



Overview
ソース: ナタリア・マーティン1, アンドリュー・J・ヴァン・アルスト1, リアノン・M・ルヴェーク1, ビクター・J・ディリタ1
1ミシガン州立大学微生物学・分子遺伝学専攻
細菌は、水平遺伝子導入として知られているプロセスで遺伝物質(デオキシリボヌクレイン酸、DNA)を交換する能力を有する。外因性DNAを組み込むことは、細菌が自然の生息地に見られる抗生物質や抗体(1)または分子の存在など、変化する環境条件に適応することを可能にする新しい遺伝的形質を獲得できるメカニズムを提供する。(2)水平遺伝子導入には、形質転換、経転移、共役の3つのメカニズムがある(3)ここでは、環境から自由なDNAを取り込む細菌の能力、形質転換に焦点を当てます。実験室では、形質転換プロセスには、4つの一般的なステップがあります:1)有能な細胞の調製、2)DNAを用いて有能な細胞のインキュベーション、3)細胞の回収、および4)形質転換剤の増殖のための細胞のめっき(図1)。
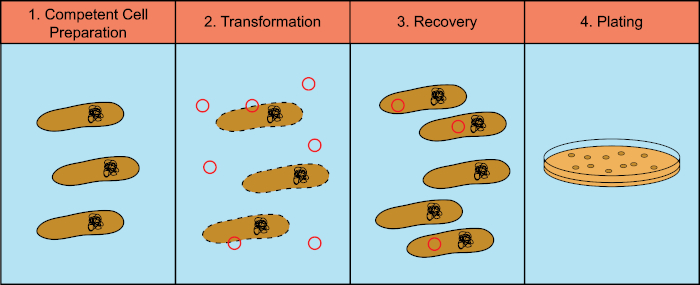
図 1: 変換プロセスの一般的な手順。形質転換プロセスには、4つの一般的なステップがあります:1)有能な細胞の調製、2)DNAによるインキュベーション、3)細胞の回収、4)形質転換剤の増殖のためのめっき細胞。
変換が起こるには、レシピエント細菌が能力と呼ばれる状態である必要があります。一部の細菌は、特定の環境条件に応じて自然に有能になる能力を持っています。しかし、他の多くの細菌は自然に有能にならないか、またはこのプロセスの条件はまだ不明です。DNAを細菌に導入する能力は、目的とするDNA分子の複数のコピーを生成し、大量のタンパク質を発現させる、クローニング手順の構成要素として、他の研究用途の範囲を持っています。分子生物学への変換の価値のために、自然な能力のための条件が不明な場合に細胞を人工的に有能にすることを目的としたいくつかのプロトコルがあります。人工的に有能な細胞を調造するには、1)細胞の化学処理を通じて、2)細胞を電気パルス(エレクトロポレーション)にさらす2つの主な方法が使用されます。前者は、DNAと細胞表面の間に引力を生み出す手順に応じて異なる化学物質を使用し、後者は電界を使用してDNA分子が入り込むことができる細菌細胞膜の細孔を生成します。化学的能力のための最も効率的なアプローチは、二価陽イオンを用いてインキュベーションであり、最も顕著なカルシウム(Ca2+)(4,5)カルシウム誘発能力は、ここで説明する手順である(6)。この方法は、主にグラム陰性細菌の形質転換に使用され、それはこのプロトコルの焦点になります。
化学的変換の手順は、細胞が化学的能力を誘発するために陽イオンにさらされる一連のステップを伴う。これらのステップは、その後、温度変化-ヒートショック-有能な細胞による外来DNAの取り込みを支持する(7)。細菌細胞の封筒は負の帯電である。大腸菌のようなグラム陰性菌では、外膜はリポ多糖(LPS)の存在により負の帯電(8)である。これにより、同様に負電荷を帯んだDNA分子の反発が生じる。化学的能力誘導において、正に帯電したカルシウムイオンは、この電荷反発を中和し、細胞表面へのDNA吸光を可能にする(9)。DNAによるカルシウム処理とインキュベーションは氷上で行われます。続いて、より高い温度(42°C)でのインキュベーション、ヒートショックが行われる。この温度の不均衡は、DNAの取り込みをさらに有利にします。細菌細胞は、ヒートショック治療に耐えるために、中指数成長期である必要があります。他の成長段階では、細菌細胞は熱に敏感になりすぎて、変換効率が大幅に低下する生存率の損失をもたらす。
