



Overview
Quelle: Ewa Bukowska-Faniband1, Tilde Andersson1, Rolf Lood1
1 Institut für Klinische Wissenschaften Lund, Abteilung für Infektionsmedizin, Biomedical Center, Universität Lund, 221 00 Lund, Schweden
Planet Erde ist ein Lebensraum für Millionen von Bakterienarten, von denen jede spezifische Eigenschaften hat. Die Identifizierung von Bakterienarten wird in der mikrobiellen Ökologie häufig verwendet, um die Artenvielfalt von Umweltproben und medizinische Mikrobiologie zur Diagnose infizierter Patienten zu bestimmen. Bakterien können mit konventionellen mikrobiologischen Methoden wie Mikroskopie, Wachstum auf bestimmten Medien, biochemischen und serologischen Tests und Antibiotika-Empfindlichkeitstests klassifiziert werden. In den letzten Jahrzehnten haben molekulare mikrobiologische Methoden die bakterielle Identifizierung revolutioniert. Eine beliebte Methode ist die 16S ribosomale RNA (rRNA) Gensequenzierung. Diese Methode ist nicht nur schneller und genauer als herkömmliche Methoden, sondern ermöglicht auch die Identifizierung von Stämmen, die unter Laborbedingungen schwer zu züchten sind. Darüber hinaus ermöglicht die Differenzierung von Stämmen auf molekularer Ebene eine Diskriminierung von phänotypisch identischen Bakterien (1-4).
16S rRNA verbindet sich mit einem Komplex von 19 Proteinen zu einer 30S-Untereinheit des bakteriellen Ribosoms (5). Es wird durch das 16S rRNA-Gen kodiert, das in allen Bakterien aufgrund seiner wesentlichen Funktion in der Ribosom-Montage vorhanden und hochkonserviert ist; Es enthält jedoch auch variable Regionen, die als Fingerabdrücke für bestimmte Arten dienen können. Diese Eigenschaften haben das 16S rRNA-Gen zu einem idealen genetischen Fragment gemacht, das bei der Identifizierung, dem Vergleich und der phylogenetischen Klassifizierung von Bakterien verwendet werden kann (6).
16S rRNA Gensequenzierung basiert auf der Polymerase-Kettenreaktion (PCR) (7-8) gefolgt von DNA-Sequenzierung (9). PCR ist eine molekularbiologische Methode, die verwendet wird, um bestimmte DNA-Fragmente durch eine Reihe von Zyklen zu verstärken, die Folgendes umfassen:
i) Denaturierung einer doppelsträngigen DNA-Vorlage
ii) Glühen von Primern (kurze Oligonukleotide), die die Vorlage ergänzen
iii) Erweiterung der Primer durch das DNA-Polymerase-Enzym, das einen neuen DNA-Strang synthetisiert
Eine schematische Übersicht über die Methode ist in Abbildung 1dargestellt.
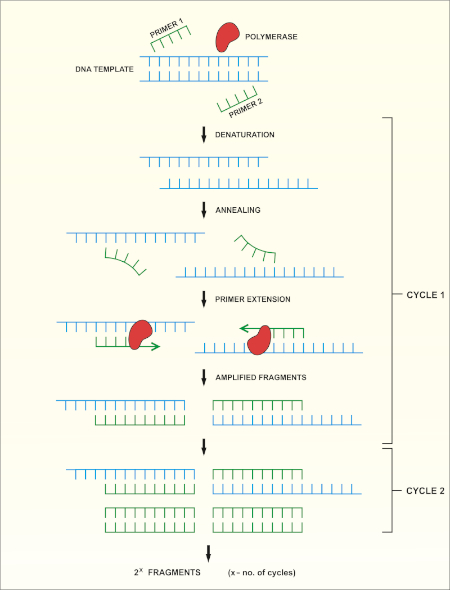
Abbildung 1: Schematische Übersicht über die PCR-Reaktion. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Es gibt mehrere Faktoren, die für eine erfolgreiche PCR-Reaktion wichtig sind, einer davon ist die Qualität der DNA-Vorlage. Die Isolierung chromosomaler DNA von Bakterien kann mit Standardprotokollen oder kommerziellen Kits durchgeführt werden. Besondere Vorsicht sollte darauf geachtet werden, DNA zu erhalten, die frei von Verunreinigungen ist, die die PCR-Reaktion hemmen können.
Konservierte Regionen des 16S rRNA-Gens ermöglichen das Design universeller Primerpaare (ein Vorwärts- und ein Rückwärtsgang), die die Zielregion in jeder Bakterienart binden und verstärken können. Die Zielregion kann in ihrer Größe variieren. Während einige Primerpaare den größten Teil des 16S rRNA-Gens verstärken können, verstärken andere nur Teile davon. Beispiele für häufig verwendete Primer sind in Tabelle 1 dargestellt und ihre Bindungsstellen sind in Abbildung 2dargestellt.
| Primername | Sequenz (5''3') | Vorwärts/ Rückwärts | verweis |
| 8F b) | AGAGTGTGATCCTGGCTCAG | vorwärts | -1 |
| 27F | AGAGTGTGATCMTGGCTCAG | vorwärts | -10 |
| 515F | GTGCCAGCMGCCGCGGTAA | vorwärts | -11 |
| 911R | GCCCCCGTCAATTCMTGA | Rückwärts | -12 |
| 1391R | GACGGGCGGTGTGTRCA | Rückwärts | -11 |
| 1492R | GGTTTTTTTACGACTT | Rückwärts | -11 |
Tabelle 1: Beispiele für Standardoligonukleotide, die bei der Amplifikation von 16S rRNA-Genen verwendet werden a).
