



Overview
资料来源:乔纳森·布莱泽1号,伊丽莎白·苏特1号,克里斯托弗·科博1号
1大学生物科学系,瓦格纳学院,1 校园路,纽约州斯塔顿岛,10301
鉴于原核生物的丰度、指数增殖倾向、种群内的物种多样性以及特定的生理需求,对原核生物的定量评估可能十分繁重。使这一挑战更加复杂的是细菌复制的四相性质(滞后、原木、静止和死亡)。准确估计微生物浓度的能力对于成功识别、隔离、培养和表征(6)是必要的。因此,微生物学家在一个多世纪以来一直采用连续稀释和各种电镀技术,在临床、工业、制药和学术实验室环境中可靠地量化细菌和病毒载量(2,4,6)。1883年,德国科学家和医生罗伯特·科赫(Robert Koch)发表了他关于致病剂(2)的著作。科赫的引种技术通常被称为现代细菌学之父,已成为全世界可培养或其他微生物的祭标标准。
连续稀释是通过连续将初始溶液(溶液0)重新悬浮成液体稀释剂(空白)的固定体积,系统地还原已知或未知的实体(溶质、有机体等)。这些空白通常由0.45%的盐水组成,虽然成分可以变化(7)。虽然实验者可以为每个稀释剂选择任意体积,但它通常是 10 的倍数,有助于对数减少样本。例如,溶液0共含有100个大肠杆菌细胞,悬浮在10 mL的营养汤中。如果去除溶液0的1mL并加入9 mL的盐碱(稀释剂1),新溶液(溶液1)将含有大肠杆菌初始浓度的1/10。在此示例中,新溶液 (溶液1) 将包含 10个大肠杆菌细胞。重复这个过程,通过去除1mL的溶液1,并将其添加到另外9 mL的盐水(稀释剂2)将产生溶液2,只包含一个大肠杆菌细胞。由于每种新溶液(9 mL稀释剂 = 1 mL溶液)总共含有10mL,我们可以得出结论,这种还原的稀释系数为10倍,或者这是10倍的序列稀释(图1)。由于我们在本例中只从 100 个细胞开始,并且我们稀释了 10 倍,因此只需两个步骤即可达到 1 个细胞的绝对最小浓度。
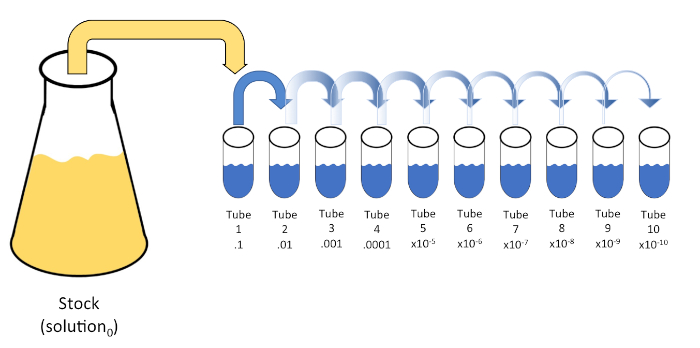
图1:库存溶液的串行稀释。在管1中加入1 mL等分的库存溶液(溶液0),其中含有9 mL的0.45%盐碱(盐分1);这种混合物的产物是溶液1。重复此操作,将新创建的解决方案1的 1 mL 等分,并将其添加到管 2 中。同位和再悬浮继续这种方式,直到达到最终管,稀释股票浓度每一步10倍。请点击此处查看此图的较大版本。
连续稀释是获得所需生物体的可管理浓度的最简单技术,辅以培养皿条纹和扩散,只是微生物学家使用的许多电镀技术中的两种。这种方法的好处是,实验者可以收获单个物种的纯菌株或与混合种群分离菌株(7)。条纹是通过将一个有机体引入固体介质(通常由甘蔗素组成)来完成的,如果适当的营养物质可用,它会生长在这种介质上。以刚性正弦模式轻轻扫过介质的无菌接种回路(使细微的条纹保持),将生物体与实验者波形的频率成比例分布。将培养皿分成三分之三或四分之一(象限条纹),并在进入菜的新区域时降低每个条纹的频率,将逐渐减少可以占据该区域的微生物数量,产生单一菌落,而不是不可量化的细菌草坪。铺板不额外稀释样品;无菌玻璃扩张器用于在整个培养皿中分配悬架介质的等分(图2)。生长在扩散板上的菌落来自单个细胞,盘上的每个菌落可以计算以估计给定悬浮液中每毫升(CFU)的菌落形成单位数,表示为CFU/mL (6) (图3)软琼脂和复制品电镀是上述技术的变化,允许分离噬菌体和突变筛选,分别(1,7)。
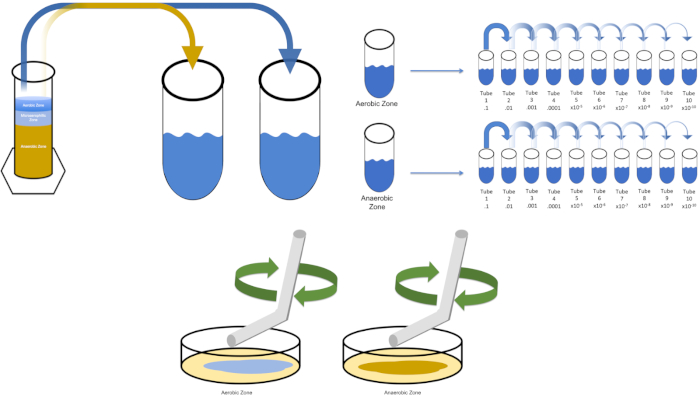