異なるDNA源を変換に使用できます。典型的には、プラスミドは、小さな円形、二本鎖DNA分子、大腸菌におけるほとんどの実験室手順における形質転換に使用される。プラスミドを形質転換後に細菌細胞に維持するには、複製の起源を含む必要があります。これにより、細菌染色体とは独立して細菌細胞内で複製することができる。すべての細菌細胞が形質転換手順中に変換されるわけではありません。したがって、形質転換は、形質転換された細胞と非形質化された細胞の混合物をもたらす。これら2つの集団を区別するために、プラスミドを獲得した細胞を同定する選択方法が用いられる。プラスミドは通常、選択可能なマーカーを含み、これは成長の利点を与える形質をコードする遺伝子である(すなわち、抗生物質または化学的または成長性の補助性からの救助に対する耐性)。形質転換後、細菌細胞は選択的培地上にめっきされ、形質転換細胞の増殖のみが可能となる。所定の抗生物質に対する耐性を与えるプラスミドで形質転換した細胞の場合、選択的培養剤はその抗生物質を含む増殖培養剤となる。選択的培養剤中に成長したコロニーが形質転換体であることを確認するためにいくつかの異なる方法を使用することができます(すなわち、プラスミドを組み込んだ)。例えば、プラスミドはプラスミド調製法(10)を用いてこれらの細胞から回収し、プラスミドサイズを確認するために消化することができる。あるいは、コロニーPCRは、目的のプラスミド(11)の存在を確認するために使用することができる。
この実験の目的は、塩化カルシウム手順(12)の適応を用いて大腸菌DH5α化学的に有能な細胞を調作し、プラスミドpUC19でそれらを変換して変換効率を決定することである。大腸菌株DH5αは、分子生物学アプリケーションで一般的に使用される株です。その遺伝子型、特にrecA1およびendA1のために、この株は挿入安定性を高め、その後の調製におけるプラスミドDNAの質を改善することを可能にする。DNAのサイズが大きくなると形質転換効率が低下するため、プラスミドpUC19はサイズが小さい(2686 bp)のでこのプロトコルで使用された(https://www.mobitec.com/cms/products/bio/04_vector_sys/standard_cloning_vectors.html参照ベクトルマップ)。pUC19はアンピシリンに対する耐性を付与し、したがって、これは選択に使用される抗生物質であった。
Procedure
このプロトコルは、塩化カルシウム手順(12)の適応を用いて有能な大腸菌DH5αの調製および形質転換について説明する。
1. セットアップ
-
機器
- 分光 光度 計
- ソルバル遠心分離機(または同等)
- ベンチトップ遠心分離機
- ヒートブロックまたは水浴
- オービタルシェーカー
- ステーナリーインキュベーター
- ゲル鋳造トレイ
- まあ櫛
- 電圧源
- ゲルボックス
- 紫外線光源
- マイクロ波
-
ソリューションと試薬
- ルリア・ベルタニ(LB)スープ(10gカゼイン酵素加水分解物、5g酵母エキス、5g塩化ナトリウム1000 mLのH2O)
- カタボライト抑圧(SOC):(2%(w/v)トリプトン、0.5%(w/v)酵母エキス、10mM NaCl、2.5mM KCl、10mM MgCl 2、10mMMgSO4、および20mMグルコース)
- CaCl2-MgCl2 (80 mM MgCl2,20 mM CaCl2)溶液.