a) Die erwarteten Längen des PCR-Produkts, das mit den verschiedenen Primerkombinationen erzeugt wird, können durch Berechnung des Abstands zwischen den Bindungsstellen für den Vorwärts- und den Reverse-Primer geschätzt werden (siehe Abbildung 2), z. B. die Größe der PCR Produkt mit Primer-Paar 8F-1492R ist 1500 bp, und für Primer-Paar 27F-911R 900 bp.
b) auch bekannt als fD1

Abbildung 2: Repräsentative Abbildung der 16S rRNA-Sequenz und der Primer-Bindungsstellen. Konservierte Bereiche sind grau gefärbt und variable Bereiche sind mit diagonalen Linien gefüllt. Um die höchste Auflösung zu ermöglichen, werden Primer 8F und 1492R (Name basierend auf der Position auf der rRNA-Sequenz) verwendet, um die gesamte Sequenz zu verstärken, was die Sequenzierung mehrerer variabler Bereiche des Gens ermöglicht. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Die Fahrradbedingungen für PCR(d. h. die Temperatur und Die Zeit, die für die Denaturierung, das Geglüht mit Primern und synthetisiert erforderlich sind) hängen von der Verwendeten-Polymerase-Art und den Eigenschaften der Primer ab. Es wird empfohlen, die Herstellerrichtlinien für eine bestimmte Polymerase zu befolgen.
Nach Abschluss des PCR-Programms werden die Produkte durch Agarose-Gel-Elektrophorese analysiert. Eine erfolgreiche PCR ergibt ein einzelnes Band in der erwarteten Größe. Das Produkt muss vor der Sequenzierung gereinigt werden, um Restgrundierungen, Desoxyribonukleotide, Polymerase und Puffer zu entfernen, die in der PCR-Reaktion vorhanden waren. Die gereinigten DNA-Fragmente werden in der Regel zur Sequenzierung an kommerzielle Sequenzierungsdienste gesendet; Einige Institutionen führen jedoch DNA-Sequenzierungen in ihren eigenen Kerneinrichtungen durch.
Die DNA-Sequenz wird automatisch aus einem DNA-Chromatogramm von einem Computer erzeugt und muss sorgfältig auf Qualität überprüft werden, da manchmal eine manuelle Bearbeitung erforderlich ist. Nach diesem Schritt wird die Gensequenz mit Sequenzen verglichen, die in der 16S rRNA-Datenbank abgelagert sind. Die Regionen mit Ähnlichkeit werden identifiziert, und die ähnlichsten Sequenzen werden geliefert.
Procedure
1. Einrichten
- Beim Umgang mit Mikroorganismen ist es erforderlich, eine gute mikrobiologische Praxis zu befolgen. Alle Mikroorganismen, insbesondere unbekannte Proben, sollten als potenzielle Krankheitserreger behandelt werden. Folgen Sie der aseptischen Technik, um eine Kontamination der Proben, der Forscher oder des Labors zu vermeiden. Waschen Sie die Hände vor und nach dem Umgang mit Bakterien, verwenden Sie Handschuhe und tragen Sie Schutzkleidung.
- Durchführung einer Risikobewertung für das Versuchsprotokoll zur genomischen DNA-Isolierung und PCR-Produktreinigung. Einige Reagenzien können schädlich sein!
- Reine Kultur ist für die 16S rRNA Sequenzierung unerlässlich. Bevor Sie zur Isolierung der genomischen DNA übergehen, stellen Sie sicher, dass das Ausgangsmaterial vollständig rein ist. Dies kann durch Streifenbeschichtung erfolgen, um einzelne Kolonien zu isolieren. Diese können auf Tellern einzeln oder bei Bedarf in Brühe weiter angebaut werden.
- Erforderliche Laborausrüstung:
- Thermischer Cycler für PCR. Die Funktion des Thermischen Cyclers besteht darin, die Temperatur entsprechend einem eingestellten Programm anzuheben und zu senken. Während der Erstellung des Programms werden Sie aufgefordert, die Temperatur- und Zeitwerte für jeden PCR-Schritt sowie die Gesamtzahl der Zyklen einzugeben.
- Agarose-Gel-Elektrophorese-System. Es wird verwendet, um DNA-Fragmente basierend auf ihrer Größe und Ladung zu trennen. In diesem Protokoll wird die Agarose-Gelelektrophorese verwendet, um die Qualität isolierter genomischer DNA- und PCR-Produkte zu visualisieren.
2. Protokoll
Hinweis: Das gezeigte Protokoll gilt für die 16S rRNA-Gensequenzierung aus einer reinen Bakterienkultur. Sie gilt nicht für metagenomische Studien.
-
Culturing Bakterien für die Isolierung der genomischen DNA (gDNA).
- Wachsen Sie Ihren Mikroorganismus auf einem geeigneten Medium. In diesem Schritt können sowohl flüssige als auch feste Medien verwendet werden. Wählen Sie Bedingungen, die das beste Wachstum erzielen. Beachten Sie bei der Planung des Experiments, dass langsam wachsende Bakterien mehrere Tage benötigen, um die späte Log/stationäre Wachstumsphase zu erreichen. In diesem Protokoll wurde Bacillus subtilis 168 in Lysogeny-Brühe (LB) über Nacht in einem Schüttel-Inkubator mit 200 Rpm, 37°C angebaut.
-
Isolierung von gDNA.
- Wenn Bakterien auf festem Medium angebaut wurden, kratzen Sie einige Zellen mit einer sterilen Schleife und setzen Sie sie in 1 ml destilliertem Wasser wieder auf
- Wenn Bakterien in flüssigem Medium angebaut wurden, verwenden Sie etwa 1,5 ml einer Nachtkultur.