图2:用于细菌枚举和菌株分离的板条纹。用识别信息(细菌名称、日期、介质)标记培养皿的底部,然后分成三份。选择适当的稀释库存样品后,采取无菌(一次性或火焰)接种回路并将其浸入试管(此处,T3)。稍微提高一侧的培养皿盖,以便只有接种回路才能进入琼脂。以锯齿形的方式将接种回路滑过介质顶部,小心不要损害琼脂。将板旋转大约 1/3rd (±118°),并降低锯齿形运动的频率。旋转最后一次,并再次降低锯齿频率。请点击此处查看此图的较大版本。
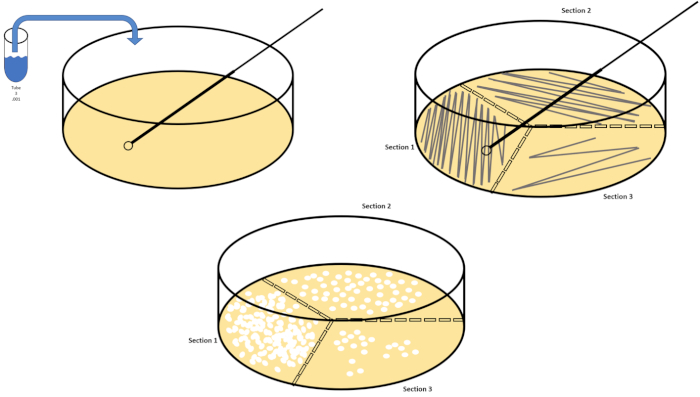
图3:铺板。1克有氧区在T1中重新悬浮,然后连续稀释。无菌玻璃或塑料一次性摊杆用于在每个菜盘中分配接种液。这与1克的厌氧区重复。请点击此处查看此图的较大版本。
与连续稀释一样,使用对数尺度来表达有机体浓度。在标准培养皿中生长的菌落数量为100mm x15mm,可通过识别孤立的生长簇进行手动枚举(或借助计算处理实现自动化)。总数少于 30 或大于 300 的计数应分别定义为计数太少(TFTC) 或计数数量过多而无法计数 (TNTC)。在后者的情况下,应进行连续稀释,以降低浓度,然后再重新处理新的培养皿。从三个单独的培养皿中识别的自足菌落的数量平均乘以稀释系数的平均值将产生CFU/mL;绘制CFU/mL的日志10与时间将揭示生物体的平均生成时间(7)。
Procedure
1. 设置
- 列出所有材料、逐步实验协议和丢弃耗材方法的流程图应写在实验室笔记本上,并保存在实验工作区附近。
- 工作空间应使用适当的防腐剂(70%乙醇)进行消毒,实验者应穿着清洁的实验室服装,防止接触异常,从而减轻污染风险。合适的服装包括但不限于实验室外套、乳胶或尼特手套、谷歌、呼吸器和合闭鞋。始终保持无菌技术至关重要。
- 准备 90 mL 0.45% 盐水。使用清洁的分级钢瓶,测量90 mL的无菌水,并将其转移到一个干净的Erlenmeyer烧瓶标记0.45%盐水。重量为 0.405 g 氯化钠(西格玛-阿尔德里希 NaCl S9888),并将其添加到标有 0.45% 盐水的烧瓶中。反复旋转,直到没有溶质可见。
- 完成后,实验者应重新消毒所有表面,并丢弃任何不需要的生物体、稀释剂、培养皿或一次性接种回路。"在洗手前,可以去除实验室服装。
2. 媒体准备
- 选择适合培养所需有机体的介质。在大多数情况下,肉汤可以促进足够的细菌生长。由于这里需要来自维诺格拉茨基协议的生物体,因此组装了由碳酸钙、硫磺、纤维素和泥浆组成的柱,并不受干扰7天。前名列分为有氧、微嗜和厌氧部分。
- 选择适合电镀感兴趣的有机体的介质。用微生物级琼脂补充液体介质通常用作凝固剂。从前称柱的有氧、微嗜热和厌氧区域采集样品时,LB 培养基/琼脂足够。注:未为这一程序采集来自微亲子区的样品。然而,这些生物应该在蜡烛罐中培养。在密封前将蜡烛引入这个培养室,可营造适合微亲亲菌扩散的低氧环境。
- 由于我们希望准备 250 mL,请使用 500 mL(或更大)Erlenmeyer 烧瓶,以防止高压灭菌时沸腾。标记一个"Broth",另一个标记"Agar"。
- 按照制造商浓度建议确定创建每个解决方案所需的介质量。LB Agar,这里使用,是结合25克/升与超纯水。我们的体积为 250 mL,需要 6.25 LB Agar/250 mL 水的解决方案。同样,LB Broth 是通过将 LB Broth 和水的相同比例组合而成的。因为它没有补充凝固剂,在冷却时不会变硬。