- M CaCl2溶液(細胞が直ちに形質転換される場合)または10%(v/v)グリセロールを含む0.1MCaCl2溶液(細胞が将来の使用のために凍結される場合)。
- LB寒天プレート
- LB寒天選択プレート(この実験では、プラスミド使用済みのアンピシリン耐性を付与するので、アンピシリン100μg/mLを含むLB寒天プレートを用いた)
- 大腸菌DH5α株
- プラスミド pUC19 DNA (100 pg/ μl)
- QIAprep スピン ミニプレップ キット (キアゲン)
- ヒンドIII制限酵素
- 1 kb プラス DNA はしご
- 低融点アガローズ
- 1X TAEバッファー(40mMトリスベース、20mM酢酸、1mM EDTA)
- 臭化エチジウム (10mg/mL)
-
一般的な安全に関する注意事項
大腸菌DH5αは、バイオセーフティレベル1(BSL1)に分類されます。このカテゴリーの微生物は、健康な成人における感染の脅威をほとんどまたは全くもたらさない。しかし、微生物の慎重な操作が必要です。
重要重要このプロトコルのすべてのステップは、無菌技術を使用して、指示がない限り氷または4°Cの温度で行われる必要があります。
2. プロトコル
- 大腸菌DH5αの凍結ストック(LBで成長した一晩培養から20%グリセロールで凍結)から、LB寒天プレート上で単離するための細菌をストリークアウトする。一晩37°C(16~20時間)でインキュベートしてください。
- 単一のコロニーをチューブ内のLBスープの3 mLに接種します。一晩37°C(16-20時間)で210 rpmで揺れを成長させる。
- 一晩培養のOD600を測定します。一晩培養を使用して、1リットルのフラスコで100 mLのLBスープをOD600=0.01に接種します。培養量がOD600=0.35(約3時間)に達するまで、分光光度計で37°CモニタリングOD600で激しく揺れる培養(210rpm)を15~20分ごとにインキュベートします。
注:変換を効率的に行うには、細菌細胞が指数関数的な成長段階にある必要があります。細胞の最大数は108細胞/mLである必要があり、大腸菌のほとんどの株はOD600=0.4に対応します。分光光度計を使用すると、OD600を測定することができ、細胞が適切な成長段階にあることを判断することができます。このプロトコルが細菌の他の株に使用される場合、この相関を決定するために、特定のOD600値でコロニー形成単位の数を決定するための較正が必要になります。 - 培養物の50mLを2本の冷たいポリプロピレン遠心分離ボトルに移します。ボトルを氷の上に20分間置き、冷まします。
- 2700g(ソルバルGSAローターで4100rpm)の遠心分離によって細胞を回収し、4°Cで10分間回収します。
- 上清を取り除く。ボトルをパッドまたはペーパータオルの上に逆さまに置くことによって、メディアの最後の痕跡を排出します。
- 各細菌ペレットをCaCl2-MgCl2(80 mM MgCl2、20 mM CaCl2)の30 mLに再ステーペンします。まず溶液の5 mLを追加し、ペレットが完全に溶解するまで慎重に旋回し、残りの25 mLの溶液を追加します。
- 手順 2.4 を繰り返します。
- 手順 2.5 を繰り返します。
- 有能な細胞が直接形質転換される場合は、チューブを慎重に旋回することにより、各細菌ペレットをCaCl 2(0.1M)氷冷溶液の2mLに再ステージングします。ペレットがこの方法で再懸濁されない場合は、上下にゆっくりとピペッティング(気泡形成を避ける)によって再懸濁する。
あるいは、有能な細胞を凍結し、後で使用するために保存することができる。有能な細胞の凍結ストックを調剤するために、10%(v/v)グリセロールを含む0.1MCaCl2溶液の2mlでペレットを再中断する。このソリューションは、氷冷である必要があります。アリコット細胞懸濁液を氷冷1.5 mLポリプロピレンチューブ(チューブ当たり160μl)に注入。有能な細胞をドライアイス/エタノール浴で直ちに凍結する。チューブを-70°Cの冷凍庫に移します。 - CaCl2-処理細胞を形質転換するには、50 μlの有能な細胞を2つの1.5mlポリプロピレンチューブのそれぞれに移します。pUC19プラスミドDNAの1 μl(100 pg)をチューブの1つに加え、DNAなしで2番目のチューブを残します(陰性対照)。穏やかに混ぜる(気泡形成を避ける)。氷の上で30分間インキュベートします。