- Pellet die Zellen durch Zentrifugation (1 Minute, 12.000 - 16.000 g), entfernen Sie den Überstand und verwenden Sie die Zellen für die gDNA-Isolierung mit einem kommerziellen Kit oder Standardprotokollen[z. B. CTAB-Gesamt-DNA-Präparat (13) oder Phenol-Chloroform-Extraktion (14)]. Hier wurde ein kommerzielles Kit verwendet, um gDNA aus 1,5 ml b. subtilis 168 Übernachtungskultur, OD600 = 1,5 zu isolieren.
Anmerkung 1: Für einige Gram-negative Bakterien kann dieser Schritt weggelassen und durch einfache Freisetzung von DNA aus Zellen durch Kochen ersetzt werden. Bakterielles Pellet in destilliertem Wasser aufbewahren und in einem Heizblock auf 100 °C für 10 Minuten inkubieren.
Anmerkung 2: Gram-positive Bakterienzellen sind schwer zu stören. Es wird daher empfohlen, eine gDNA-Isolationsmethode oder ein Kit zu wählen, das der Isolierung von dieser Gruppe von Bakterien gewidmet ist.
-
gDNA-Qualitätsprüfung.
- Überprüfen Sie die Qualität der isolierten gDNA durch Agarose-Gel-Elektrophorese. Mischen Sie zunächst 5 l der isolierten gDNA mit 1 l des Ladefarbstoffs (6x), und laden Sie die Probe auf ein 0,8% Agarose-Gel, das ein DNA-Färbereagenz enthält.
- Laden Sie einen molekularen Massenstandard und führen Sie die Elektrophorese aus, bis die Farbfront den Boden des Gels erreicht.
- Sobald die Elektrophorese abgeschlossen ist, visualisieren Sie das Gel auf einem geeigneten Transilluminator (entweder UV oder blaues Licht). gDNA erscheint als dickes hochmolekulares Band (über 10 kb). Ein Beispiel für die gDNA-Qualitätsprüfung ist in Abbildung 3dargestellt.
- Wenn die gDNA die Qualitätskontrolle besteht(d.h.das hochmolekulare Band vorhanden ist und es wenig bis gar keine Verschmierung der gDNA gibt), verdünnen Sie Ihre gDNA seriell, indem Sie zuerst 3 Mikrozentrifugenrohre wie folgt etikettieren: "10x", "100x" und "1000x".
- Pipette 90 l steriles destilliertes Wasser in jede der 3 Rohre.
- Nehmen Sie 10 l der gDNA-Lösung und fügen Sie sie dem Mitrohr mit der Aufschrift "10x" hinzu.
- Pipet das gesamte Volumen(d.h. 100 l) gründlich nach oben und unten, um sicherzustellen, dass die Lösung gleichmäßig gemischt wird. Nehmen Sie dann 10 l der Lösung aus diesem Rohr und übertragen Sie sie in das Rohr mit der Aufschrift "100x".
- Mischen Sie wie zuvor beschrieben und wiederholen Sie das gleiche Verfahren, indem Sie 10 l der Lösung von Rohr "100x" auf das Rohr "1000x" übertragen. Diese Verdünnungen werden als Vorlage in der PCR-Reaktion verwendet.

Abbildung 3: Agarose Gel Elektrophorese von gDNA isoliert aus Bacillus subtilis. Spur 1: M - Molekularmassenmarker (von oben nach unten: 10000 bp, 8000 bp, 6000 bp, 5000 bp, 4000 bp, 3500 bp, 3000 bp, 2500 bp, 2000 bp, 1500 bp, 1000 bp). Lane 2: gDNA - genomische DNA isoliert von Bacillus subtilis. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
-
Verstärkung des 16S rRNA-Gens durch PCR.
Hinweis: Das nachstehende PCR-Protokoll ist für ein bestimmtes DNA-Polymerase- und Primerpaar 8F - 1492R optimiert (siehe Tabelle 1). Für jedes Polymerase- und Primerpaar ist eine Optimierung des Protokolls erforderlich.- Alle Reagenzien auf Eis auftauen.
- Bereiten Sie den PCR-Mastermix vor, wie in Tabelle 2 dargestellt. Da die DNA-Polymerase bei Raumtemperatur aktiv ist, muss die Reaktionseinrichtung auf Eis durchgeführt werden, d.h. die PCR-Röhren und die Reaktionskomponenten sollten ständig auf Eis gehalten werden. Bereiten Sie eine Reaktion pro gDNA-Probe und eine Reaktion für negative Kontrolle vor. Negativsteuerung ist ein PCR-Mix ohne gDNA-Vorlage und wird verwendet, um sicherzustellen, dass die anderen Komponenten der Reaktion nicht kontaminiert sind.
Hinweis: Bei mehreren Proben wird üblicherweise ein Master-Mix vorbereitet. Master Mix ist eine Lösung, die alle Reaktionskomponenten außer der Vorlage enthält. Es hilft, wiederholte Pipetten zu vermeiden, Pipettierfehler zu vermeiden und sorgt für eine hohe Konsistenz zwischen den Proben. Um den Master-Mix vorzubereiten, multiplizieren Sie das Volumen der einzelnen Komponenten (mit Ausnahme der DNA-Vorlage) mit der Anzahl der getesteten Proben. Mischen Sie alle Komponenten in Mikrozentrifugenrohr und Pipetten Sie das gesamte Volumen nach oben und unten mehrmals. - Aliquot 49 l des Master-Mixes in die einzelnen PCR-Röhren.
