondaire en égouttant les sections et lavez-les avec 1X PBS 2 fois pendant 5 min chacune.
Développement des couleurs
- Visualisation à l'aide de HRP : Ajouter 100 l de réactif de complexe avidin-biotine (ABC) et incuber les sections dans l'obscurité pendant 30 min à température ambiante
Note: Les anticorps étiquetés fluorescents peuvent également être utilisés et visualisés à l'aide d'un microscope approprié. - Laver les diapositives avec 1X PBS 2 fois pendant 5 min chacun.
- Développer les diapositives en couvant les sections dans 100 OL de DAB pendant jusqu'à 5 min.
- Arrêter le développement en ajoutant de l'eau distillée (dH2O) pendant 5 min à température ambiante.
Contre-coloration (si désiré)
- Immerger brièvement les diapositives dans Harris Hematoxylin Solution (ou 0,5 % de méthyle vert en 0,1 m d'acétate de sodium (pH 4,2)) pendant 10 min.
- Rincer le contre-tache en lavant les lames en dH2O deux fois pendant 5 min chacune.
déshydratation
- Immerger les lames dans 95 % d'éthanol 2 fois pendant 5 min chacune.
- Immerger les lames dans 100% d'éthanol 2 fois pendant 5 min chacun.
- Immerger les lames dans 100% xylène 2 fois pendant 5 min chacun.
- Blot les diapositives avec un essuie-tout.
Application de montage et de couverture
- Ajoutez une goutte de support de montage comme le mont Organo-Limonene aux toboggans et placez une couverture sur les sections.
Analyse microscopique
- Observez les sections tachées au microscope approprié pour analyse. Ici, un microscope léger standard a été utilisé pour l'observation et un appareil photo numérique monté a été utilisé pour l'imagerie.
L'immunocytochimie et l'immunohistochimie sont des méthodes de coloration pour une protéine d'intérêt dans les cellules et les tissus cultivés, respectivement. Le principe de base des deux techniques connexes consiste à utiliser des anticorps spécifiques marqués avec un système de détection pour identifier et visualiser la protéine et déterminer son emplacement dans les cellules et les tissus, ainsi que les niveaux relatifs. Le processus dans l'une ou l'autre expérience commence par la préparation de l'échantillon.
Pour l'immunocyctochimie, qui visualise spécifiquement l'emplacement des protéines ou des antigènes dans les cellules, cela implique trois étapes. La première étape consiste à placage, qui consiste à cultiver les cellules dans les supports de croissance sur un glissement de couverture ou une glissière, généralement, dans les puits d'une plaque de culture. Ceci est suivi par la fixation, où un agent précipitamment ou de liaison croisée comme le paraformaldéhyde est ajouté aux cellules pour préserver l'intégrité structurale des protéines et empêcher l'activité enzymatique de les dégrader. La dernière étape est la perméabilisation, qui consiste à ajouter un détergent pour rendre les membranes cellulaires perméables pour la coloration.
Dans la méthode de contrepartie, l'immunohistochimie, les protéines ou les antigènes sont visualisés dans les tissus et la préparation de l'échantillon comporte cinq étapes. Tout d'abord, le tissu entier est soumis à la fixation, généralement avec du paraformaldéhyde. Ceci est suivi par l'intégration du tissu dans un bloc de paraffine, puis la section de ce bloc à l'aide d'une machine appelée microtome pour couper le tissu en fines tranches qui peuvent être placés sur des diapositives. Ensuite, les diapositives sont soumises à la déparaffinisation, ou l'enlèvement de la paraffine autour de la tranche de tissu. Ensuite, une étape facultative de récupération d'antigène peut être effectuée. Cela peut être fait en utilisant de la chaleur ou des enzymes pour démasquer les épitopes qui ont été reconnectés pendant la fixation les rendant disponibles pour la fixation des anticorps. Après la préparation appropriée de l'échantillon, un anticorps primaire spécifique à la cible est ajouté à l'échantillon de cellules ou de tissus. Cet anticorps primaire doit se lier à la protéine d'intérêt. Ensuite, un anticorps secondaire est ajouté, qui détecte et se lie à l'anticorps primaire. Cet anticorps secondaire est conjugué à une enzyme appelée HRP ou peut se lier à celle-ci. Lorsque son substrat spécifique, le DAB, est ajouté, HRP convertit cela en un précipité brun insoluble. Cette tache brune marque l'emplacement de la protéine cible. Les diapositives sont également tachées d'hématoxylin, qui étiquette les noyaux en bleu et fournit un point de référence spatial pour déterminer la localisation subcellulaire. Après cela, des supports de montage sont ajoutés à la diapositive, suivis d'un bordereau de couverture afin de sceller et de préserver l'échantillon taché. Enfin, les diapositives peuvent être représentées sur un microscope léger.