注:10μL以下の体積のDNAは50ng以下で、変換に使用する必要があります。 - チューブをヒートブロックに移し、42°Cで45度正確にインキュベートします。
注:ヒートショックは重要なステップです。温度またはインキュベーション時間を超えないようにしてください。 - 容易に氷に管を移す。2分間インキュベートします。
- 950 μLのSOC培地を加え、37°Cで1時間チューブをインキュベートし、プラスミドにコードされた抗生物質耐性マーカーを回収して発現させます。
- SOCで1000 μLの細胞懸濁液の10μLを希釈し(1/100希釈)、SOCで1000μLの細胞懸濁液の100μL(1/10希釈)。希釈のプレート100μlは、コントロールと同様に、選択的プレート上に、へらを用いて広がる。通常、1/100および1/10希釈のメッキ100 μLは、プレートあたり十分な数のコロニー形成単位(cfu)をもたらすでしょう。理想的には、この数は30~300 cfuの範囲で、十分なコロニーがあるが互いに分離している必要があります。ただし、cfu の数は変換効率によって異なります (データ分析と結果セクションを参照)。
- プレートを37°Cでインキュベートします。形質転換されたコロニーは12〜16時間で現れるはずです(この範囲は細胞株と選択方法によって異なります)。ネガティブコントロールでコロニーが成長してはなりません。
- 変換のために得られたcfu/プレートをカウントします(表1)。
- pUC19プラスミドを持つ形質転換体を確認するために、プラスミド調製およびその後の消化が行われる。この目的のために、チューブ内のLBスープの3ミリリットルに単一のコロニーを接種します。一晩37°C(16-20時間)で210 rpmで揺れを成長させる。
- メーカーの指示に従って、QIAprepスピンミニプレップキットを使用してプラスミド製剤を準備します。
- 精製したpUC19の1μgを37°Cで1時間37°Cで消化する。
注:pUC19複数のクローニング部位で切断する酵素は、このステップに使用できます。
| コンポーネント | 量 |
| 10X 制限ダイジェスト バッファ | 2.5 μl |
| プラスミド pUC19 | 1 μg |
| ヒンドIii | 1 μl |
| H2O | 20.5 μl (25 μl まで) |
- 分子量はしご、消化されたpUC19 DNAおよび同量の未消化pUC19 DNAを1%のアガロースゲルで95Vで1時間の臭化物を含む。
注:時間と電圧は、使用する機器によって異なります。 - 紫外線の下でゲルを視覚化します。消化されたpUC19 DNA(図2)の大きさを比較する(データ分析と結果セクションを参照)。
各特定の変換実験の目標に従って変換を検証するために必要な手順を進めます。

図2:形質転換DH5α細胞からの回収プラスミドDNAの消化。プラスミドDNAは形質転換されたDH5α細胞から回収し、ヒンdIIIで消化し、1%のアガロースゲルで走り、UV源で可視化した(ステップ2.19~2.22)。
3. データ分析と結果
形質転換効率を計算するには、細胞が細胞外DNAをどの程度よく取り上げたかを示す指標で、形質転換で得られたコロニーをカウントする必要があります。
| 希釈 | Cfu |
| 1/100 | 34 |
| 1/10 | 246 |
表1:コロニー形成単位(cfu)は、形質転換実験からカウントされた。
形質転換効率(TE)は、1μgのプラスミドを所定の容積の有能な細胞に変換することから生じるcfuの数の尺度である。多くのパラメータは、プラスミドサイズ、細胞遺伝子型、能力準備中の成長段階、変換方法など、変換効率に影響を与えます。TE を計算する際には、めっき前に行われた希釈(もしある場合)を考慮し、cfuの総数の計算に組み込むことが重要です。変換効率 (TE) は、次の式で計算されます。