- Fügen Sie 1 L-Vorlage in Rohre mit Master-Mix. Für die negative Kontrolle fügen Sie 1 l steriles Wasser hinzu. Um sicherzustellen, dass die Komponenten gut gemischt sind, leiten Sie die Mischung vorsichtig auf und ab, 10 Mal mit einer Pipette, die auf 30-50 l eingestellt ist.
- Stellen Sie die PCR-Maschine mit dem in Tabelle 3 gezeigten Programm ein.
- Legen Sie die Rohre in die PCR-Maschine und starten Sie das Programm.
- Sobald das Programm abgeschlossen ist, untersuchen Sie die Qualität Ihres PCR-Produkts durch Agarose-Gel-Elektrophorese.
- Eine erfolgreiche PCR-Reaktion mit dem 8F-1492R Primer-Paar ergibt ein einzelnes Band von ca. 1,5 kb (Abbildung 4). Wenn andere Bänder (d. h. unspezifische Produkte) vorhanden sind, optimieren Sie das PCR-Programm, indem Sie die Glühtemperatur anpassen. Wenn ein einzelnes Band mit der erwarteten Größe vorhanden ist, fahren Sie mit dem nächsten Schritt fort. Hier ergab die PCR-Reaktion mit 100-fach verdünnter gDNA-Schablone das beste Produkt, da sie ein scharfes Band von erwarteter Größe hatte und unspezifische Produkte fehlte. Daher wurde es gewählt, gereinigt und zur Sequenzierung geschickt zu werden.
- Vor der Sequenzierung muss das Produkt von Restgrundierungen, Desoxyribonukleotiden, Polymerase und Puffer gereinigt werden, die in der PCR-Reaktion vorhanden waren. Die PCR-Produkte können mit einem kommerziellen PCR-Reinigungskit isoliert werden. Die PCR-Reaktion wird auf eine Säule geladen, die eine DNA-bindende Matrix enthält. Das PCR-Produkt bindet an die Spalte, während andere Komponenten durch die Spalte fließen. Die Säule wird dann mit Waschpuffer gewaschen, und schließlich wird die DNA im Puffer der Wahl eluiert. Vergewissern Sie sich, dass der mit dem Kit ergänzte Elutionspuffer mit der Sequenzierung kompatibel ist.
- Senden Sie das gereinigte PCR-Produkt zur DNA-Sequenzierung. Befolgen Sie die Richtlinien für die Einreichung von Sequenzierungsmustern in der gewählten Sequenzierungseinrichtung. Für die beste Sequenzabdeckung verwenden Sie die PCR-Verstärkungsprimer (die gleichen wie in Abschnitt 2.4.1) als Sequenzierungsprimer. Hier wurden die Primer 8F und 1492R zur Sequenzierung des PCR-Produkts verwendet.
| teil | Endgültige Konzentration | Volumen pro Reaktion | Volumen pro x-Reaktionen (Master-Mix) |
| 5x Reaktionspuffer | 1x | 10 l | 10 l x |
| 10 mM dNTPs | 200 m | 1 L | 1 l x |
| 10 M Primer 8F | 0,5 m | 2,5 l | 2,5 x x |
| 10 M Primer 1492R | 0,5 m | 2,5 l | 2,5 x x |
| Phusion-Polymerase | 1 Einheit | 0,5 l | 0,5 x x |
| Vorlagen-DNA * | - | 1 L | - |
| ddH2O | - | 32,5 l | 32,5 x x |
| Gesamtvolumen | 50 l | 49 x x |
Tabelle 2: PCR-Reaktionskomponenten. * Verwenden Sie die 10x, 100x oder 1000x verdünnte gDNA ab Schritt 2.3.
| schritt | temperatur | zeit | Zyklen |
| Anfängliche Denaturierung | 98°C | 30 Sek. | |
| Denaturierung | 98°C | 10 Sek. | 25-30 |
| Glühen | 60°C | 30 Sek. | |
| anbau | 72°C | 45 Sek. | |
| Endgültige Verlängerung | 72°C | 7 Min. | |
| halten | 4°C | ∞ |
Tabelle 3: PCR-Programm zur Amplifikation des 16S rRNA-Gens.

Abbildung 4: Agarose-Gel-Elektrophorese von PCR-Produkten verstärkt mit Primer 8F und 1492R und gDNA als Vorlage. Die gDNA-Probe von B. subtilis (siehe Abbildung 3) wurde 10, 100 und 1000 Mal verdünnt, um das beste Ergebnis zu testen. Spur 1: M - Molekularmassenmarker (von oben nach unten: 10000 bp, 8000 bp, 6000 bp, 5000 bp, 4000 bp, 3500 bp, 3000 bp, 2500 bp, 2000 bp, 1500 bp, 1000 bp, 750 bp, 500 bp, 250 bp). Spur 2: PCR-Reaktion mit 10x verdünnter Schablone. Spur 3: PCR-Reaktion mit 100-fach verdünnter Schablone. Spur 4: PCR-Reaktion mit 1000x verdünnter Schablone. Spur 5: (C-) - Negativkontrolle (Reaktion ohne DNA-Vorlage). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
3. Datenanalyse und Ergebnisse
Hinweis: Das PCR-Produkt wird mit den Vorwärts-Primern (hier 8F) und den umgekehrten (hier 1492R) Primern sequenziert. Daher werden zwei Sätze von Datensequenzen generiert, einer für den Vorwärts und einer für den umgekehrten Primer. Für jede Sequenz werden mindestens zwei Dateitypen erzeugt: i) eine Textdatei mit der DNA-Sequenz und ii) ein DNA-Chromatogramm, das die Qualität des Sequenzierungslaufs anzeigt.