- 称量介质,并将其与水混合,其比例符合制造商的建议。在标有"Agar"的烧瓶中加入 6.25 克 LB Agar,在标有"Broth"的烧瓶中加入 6.25 克 LB Broth。在每个烧瓶中加入250 mL超纯水。
- 将铝箔包裹在每个烧瓶上,并使用高压灭菌器在 121°C、15 psi 下对介质进行至少 15 分钟的消毒。
- 使用耐热手套或垫,在循环完成后从高压釜中取出烧瓶,并将其放入 40-50°C 水浴中。
- 达到适当温度后,将标有"Broth"的烧瓶内的物品倒入 250 mL Erlenmeyer 或圆底烧瓶中。标注 250 mL 烧瓶"溶液0"。
- 获取 10,100mm x 15 mm 无菌培养皿,并标记它们的日期、名称、使用的介质类型和生物体的 Winogradsky 柱区。
- 从水浴中取出标有"Agar"的烧瓶,开始倒入 10 个培养皿中。每道菜的加餐不应超过15 mL。这也可以使用移液器和 25 mL 血清移液器执行,以提高精度。使用无菌移液器尖端去除任何气泡,然后用板盖盖住,留过夜凝固。
3. 稀释剂制备
- 准备 10 个能够储存在机架中的 20 mL 或更多的试管,并贴上 T1-T10 标签。每个管号与其对应的稀释系数一致(即 T4 = 1x10-4或 0.0001 或 1/10,000 的库存浓度)。
- 将 9 mL 的 0.45% 盐水放入 10 个试管中。
- 盐碱坯现在已准备好用高压灭菌器消毒。使用铝箔覆盖 10 个试管中的每一个,然后将它们转移到与高压灭菌器兼容的试管机架上。在 121°C、15 psi 下至少消毒 15 分钟。
- 使用耐热手套取下毛坯,然后冷却。盖上盖子,在4°C下存放,直到需要当管达到室温时,或当冷却到触摸。
4. 培育目标有机体
- 用先前条纹的板或冷冻股票的50 μL的单一菌群接种"溶液0"。通过在37°C的孵化器中放置接种的"溶液0",让目标生物体有时间进行复制(如有必要)。(注意:应盖住烧瓶以防止污染。如果目标生物体有氧运动,请使用无菌纱布和棉塞来防止污染。如果评估维诺格拉茨基柱的区域,只需从每个所需区域中取出 1 克(本研究目的为有氧和无氧),并在 T1 中重新悬浮,然后再继续步骤 5.3。
5. 串行稀释
- 从培养箱中获取标有"营养汤"的烧瓶,然后剧烈摇动。
- 管液1 mL的"溶液0"进入标有T1的试管。漩涡T1。如果评估维诺格拉茨基的外植,称体重1克的所需区域,并在涡旋前将其添加到T1。(注意:这里使用 1 mL 是为了简单起见 - 也可以使用较小或更大的稀释剂。
- 从试管 T1 中取出 1 mL 并将其添加到试管 T2 中。漩涡T2。
- 从试管 T2 中取出 1 mL 并将其添加到试管 T3 中。漩涡T3。
- 从试管 T3 中取出 1 mL 并将其添加到试管 T4 中。漩涡T4。
- 从试管 T4 中取出 1 mL 并将其添加到试管 T5 中。漩涡T5。
- 从试管 T5 中取出 1 mL 并将其添加到试管 T6 中。漩涡T6。
- 从试管 T6 中取出 1 mL 并将其添加到试管 T7 中。漩涡T7。
- 从试管 T7 中取出 1 mL 并将其添加到试管 T8 中。漩涡T8。
- 从试管 T8 中取出 1 mL 并将其添加到试管 T9 中。漩涡T9。
- 从试管 T9 中取出 1 mL 并将其添加到试管 T10 中。
6. 铺板
- 将T1稀释的样品直接移至培养皿中。此步骤可以,但不需要,重复为每个管。
- 获得无菌的一次性扩散杆或火焰消毒玻璃扩散棒。在顺时针/逆时针运动中,滑动摊杆的水平部分,将样品均匀地分布在培养皿中。
- 对要评估的维诺格拉茨基列的每个区域重复上述步骤。