Dans cette vidéo, vous observerez la technique de préparation de l'échantillon pour les cellules plaquées et les sections tissulaires, suivie de l'immunostaining des sections tissulaires.
Tout d'abord, les cellules d'intérêt doivent être assises sur des couvertures. Pour ce faire, en travaillant dans une hotte de culture de tissu, placez des couvertures individuelles dans les puits d'une plaque de 24 puits. Ensuite, fermez la ceinture et allumez la lumière UV pour stériliser les couvertures pendant au moins 15 minutes. Ensuite, éteignez la lumière UV. Pour soulever les cellules d'intérêt d'un plat confluent de 10 centimètres, aspirer le support, laver brièvement avec du PBS, et ajouter la trypsine aux cellules pendant 2 minutes. Ensuite, appuyez sur le côté de la plaque pour s'assurer que les cellules se sont détachées et neutraliser la trypsine avec des médias. Ensuite, ajoutez 0. 5 ml de la suspension cellulaire dans chaque puits, en veillant à couvrir les couvertures. Placez la plaque dans un incubateur de CO2 humidifié et laissez les cellules se développer à 37 degrés Celsius jusqu'à ce qu'elles soient de 50 à 70 % de confluents.
Une fois que les cellules atteignent la confluence optimale, aspirez le milieu de culture de chaque puits, puis fixez les cellules en les incuber dans. 5 ml de paraformaldéhyde dilué à 4 % en 1X PBS pendant 20 minutes à température ambiante. Après avoir enlevé le fixatif, rincer les cellules ajouter 1 ml de 1X PBS sur chaque coverslip. Immédiatement aspirer le PBS, puis répéter le rinçant 2 fois de plus pour un total de 3 lavages.
Maintenant, perméabliser les cellules en ajoutant 0,5 ml de 0,1% Triton X-100 en 1X PBS à chaque puits. Laisser l'assiette à température ambiante pendant 15 minutes. Aspirer le tampon de perméabilisation, puis rincer les cellules en ajoutant 1 ml de 1X PBS dans chaque puits. Immédiatement aspirer hors du PBS et répéter le rinçant 2 fois de plus pour un total de 3 lavages. Maintenant que les cellules sur les couvertures sont fixes et perméabilisées, procédez à la procédure de coloration démontrée pour l'exemple suivant d'immunohistochimie à l'exception que les incubations devraient être exécutées dans les puits de la plaque de 24 puits plutôt que directement sur une diapositive de section de tissu.
Pour commencer, obtenir des sections de tissus préparées, fixées à la formaline et à la paraffine. Déparaffiniser les diapositives en les plaçant dans un toboggan, puis les plonger complètement dans 250 ml de xylène 100%. Laisser les lames incuber pendant 5 minutes dans le xylène. Ensuite, retirez les lames du contenant, essuyez-les avec un essuie-tout et placez-les dans un nouveau bain de xylène dans un contenant frais pendant 5 minutes supplémentaires.
Ensuite, réhydratez les sections dans une série de solutions d'éthanol gradué s'ensuivent avec 100 % d'éthanol pendant 3 minutes. Essuyez le toboggan à l'eau avec un essuie-tout et transférez les lames dans un nouveau contenant d'éthanol à 100 % pendant encore 3 minutes. Poursuivez ce cycle de lavage, de séchage avec un essuie-tout et transférez les glissières dans un nouveau bain suivant les concentrations indiquées d'éthanol pour le temps spécifié. Après le lavage final à l'éthanol, essuyez la grille à l'aide d'un essuie-tout et incubez les glissières dans 100 ml de peroxyde d'hydrogène de 0,3 % pendant 30 minutes à température ambiante afin de bloquer toute activité endogène de peroxidase. Laver les toboggans en 250 ml de 1X PBS pendant 5 minutes. Répétez ce lavage dans un récipient de 1X PBS frais pendant 5 minutes supplémentaires.