- Öffnen Sie für die Vorwärtsgrundierung das Chromatogramm, und untersuchen Sie die Sequenz sorgfältig. Ein ideales Chromatogramm für eine Qualitätssequenz sollte gleichmäßig eimierte Spitzen und wenig oder gar keine Hintergrundsignale haben (Abbildung 5A).
- Wenn das Chromatogramm nicht hochwertig ist, sollte die Sequenz verworfen oder die Sequenztextdatei entsprechend den folgenden Kriterien überarbeitet werden:
- Das Vorhandensein von Doppelspitzen im gesamten Chromatogramm weist auf das Vorhandensein mehrerer DNA-Vorlagen hin. Dies kann der Fall sein, wenn die Bakterienkultur nicht rein war. Eine solche Sequenz sollte verworfen werden (Abbildung 5B).
- Ein mehrdeutiges Chromatogramm kann aus dem Vorhandensein unterschiedlicher farbiger Spitzen an der gleichen Stelle entstehen. Einer der häufigsten Fehler ist das Vorhandensein von zwei verschiedenen farbigen Spitzen in der gleichen Position und unsachgemäße Zuweisung der Basen durch die Sequenzierungssoftware (Abbildung 5C). Korrigieren Sie alle falsch zugewiesenen Nukleotide manuell und bearbeiten Sie sie in der Textdatei.
- Chromatogramme mit niedriger Auflösung können zu "breiten Spitzen" führen, die in diesen Regionen häufig zu Fehlzählungen der Nukleotide führen (Abbildung 5D). Dieser Fehler ist schwer zu korrigieren, und daher sollten mögliche Inkongruenzen im weiteren Ausrichtungsschritt nicht als zuverlässig behandelt werden.
- Schlechte Chromatogramm-Lesequalität und das Vorhandensein mehrerer Spitzen ist häufig an den 5' und 3' Enden der Sequenz zu sehen. Einige Sequenzer-Software entfernt diese minderwertigen Fragmente automatisch(Abbildung 5E), und die Nukleotide sind nicht in der Textdatei enthalten. Wenn Ihre Sequenz nicht automatisch abgeschnitten wurde, bestimmen Sie die minderwertigen Fragmente(z.B. schwaches Signal, überlappende Spitzen, Auflösungsverlust) an den Enden und entfernen Sie die entsprechenden Basen aus der Textdatei.

Abbildung 5: Beispiele für die Fehlerbehebung bei der DNA-Sequenzierung. A) Ein Beispiel für eine hochwertige Chromatogrammsequenz (gleichmäßig verteilte, eindeutige Spitzen). B) Schlechte Qualität Sequenz, die in der Regel am Anfang des Chromatogramms auftritt. Der Grauzonenbereich gilt als minderwertig und wird von der Sequenzierungssoftware automatisch entfernt. Weitere Basen können manuell getrimmt werden. C) Vorhandensein von Doppelspitzen (durch Pfeile angezeigt). Ein Nukleotid, das durch den roten Pfeil angezeigt wird, wurde vom Sequenzer als "T" (roter Peak) gelesen, aber der blaue Peak ist stärker, und es kann auch als "C" interpretiert werden. D) Überlappende Spitzen weisen auf eine DNA-Kontamination hin(d. h. mehr als eine Schablone). E) Verlust der Auflösung und so genannte "breite Spitzen" (gekennzeichnet durch Rechteck), die zuverlässige Basisrufe verhindern. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
- Wiederholen Sie 3.1 und 3.2 für die umgekehrte Grundierung.
- Zusammenstellen Sie schließlich die Vorwärts- und Rückwärtssequenzen in einer zusammenhängenden Sequenz. Ein guter Sequenzierungslauf ergibt eine Sequenz von bis zu 1100 bp. Wenn man bedenkt, dass das PCR-Produkt 1500 bp lang ist, sollten sich die Sequenzen, die mit Vorwärts- und Reverse-Primern erhalten wurden, teilweise überlappen.
- Zusammenführen Sie die beiden Sequenzen mit dem DNA-Sequenzbauprogramm, z.B. einem freien Werkzeug wie CAP3 (http://doua.prabi.fr/software/cap3) (15).
- Fügen Sie die beiden Sequenzen im FASTA-Format in das angegebene Feld ein. Klicken Sie auf die Schaltfläche "Absenden" und warten Sie, bis die Ergebnisse zurückgegeben werden.
- Um die montierte Sequenz anzuzeigen, drücken Sie "Contigs" in der Ergebnis-Registerkarte. Um die Details der Ausrichtung anzuzeigen, drücken Sie "Montagedetails".
Anmerkung 1: Wenn CAP3-Software für die Contig-Assembly verwendet wird, ist es nicht erforderlich, die umgekehrte Primersequenz in Reverse-Komplementär zu konvertieren. Dieser Schritt kann jedoch erforderlich sein, wenn ein anderes Programm verwendet wird.
Anmerkung 2: Das FASTA-Format ist ein textbasiertes Format, das die Nukleotidsequenz darstellt. Die erste Zeile (die Beschreibungszeile) in einer FASTA-Datei beginnt mit dem Symbol ">", gefolgt vom Namen oder einem eindeutigen Bezeichner der Sequenz. Der Beschreibungszeile folgt die Nukleotidsequenz. Fügen Sie Ihre Sequenzen im folgenden Format ein:
>sequenz_frw_primer
Fügen Sie Ihre Sequenz aus der Textdatei hier ein
>sequence_rvs_primer
Fügen Sie Ihre Sequenz aus der Textdatei hier ein
- Führen Sie eine Datenbanksuche durch, indem Sie die Website für das Basic Local Alignment Search Tool (BLAST; https://blast.ncbi.nlm.nih.gov/Blast.cgi) besuchen.