まず、この例では0.0001μgのDNAのμgでcfuを分割します。次に、結果を希釈係数で除算します。この例では、1/10希釈を用いて、1ml溶液の100μLをめっきした(希釈:1/10×100μL/1000 μL=0.01)。

細菌は非常に適応可能であり、この適応を容易にする1つのメカニズムは、外部DNA分子を取り込む能力である。細菌が取り込むことができるDNAの1つのタイプはプラスミドと呼ばれ、抗生物質耐性遺伝子などの有用な情報を頻繁に含むDNAの円形部分である。外部ソースから組み込まれた新しい遺伝情報によって細菌が改変される過程を、形質転換と呼ばれる。大腸菌または大腸菌を用いて実験室で簡単に変換を行うことができる。
形質転換するためには、まず大腸菌細胞を有能にしなければならず、それは環境からDNA分子を取り込むことができることを意味する。これを達成するためのプロトコルは、驚くほど簡単で、塩化カルシウム溶液中の細胞の短いインキュベーションである。このインキュベーションは、細胞がDNA分子に透過性になる原因となります。細胞が遠心分離によってペレット化された後、上清が除去される。プラスミドDNAが有能な細胞に追加されるようになりました。DNAで細胞をインキュベートした後、ミックスは一時的に摂氏42度に加熱され、その後、氷上で急速に冷却されます。このヒートショックにより、DNAが細胞の壁や膜を横切って転送されます。細胞は、その後、新鮮な媒体でインキュベートされます。その後、細菌は37度に配置され、膜を再シールし、耐性タンパク質を発現させることができます。
プラスミド中に取り込んだ細胞は、DNAを忠実にコピーして子孫に渡し、抗生物質耐性メディエーターを含む、それによってコードされる可能性のあるタンパク質を発現します。これらの耐性遺伝子は、プラスミドを取り込んでいない細胞が耐性遺伝子産物を発現しないため、正常に形質転換された細菌を同定するための選択可能なマーカーとして使用することができる。これは、細胞が適切な抗生物質を含む固体培地でめっきされると、プラスミドを取り込んだ細胞だけが成長することを意味します。増殖コロニーにおける細胞の形質転換は、試料からDNAを抽出する前に収率を高めるために一晩液体媒体でそれらの細胞を培養することによってさらに確認することができる。DNAを単離すると、診断制限酵素ダイジェストを行うことができます。制限酵素は予測可能な場所でDNAを切断するので、これらのダイジェストをゲル上で実行すると、所望のプラスミドが正常に形質転換された場合、予測可能なパターンを示す必要があります。例えば、pUC19を調製し、制限酵素HindIIIで切断した場合、2686ヌクレオチドの単一バンドがゲル上に見られる必要があります。
この研究室では、pUC19で大腸菌株DH-5 αを変換し、DNAゲル電気泳動による正常な形質転換を確認します。
手順を開始する前に、ラボコートや手袋を含む適切な個人用保護具を着用してください。次に、70%のエタノールでワークスペースを殺菌します。
さて、無菌LB寒天プレートに細菌のループフルを堆積させ、新しいループで細菌をストリークすることによって、化学的に有能な細胞を調べます。その後、一晩37°Cでプレートをインキュベートします。翌日、再び70%エタノールでベンチトップを殺菌し、インキュベーターからプレートを取り出します。
単一の、よく分離されたコロニーを無菌ループを持つチューブ内のLBスープの3ミリリットルに接種する。その後、210 RPMで揺れで、一晩で摂氏37度で培養を成長させます。翌日、分光光度計で一晩培養の光学密度を測定する。次いで、1リットルフラスコに100ミリリットルのLBスープを加え、光学密度0で一晩培養して接種する。01. 今、揺れで37°Cで培養し、培養が指数関数的な成長段階に達するまで15〜20分ごとにOD600をチェックします。