- 在37°C培养箱中孵育板24小时。对于厌氧生物,请使用厌氧室。
7. 条纹
- 选择目标生物体的适当稀释。例如,溶液4将产生初始浓度的1/10,000稀释。通常,对 1/1,000th (T3/解决方案)、1/1,000,000th (T6/溶液6)和 1/1,000,000(T9/溶液9)的稀释被评估为列举微生物。
- 使用塑料无菌一次性接种回路或受火时间不少于 10 秒的可重复使用的金属接种回路,从步骤 5 浸入所需的溶液中。校准的接种回路应传输 0.01 mL。(注意:从热中取出后,不要让火焰循环立即接触细菌)
- 稍微提高一侧的培养皿盖,以便只有接种回路才能进入琼脂。以锯齿形的方式将接种回路滑过介质顶部,小心不要损害琼脂。放下培养皿盖。
- 使用新的一次性接种回路或重新消毒可重复使用的回路。
- 将板旋转大约 1/3rd (±118°),并降低锯齿形运动的频率。
- 同样,使用新的一次性循环或在旋转最后一次之前对金属回路进行重新消毒,并再次降低锯齿形频率。放下培养皿盖。
- 重复步骤7.2 - 7.6,直到至少三个培养皿被条纹三个不同的稀释,使用一个新的一次性循环或重新燃烧一个可重复使用的循环(图2)。
- 将带条纹的培养皿放在 37°C 培养箱中过夜。对于厌氧生物,使用厌氧室。
8. 数据分析和结果
- 文化是从7天维诺格拉茨基柱的氧和缺氧区收获的。这些区域分别适用于异营养化和铁氧化性厌氧。
- 在LB Agar板条纹或扩散之前,柱外植物被连续稀释。
- 条纹揭示了来自每个被评估的维诺格拉茨基区的混合种群。铺板也产生了类似的结果。
- 要计算 CFU/mL 或 CFU/g,从三个板中平均计数菌落数。将平均菌落数乘以稀释系数,再除以数量。例如,如果在接种溶液6(T6)0.1 mL的板上平均计算65个菌落,则前面描述的公式等于650,000,000 CFU/mL。
- 现在可以从每个板块中选择孤立的菌落,用于浓缩测定,以确定物种特性。
有时,为了识别和研究细菌,我们首先需要从样本中分离和丰富细菌。例如,从维诺格拉茨基柱获得的样本是混合的,这意味着它们包含多种细菌或菌株,因此研究单个细菌或列举存在的不同种类可能具有挑战性。为此,通常采用连续稀释和电镀技术可靠地量化细菌负荷和分离单个菌落。
连续稀释是一个过程,通过这一过程,通过连续悬浮在固定体积的液体稀释剂中,生物体(本例中所示的细菌)的浓度被系统地降低。通常稀释剂的体积是10的倍数,以促进样本生物体的对数减少。例如,首先从维诺格拉茨基感兴趣区域去除一克沉积物,并加入到10毫升适当的液体介质中。然后,将一毫升的这种第一稀释剂添加到另一个含有九毫升介质的管中。这个过程可以重复,直到几个不同浓度的细菌已经准备。在这个例子中,连续稀释是枚举细菌的关键,因为来自维诺格拉茨基柱的混合样品含有未知(通常是大量的)细菌。
接下来,条纹电镀和扩散电镀可分别在样品中分离和枚举细菌。条纹是通过将稀释的样品引入固体介质的一部分,辅以营养,这是分为三分之二。然后,此接种以锯齿形模式分布在板的每三分之一上。由于板的不同部分有条纹,与前一个样品仅交叉一次,因此样品分布得更薄。这意味着您可能只需要从一个稀释中条纹,在后面的部分实现单个殖民地。孵化后,条纹板允许观察菌落形态,这些信息可以帮助区分不同的细菌物种。