- Wählen Sie das Werkzeug "Nukleotid BLAST", um Ihre Sequenz mit der Datenbank zu vergleichen.
- Geben Sie Ihre Sequenz (das in 3.5 zusammengebaute Kontus) in das Textfeld "Abfragesequenz" ein und wählen Sie dann im Scroll-Down-Menü die Datenbank "16S rRNA-Sequenzen (Bakterien und Archea)" aus.
- Drücken Sie die Taste "BLAST" am unteren Rand der Seite. Die ähnlichsten Sequenzen werden zurückgegeben. Ein BLAST-Beispiel ist in Abbildung 6dargestellt. Im vorgestellten Experiment ist der Top-Hit B. subtilis Stamm 168, der eine 100%ige Identität mit der in der BLAST-Datenbank verfügbaren Sequenz zeigt.
- Wenn der obere Treffer keine 100%ige Identität anzeigt, gehen Sie zur Ausrichtung, und überprüfen Sie, ob es nicht stimmt. Wenn Sie auf den Top-Hit klicken, werden Sie zu den Details der Ausrichtung weitergeleitet. Ausgerichtete Nukleotide werden durch kurze vertikale Linien verbunden, während nicht übereinstimmende Nukleotide einen Abstand zwischen ihnen haben. Kehren Sie zum Chromatogramm zurück, das Sie von der Sequenzierungsfirma erhalten haben, und überarbeiten Sie die Sequenz erneut mit dem Fokus auf die nicht übereinstimmende Region. Korrigieren Sie die Reihenfolge, wenn weitere Fehler gefunden werden. Führen Sie BLAST erneut mit der korrigierten Sequenz aus.

Abbildung 6: Beispiel für das Nukleotid BLAST-Ergebnis. 16S rRNA Gensequenz aus rein Kultur von B. subtilis 168 wurde als Abfragesequenz verwendet. Der Top-Hit zeigt wie erwartet 100% Identität (unterstrichen) zum B. subtilis-Stamm 168. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Die Erde ist die Heimat von Millionen von Bakterienarten, jede mit einzigartigen Eigenschaften. Die Identifizierung dieser Arten ist bei der Bewertung von Umweltproben von entscheidender Bedeutung. Ärzte müssen auch verschiedene Bakterienarten unterscheiden, um infizierte Patienten zu diagnostizieren.
Um Bakterien zu identifizieren, können eine Vielzahl von Techniken eingesetzt werden, einschließlich mikroskopischer Beobachtung der Morphologie oder Deswachstum auf einem bestimmten Medium, um koloniemorphologische zu beobachten. Genetische Analyse, eine andere Technik zur Identifizierung von Bakterien hat in den letzten Jahren an Popularität gewonnen, zum Teil aufgrund der 16S ribosomalen RNA-Gensequenzierung.
Das bakterielle Ribosom ist ein Protein-RNA-Komplex, der aus zwei Untereinheiten besteht. Die 30S-Untereinheit, die kleinere dieser beiden Untereinheiten, enthält 16S rRNA, die durch das 16S rRNA-Gen in der genomischen DNA kodiert wird. Spezifische Regionen von 16S rRNA sind aufgrund ihrer wesentlichen Funktion in der Ribosom-Montage hochkonserviert. Während andere Regionen, weniger wichtig für die Funktion, kann zwischen Bakterienarten variieren. Die variablen Regionen in 16S rRNA können als einzigartige molekulare Fingerabdrücke für Bakterienarten dienen, so dass wir phänotypisch identische Stämme unterscheiden können.
Nach Erhalt einer Qualitätsprobe von gDNA kann PCR des 16S rRNA-Codierungsgens beginnen. PCR ist eine häufig verwendete molekularbiologische Methode, die aus Zyklen der Denaturierung der doppelsträngigen DNA-Vorlage, dem Glühen von universellen Primerpaaren, die hochkonservierte Regionen des Gens verstärken, und der Erweiterung von Primern durch DNA-Polymerase besteht. Während einige Primer den größten Teil des 16S rRNA-Codierungsgens verstärken, verstärken andere nur Fragmente davon. Nach pcR können die Produkte mittels Agarose-Gel-Elektrophorese analysiert werden. Wenn die Amplifikation erfolgreich war, sollte das Gel ein einzelnes Band von einer erwarteten Größe enthalten, abhängig vom verwendeten Primerpaar, bis zu 1500bp, der ungefähren Länge des 16S rRNA-Gens.
Nach der Reinigung und Sequenzierung können die erhaltenen Sequenzen dann in die BLAST-Datenbank eingegeben und mit Referenzsequenzen von 16S rRNA verglichen werden. Da diese Datenbank Übereinstimmungen auf der Grundlage der höchsten Ähnlichkeit zurückgibt, ermöglicht dies die Bestätigung der Identität der Bakterien von Interesse. In diesem Video beobachten Sie die 16S rRNA-Gensequenzierung, einschließlich PCR, DNA-Sequenzanalyse und -bearbeitung, Sequenzmontage und Datenbanksuche.
Beim Umgang mit Mikroorganismen ist es wichtig, eine gute mikrobiologische Praxis zu befolgen, einschließlich der Verwendung aseptischer Technik und des Tragens geeigneter persönlicher Schutzausrüstung. Nach durchführung einer geeigneten Risikobewertung für den Mikroorganismus oder die Umweltprobe von Interesse, erhalten Sie eine Testkultur. In diesem Beispiel wird eine reine Kultur von Bacillus subtilis verwendet.