Ensuite, effectuez la récupération d'antigène en immergeant les diapositives dans 250 ml de tampon de citrate IHC au pH 6.0 et en les faisant bouillir pendant 20 minutes. Ensuite, passez au protocole de coloration.
Pour commencer le processus de coloration pour iHC, encerclez les sections avec un stylo hydrophobe pour identifier la zone minimale que le tampon doit couvrir. Ensuite, utilisez une pipette pour placer 100 microlitres de tampon de blocage, qui dans cette expérience est sérum de cheval dilué en 1X PBS, sur la section. Incuber les toboggans pendant 1 heure à température ambiante. Après cela, retirez le tampon de blocage à l'aide d'une pipette.
Ensuite, diluer l'anticorps primaire et bloquer tampon à une dilution 1:100 en ajoutant 990 microlitres de sérum de cheval dilué dans 1X PBS dans un 1. 5 mL eppendorf tube, suivie de 10 microlitres de l'anticorps primaire. Ajouter 100 microlitres de l'anticorps primaire dilué à chaque section, et couver les diapositives pendant 30 minutes à température ambiante. Lorsque la minuterie retentit, égouttez l'anticorps primaire de chaque diapositive, puis lavez-les en 250 ml de 1X PBS pendant 5 minutes. Répétez ce lavage une fois de plus en utilisant frais 1X PBS.
Tandis que les glissières se lavent dans 1X PBS, diluent l'anticorps secondaire à une dilution 1:200 en ajoutant 995 microlitres de tampon de blocage à un tube de 1,5 ml suivi de 5 microlitres de l'anticorps secondaire, qui dans ce cas est biotinylated cheval anti-souris IGG. Ajouter 100 microlitres de l'anticorps secondaire dilué à chaque section, puis couver les diapositives pendant 30 minutes à température ambiante. Après 30 minutes, retirez l'anticorps secondaire en le vidant des sections, puis lavez les glissières en 250 ml de 1X PBS pendant 5 minutes. Répétez ce lavage à l'aide de 1X PBS frais.
Maintenant, ajoutez 100 microlitres de réactif complexe avidin-biotine, et incubez les sections dans l'obscurité pendant 30 minutes à température ambiante. Ensuite, lavez les toboggans en les immergeant dans 250 ml de 1X PBS pendant 5 minutes. Semblable aux étapes de lavage précédentes, répétez ce lavage une fois de plus en utilisant frais 1X PBS. Ensuite, développer les diapositives en couvant les sections dans 100 microlitres de DAB pour un jusqu'à 5 minutes. Arrêtez le développement en immergeant les sections dans 250 ml d'eau distillée pendant 5 minutes.
Maintenant, les diapositives peuvent être contre-tachées, si désiré. Pour ce faire, trempez brièvement les diapositives dans 250 ml de Harris Hematoxylin Solution. Rincer la contre-tache en lavant les lames dans 250 ml d'eau distillée pendant 5 minutes. Répétez ce lavage 1 fois de plus à l'aide d'eau fraîche distillée. Ensuite, déshydratez les sections. Pour ce faire, d'abord couver les diapositives dans 95% d'éthanol pendant 5 minutes. Blot les diapositives sur un essuie-tout, et les transférer dans un nouveau récipient d'éthanol frais 95% pendant un autre 5 minutes. Poursuivre le cycle de lavage, de ballonnement avec un essuie-tout et de transférer les glissières dans un nouveau bain, en suivant les solutions indiquées pendant 5 minutes chacune.
Après l'incubation finale, épongez les diapositives avec un essuie-tout, puis ajoutez une goutte de support de montage, comme le mont Organo-Limonene, aux diapositives. Maintenant, placez un bordereau sur les sections, en prenant soin de ne pas piéger les bulles d'air. Les diapositives sont maintenant prêtes à être observées au microscope pour analyse.