或者,如果主要目标是枚举细菌在样品传播电镀可以使用。在铺面电镀中,单个样品的等分均匀地分布在固体介质的整个表面上。通常,由于我们不知道混合样品中的细菌数量,因此为每个稀释剂或其中具有代表性的样本制作了一个扩散板。孵育后,可以使用这些扩散板进行枚举。任何菌落计数小于30的板都应该丢弃,因为小盘会有更大的误差。同样,任何超过300的计数都应该被丢弃,因为殖民地拥挤和重叠可能导致对殖民地计数的低估。如果记录每个剩余菜肴的菌落计数,并乘以稀释系数,然后除以镀料,则每毫升悬浮液产生菌群形成单位或 CFU。在本视频中,您将学习如何通过连续稀释、扩散电镀和条纹电镀,对含有已知细菌的样品以及维诺格拉茨基柱各个区域所含的微生物群落进行定性和定量评估。
首先,穿上任何适当的个人防护装备,包括实验室外套、手套和护目镜。接下来,用 70% 乙醇对工作空间进行消毒,然后擦拭表面。接下来,收集两个500毫升的Erlenmeyer烧瓶,并贴上一个汤和一个琼脂的标签。要制备 LB 琼脂溶液,在贴有标签的烧瓶中混合约 6.25 克 LB 琼脂、3 克技术琼脂和 250 毫升蒸馏水。
然后,通过组合 2 准备 LB 肉汤。5 克 LB 介质和 100 毫升蒸馏水,在标有肉汤的烧瓶中。高压灭菌瓶后,使用耐热手套将烧瓶从高压灭菌器中取出,并将其放入 40 至 50 摄氏度的水浴中。一旦烧瓶温度达到 50 摄氏度,请仔细准备三个 100 毫升的肉汤溶液等分,并将每个等分溶液标记为零。接下来,收集10个无菌培养皿,并标记它们的日期,名称,使用的介质类型,和维诺格拉茨基柱区,生物体将从中收获。从琼脂瓶中移入每个培养皿中的15毫升琼脂。然后,使用移液器尖端去除任何气泡,更换板盖,并让他们在台面上过夜凝固。
第二天,用70%的乙醇擦拭台面。接下来,标记 10 20 毫升试管 T1 到 T10,并将其放入机架中。将9毫升的0.45%盐水放入每个管中。现在,用盖子松散地覆盖10个试管中的每一个,并将其转移到与高压灭菌器兼容的试管机架上。循环完成后,使用耐热手套去除盐水坯,使其冷却。将管在室温下存放,直到达到约 22 摄氏度。
为了培育已知的目标生物体,本例中的大肠杆菌,用先前条纹的板中单一菌群接种100毫升溶液零。然后,覆盖管子,并在37摄氏度的温度下孵育。要评估维诺格拉茨基柱的区域,请从有氧区向 T1 添加大约一克材料,然后通过涡旋重新悬浮。然后,用来自厌氧区的一克材料重复此过程。
从培养箱中取出含有大肠杆菌溶液的管,并将其摇动。然后,将一毫升溶液移液放入T1试管和涡旋中混合。从 T1 中取出一毫升溶液并将其转移到 T2,涡旋混合。通过管 T10 重复此过程。要评估维诺格拉茨基柱的有氧和厌氧区,请从先前准备的 T1 管中去除一毫升溶液并将其转移到相应的 T2 管。然后,继续通过 T10 管进行串行稀释,如前面所示。
为了铺板,移液器100微升的稀释样品从每个T3管到相应的培养皿。然后,使用无菌扩张杆将样品轻轻地分发到培养皿上,并更换盘盖。如前所述,对 T6 和 T9 稀释重复此过程。在37摄氏度的培养箱中孵育含有有氧生物的板24小时。在设定为 37 摄氏度的厌氧室中孵育含有厌氧生物的板,24 小时。第二天,从培养箱和厌氧室中取出T3、T6和T9稀释板,并将其转移到工作台顶部。一次使用一个板,以锯齿形模式在介质顶部滑动无菌接种回路。然后,更换培养皿盖。接下来,将板旋转 1/3 并消毒循环,以减少先前制作的锯齿形图案的频率。再次,在消毒循环后,将板旋转 1/3,最后一次降低锯齿形图案的频率,然后更换盖子。如前所述,对其余板重复此条纹方法。然后,将含有有氧生物的带条纹板放在37摄氏度的培养箱中过夜,在厌氧室中设置有氧生物的带条纹板过夜至37摄氏度。