Zunächst wachsen Sie Ihren Mikroorganismus auf einem geeigneten Medium unter den entsprechenden Bedingungen an. In diesem Beispiel wird Bacillus subtilis 168 in LB-Brühe über Nacht in einem Schüttel-Inkubator angebaut, der auf 200 Umdrehungen pro Minute bei 37 Grad Celsius eingestellt ist. Als nächstes verwenden Sie ein kommerziell erhältliches Kit, um genomische DNA oder gDNA aus 1,5 Millilitern der B. subtilis Nachtkultur zu isolieren.
Um die Qualität der isolierten DNA zu überprüfen, mischen Sie zunächst fünf Mikroliter der isolierten gDNA mit einem Mikroliter DNA-Gel-Ladefarbstoff. Dann laden Sie die Probe auf ein 0,8% Agarose-Gel, das DNA-Färbungsreagenz enthält, wie SYBR safe oder EtBr. Danach einen ein Kilobase-Molekularmassenstandard auf das Gel laden und die Elektrophorese so lange ausführen, bis der Vorderfarbstoff etwa 0,5 Zentimeter vom Boden des Gels entfernt ist. Sobald die Gelelektrophorese abgeschlossen ist, visualisieren Sie das Gel auf einem blauen Lichttransilluminator. Die gDNA sollte als dickes Band erscheinen, über 10 Kilobase in der Größe und haben minimale Verschmierung.
Um anschließend serielle Verdünnungen der gDNA zu erstellen, beschriften Sie drei Mikrozentrifugenröhrchen als 10X, 100X und 1000X. Verwenden Sie dann eine Pipette, um 90 Mikroliter steriles destilliertes Wasser in jede der Rohre zu geben. Als nächstes fügen Sie 10 Mikroliter der gDNA-Lösung in die 10X-Röhre. Pipette das gesamte Volumen nach oben und unten, um sicherzustellen, dass die Lösung gründlich gemischt wird. Entfernen Sie dann 10 Mikroliter der Lösung aus dem 10X-Rohr und übertragen Sie diese in das 100X-Rohr. Mischen Sie die Lösung wie zuvor beschrieben. Schließlich übertragen Sie 10 Mikroliter der Lösung in der 100X-Röhre, auf die 1000X-Röhre.
Um das PCR-Protokoll zu beginnen, tauen Sie die notwendigen Reagenzien auf Eis auf. Bereiten Sie dann den PCR-Master-Mix vor. Da die DNA-Polymerase bei Raumtemperatur aktiv ist, muss die Reaktion auf Eis erfolgen. Aliquot 49 Mikroliter des Master-Mix in jede der PCR-Röhren. Dann fügen Sie einen Mikroliter Schablone zu jedem der Versuchsröhrchen und einen Mikroliter sterilen Wassers in das negative Kontrollrohr, Pipettieren nach oben und unten, um zu mischen. Danach stellen Sie die PCR-Maschine entsprechend dem in der Tabelle beschriebenen Programm ein. Legen Sie die Rohre in den Thermocycler und starten Sie das Programm.
Sobald das Programm abgeschlossen ist, untersuchen Sie die Qualität Ihres Produkts durch Agarose-Gel-Elektrophorese, wie zuvor gezeigt. Eine erfolgreiche Reaktion mit dem beschriebenen Protokoll sollte zu einem einzigen Band von ca. 1,5 Kilobase führen. In diesem Beispiel lieferte die Probe, die 100X verdünnte gDNA enthielt, das produkthöchste Qualität. Als nächstes reinigen Sie das beste PCR-Produkt, in diesem Fall die 100X gDNA, mit einem handelsüblichen Kit. Jetzt kann das PCR-Produkt zur Sequenzierung gesendet werden.
In diesem Beispiel wird das PCR-Produkt mit Vorwärts- und Reverse-Primern sequenziert. So werden zwei Datensätze erzeugt, die jeweils eine DNA-Sequenz und ein DNA-Chromatogramm enthalten: einer für den Vorwärtsprimer und der andere für den umgekehrten Primer. Untersuchen Sie zunächst die Chromatogramme, die aus jedem Primer erzeugt werden. Ein ideales Chromatogramm sollte gleichmäßig verteilte Spitzen mit wenig bis gar keinen Hintergrundsignalen haben.
Wenn die Chromatogramme doppelte Spitzen anzeigen, können mehrere DNA-Vorlagen in den PCR-Produkten vorhanden gewesen sein, und die Sequenz sollte verworfen werden. Wenn die Chromatogramme Spitzen in verschiedenen Farben an der gleichen Stelle enthielten, wurde die Sequenzierungssoftware wahrscheinlich falsch als Nukleotide bezeichnet. Dieser Fehler kann manuell in der Textdatei identifiziert und korrigiert werden. Das Vorhandensein breiter Spitzen im Chromatogramm deutet auf einen Auflösungsverlust hin, der zu einer Fehlzählung der Nukleotide in den zugehörigen Regionen führt. Dieser Fehler ist schwer zu korrigieren, und Inkongruenzen in einem der nachfolgenden Schritte sollten als unzuverlässig behandelt werden. Schlechte Chromatogramm-Lesequalität, die durch das Vorhandensein mehrerer Spitzen angezeigt wird, tritt in der Regel an den fünf Prim- und drei Primenden der Sequenz auf. Einige Sequenzierungsprogramme entfernen diese Abschnitte mit geringer Qualität automatisch. Wenn Ihre Sequenz nicht automatisch abgeschnitten wurde, identifizieren Sie die Fragmente niedriger Qualität, und entfernen Sie deren entsprechende Basen aus der Textdatei.