Pour observer les sections tachées, utilisez un microscope léger standard pour visualiser la tache, et un appareil photo numérique pour capturer l'image. Dans cet exemple particulier d'IHC, les tissus de rate de type sauvage et spontanés, double-transgéniques, ou Souris DTG, sont comparés pour étudier l'expression dyclin D1 dans le lymphome. Les tissus étaient paraffins-incorporés, sectionnés, et souillés avec l'anticorps anticyclin D1, et imaged au grossissement 20X. Cyclin D1 cellules exprimantes sont indiqués par la couleur brun-rougeâtre sur le fond du tissu bleu. En comparant les intensités de coloration parmi les images des différentes souris, les rates non agrandies ont des quantités relativement faibles d'expression Cyclin D1 quel que soit le génotype de la souris. En revanche, la rate agrandie de la souris DTG, montre une coloration redâtre-brun accrue indiquant une corrélation entre le développement du cancer et l'expression Cyclin D1 dans ce modèle de souris.
Subscription Required. Please recommend JoVE to your librarian.
Results
Le CSI et la CCI ont une vaste gamme d'applications. Par exemple, une utilisation de IHC est d'examiner l'expression des oncogènes dans les modèles spontanés de souris du développement de tumeur. Dans la figure 2, nous avons entrepris de déterminer si l'expression de la cycline D1 a été augmentée dans les rates élargies dans un modèle spontané de souris de développement de lymphome. Des échantillons de tissu splénique ont été fixés dans le paraformaldéhyde, incorporés dans le paraffine, sectionnés, souillés à l'aide d'un anticorps anti-cyclin D1 (dilué 1:200 dans le tampon de blocage), puis les sections ont été imaged à 20X grossissement. Cyclin D1 cellules exprimantes sont indiqués par la couleur brun-rougeâtre sur le fond du tissu bleu. Ces résultats suggèrent que l'expression de cyclin D1 ait été augmentée dans les rates agrandies, indiquant une corrélation entre le développement de cancer et l'expression de cyclin D1 dans ce modèle.

Figure 2 : Expression splénique Cyclin D1 dans un modèle de lymphome à double transgénique spontané (DTG). Une image du tissu splénique souillé avec un anticorps primaire anti-Cyclin D1, contresifié avec le vert de méthyle, et visualisé utilisant un anticorps secondaire biotinylated et le réactif d'ABC activé avec le substrat de DAB. La couleur rouge-brun représente des endroits où l'anticorps a lié indiquant la présence de Cyclin D1 exprimant des cellules tumorales dans la structure du tissu splénique qui a été contre-taché bleu. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
L'immunohistochimie (IHC) et l'immunocytochimie (ICC) sont des techniques utilisées pour visualiser l'expression et la localisation d'antigènes spécifiques à l'aide d'anticorps. Les tissus sont d'abord coupés en fines sections qui maintiennent la morphologie tissulaire et placés sur une lame. Les anticorps sont ensuite ajoutés et lieront l'antigène d'intérêt et sont équipés d'une étiquette spécifique qui leur permet d'être visualisés au microscope. Ainsi, à travers ce concept de base, la distribution des antigènes dans le contexte de la structure tissulaire peut être visualisée et étudiée. Cependant, bien que le concept global soit fondamental, il existe de multiples approches et variations différentes qui ont été développées qui augmentent à la fois la complexité et l'utilité de ces techniques. Ce document a couvert le concept de base du CSI et de la CPI, les principales décisions qui doivent être prises en considération lors de l'utilisation de ces techniques, et un protocole détaillé étape par étape. Les images produites par l'IHC et l'ICC sont généralement le produit final et peuvent être publiées comme c'est le cas pour mettre en évidence des différences évidentes dans les quantités ou la distribution des taches entre les différentes conditions.
Subscription Required. Please recommend JoVE to your librarian.
References
- Coons, A. H. Creech, H. J., Jones, N. and Berliner, E. The Demonstration of Pneumococcal Antigen in Tissues by the Use of Fluorescent Antibody, The Journal of Immunology, 45 (3), 159-170 (1942).
- Ripoli, F. L., Mohr, A., Hammer, S. C., Willenbrock, S., Hewicker-Trautwein, M., Hennecke, S., Escobar, H. M. and Nolte, I. A comparison of fresh frozen vs. Formalin-fixed, paraffin-embedded specimens of canine mammary tumors via branched-DNA assay. International Journal of Molecular Sciences, 17 (5) (2016).