文化是从七天维诺格拉茨基柱的有氧和厌氧区收获的。然后,在LB琼脂板上条纹和扩散之前,这些文化被连续稀释。条纹揭示了来自每个评估的维诺格拉茨基区的混合种群,并且扩散板产生了类似的结果。从混合种群条纹的板将导致不同形状、大小、纹理和颜色的细菌菌落。相比之下,含有已知有机体的条纹和扩散板大肠杆菌则表现出同源种群。通常,最好使用具有相同样本和稀释因子的三个板的平均菌群计数来计算每毫升的CCF。将平均菌落数乘以稀释系数,再除以数量。最后,从每个板块中选择的分离菌落可用于进一步浓缩测定,以确定物种特性。
Applications and Summary
通过电镀进行细菌枚举和菌株分离需要目标生物体的可管理浓度。因此,成功的电镀取决于连续稀释。因此,上述技术仍然是微生物检查和实验的基石。虽然设计简单,但实验者可以修改稀释因子和电镀技术,以在不损害每种方法完整性的情况下支持结果。绘制细菌生长的四个阶段在描述所需微生物时非常有用。这些阶段、滞后、对数、静止和死亡,以细菌复制的变化为标志。滞后阶段由于生理适应而生长缓慢,对数阶段是活细胞呈指数级上升的最大增殖期,由于环境限制和毒素的积累,则达到固定期,在死亡阶段之前,细胞计数开始下降。这可以通过连续稀释(或 1 步稀释以避免混淆)解决方案0每小时共 8 小时来实现,从时间0开始(每次稀释后应将解决方案0返回到摇动培养箱)。计算时间0的单个稀释剂的 CFU/ml 的日志10,并在 Y 轴上绘制。对采样时间1重复此计算(确保使用与时间0相同的稀释系数计算 CFU/mL )。重复,直到每次 (时间1- 时间8) 在 X 轴上绘制。
References
- Allen, M.E., Gyure, R.A. (2013) An Undergraduate Laboratory Activity Demonstrating Bacteriophage Specificity. Journal of Microbial Biological Education 14: 84-92.
- Ben-David, A., Davidson, C.E. (2014) Estimation Method for Serial Dilution Experiments. Journal of Microbiological Methods 107:214-221.
- Goldman, E., Green, L.H. (2008) Practical Handbook of Microbiology.
- Koch, R. (1883) New Research Methods for Detection of Microcosms in Soil, Air and Water.
- Lederberg, J., Lederberg, E.M. (1952) Replica Plating and Indirect Selection of Bacterial Mutants. Journal of Bacteriology 63:399-406
- Pepper, I., Gerba, C., Ikner, L. (2019) Bacterial Growth Curve Analysis and its Environmental Changes. JoVE Science Education Database. Environmental Microbiology.
- Sanders., E.R. (2012) Aseptic Laboratory Technique: Plating Methods. JoVE 63:e3063.