Verwenden Sie ein DNA-Montageprogramm, um die beiden Primersequenzen in einer kontinuierlichen Sequenz zusammenzubauen. Denken Sie daran, dass Sequenzen, die mit Vorwärts- und Rückwärtsprimern erhalten wurden, sich teilweise überlappen sollten. Fügen Sie im DNA-Montageprogramm die beiden Sequenzen im FASTA-Format in die entsprechende Box ein. Klicken Sie dann auf die Schaltfläche Senden, und warten Sie, bis das Programm die Ergebnisse zurückgibt.
Um die zusammengebaute Sequenz anzuzeigen, klicken Sie auf Contigs in der Ergebnis-Registerkarte. Um dann die Details der Ausrichtung anzuzeigen, wählen Sie Die Baugruppendetails aus. Navigieren Sie zur Website für das grundlegende Suchwerkzeug für lokale Ausrichtungen (BLAST), und wählen Sie das Nukleotid-BLAST-Tool aus, um ihre Sequenz mit der Datenbank zu vergleichen. Geben Sie Ihre Sequenz in das Textfeld der Abfragesequenz ein und wählen Sie die entsprechende Datenbank im Bildlauf-Down-Menü aus. Klicken Sie schließlich auf die SCHALTFLÄCHE BLAST am unteren Rand der Seite, und warten Sie, bis das Tool die ähnlichsten Sequenzen aus der Datenbank zurückgibt.
In diesem Beispiel ist der Top-Treffer B. subtilis-Stamm 168, der eine 100%ige Identität mit der Sequenz in der BLAST-Datenbank anzeigt. Wenn der Top-Treffer nicht 100% Identität zu Ihrer erwarteten Art oder Sorte zeigt, klicken Sie auf die Sequenz, die Ihrer Abfrage am ehesten entspricht, um die Details der Ausrichtung anzuzeigen. Ausgerichtete Nukleotide werden durch kurze vertikale Linien verbunden, und nicht übereinstimmende Nukleotide haben Lücken zwischen ihnen. Konzentrieren Sie sich auf die identifizierten nicht übereinstimmenden Regionen, überarbeiten Sie die Reihenfolge und wiederholen Sie die BLAST-Suche, falls gewünscht.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
Die Identifizierung von Bakterienarten ist wichtig für verschiedene Forscher, sowie für diejenigen im Gesundheitswesen. 16S rRNA-Sequenzierung wurde ursprünglich von Forschern verwendet, um phylogenetische Beziehungen zwischen Bakterien zu bestimmen. Im Laufe der Zeit wurde es in metagenomischen Studien implementiert, um die biologische Vielfalt von Umweltproben und in klinischen Laboratorien als Methode zur Identifizierung potenzieller Krankheitserreger zu bestimmen. Es ermöglicht eine schnelle und genaue Identifizierung von Bakterien in klinischen Proben, was eine frühere Diagnose und eine schnellere Behandlung von Patienten erleichtert.
Subscription Required. Please recommend JoVE to your librarian.
References
- Weisburg, W.G., Barns, S.M., Pelletier, D.A. and Lane D.J. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 173 (2): 697-703. (1991)
- Drancourt, M., Bollet, C., Carlioz, A., Martelin, R., Gayral, J.P., Raoult D. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol. 38 (10):3623-3630. (2000)
- Woo, P.C., Lau, S.K., Teng, J.L., Tse, H., Yuen, K.Y. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect. 14 (10):908-934. (2008)
- Tang, Y.W., Ellis, N.M., Hopkins, M.K., Smith, D.H., Dodge, D.E., Persing, D.H. Comparison of phenotypic and genotypic techniques for identification of unusual aerobic pathogenic gram-negative bacilli. J Clin Microbiol. 36 (12):3674-3679. (1998)
- Tsiboli, P., Herfurth, E., Choli, T. Purification and characterization of the 30S ribosomal proteins from the bacterium Thermus thermophilus. Eur J Biochem. 226 (1):169-177. (1994)
- Woese, C.R. Bacterial evolution. Microbiol Rev. 51 (2):221-271. (1987)
- Bartlett, J.M., Stirling, D. A short history of the polymerase chain reaction. Methods Mol Biol. 226:3-6. (2003)
- Wilson, K.H., Blitchington, R.B., Greene, R.C. Amplification of bacterial 16S ribosomal DNA with polymerase chain reaction. J Clin Microbiol. 28 (9):1942-1946. (1990)
- Shendure, J., Balasubramanian, S., Church, G.M., Gilbert, W., Rogers, J., Schloss, J.A., Waterston, R.H. (2017) DNA sequencing at 40: past, present and future. Nature. 550:345-353.
- Lane, D.J. 16S/23S rRNA sequencing. (1991) In Nucleic acid techniques in bacterial systematics. (Goodfellow, M. and Stackebrandt, E., eds.) p.115-175. Wiley and Sons, Chichester, United Kingdom.
- Turner, S., Pryer, K.M., Miao, V.P., Palmer, J.D. (1999) Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryot Microbiol. 46:327-338.
- Fredricks, D.N., Relman, D.A. (1998) Improved amplification of microbial DNA from blood cultures by removal of the PCR inhibitor sodium polyanetholesulfonate. J Clin Microbiol. 36:2810-2816.
- Wilson, K. Preparation of genomic DNA from bacteria. (2001) Curr Protoc Mol Biol. Chapter 2:Unit 2.4.
- Wright, M. H., Adelskov, J., Greene, A.C. (2017) Bacterial DNA extraction using individual enzymes and phenol/chloroform separation. J Microbiol Biol Educ. 18:18.2.48.
- Huang, X., Madan, A. (1999). CAP3: A DNA sequence assembly program. Genome Res. 9:868-877.