約3時間後、培養物50ミリリットルを2本の冷たいポリプロピレンボトルに移します。その後、ボトルを氷の上に戻し、20分間冷まします。次に、遠心分離を介して細胞を回復します。上清を捨て、ボトルをペーパータオルの上に逆さまに置きます。次に、冷たい塩化カルシウム塩化マグネシウム溶液の5ミリリットルで細菌ペレットを再中断し、ペレットが完全に溶解するまで慎重に旋回します。次に、溶解した細菌ペレットに溶液の別の25ミリリットルを添加する。前に示したように、他の細菌ペレットを再中断します。この後、遠心分離を繰り返し、上清を取り除きます。
有能な細胞が直接形質転換される場合は、チューブを慎重に旋回することにより、氷冷0.1モル塩化カルシウム溶液の2ミリリットルに各細菌ペレットを再停止します。形質転換手順を開始するには、50マイクロリットルの有能な細胞を2つの標識された1.5ミリリットルポリプロピレンチューブに移します。次に、pUC19プラスミドDNAの1マイクロリットルをチューブの1つに添加する。泡の形成を避け、穏やかに混合し、氷の上で30分間両方のチューブをインキュベートします。インキュベーション後、チューブをヒートブロックに移し、42°Cで45秒間インキュベートします。すぐにチューブを氷に移し、2分間インキュベートします。次に、各チューブに950マイクロリットルのSOC培地を追加し、37°Cで1時間インキュベートして細菌が回復し、プラスミドにコードされた抗生物質耐性マーカーを発現させます。
1~100°Cの希釈を行うには、990マイクロリットルのSOCメディアと10マイクロリットルのセルサスペンションを1.5ミリリットルのチューブに加えます。次いで、1.5ミリリットルのチューブに900マイクロリットルのSOC培体と100マイクロリットルの細胞懸濁液を加えて1~10希釈する。次に、希釈された細胞懸濁液の100マイクロリットルと負の対照の100マイクロリットルをプレートし、拡散機を用いてアンピシリンを含む別々の選択的プレート上に、12~16時間37°Cでプレートをインキュベートする。インキュベーション後、変換によって得られたコロニー形成単位(CFE)をプレートごとにカウントし、これらのデータを記録する。形質転換体にpUC19プラスミドがあることを確認するには、無菌ループを持つプレートから単一の、よく分離されたコロニーを選び、LBスープの3ミリリットルを含むチューブに導入します。その後、一晩、揺れで摂氏37度で培養をインキュベートします。翌日、DNAミニ準備キットを使用して、製造元の指示に従って、培養物の3ミリリットルからDNAを分離します。DNAミニ準備を完了した後、1時間37°Cの制限酵素で精製pUC19の1マイクログラムを消化する。さて、分子量はしごの20マイクロリットル、消化されたプラスミドDNAの1マイクログラム、および1マイクログラムの未消化プラスミドDNAを、1ミリリットル当たり1マイクログラムを含む1%のアガロースゲルの連続したウェルに積み込む。その後、95ボルトで1時間ゲルを実行します。最後に、UVイルミレータでゲルを視覚化します。
この実験では、大腸菌DH5アルファ化学的に有能な細胞を塩化カルシウム手順の適応を用いて調製し、次いでプラスミドpUC19で形質転換効率を決定した。変換効率を計算するには、記録された CFU カウントを 100 で 1 回、10 希釈で 1 回、CFU が 30 ~ 300 の他の希釈を使用します。まず、記録されたCFUカウントは、この例では246を、DNAの量で割って、ここで.0001マイクログラム、めっきされた。次に、この数値は、マイクログラムあたりのCFUの変換効率を与えるために使用される希釈係数で割られる。この例では、1~10の希釈を用いて、1ミリリットル溶液の100マイクロリットルをめっきし、最終的な希釈係数を0.01にした。未消化プラスミドレーンでは、円形DNAは、様々な明るさの2つまたは3つの異なるバンドとして現れることがあります。これは、円形の切断されていないDNAは、スーパーコイル状、開いた円、またはより線形など、いくつかの異なる立体構造状態に存在し、これらの各々が異なる速度でゲルを通過する可能性があるためです。回収されたプラスミドDNA消化の分析は、使用されるプラスミドがpUC19 DNA、2,686塩基対の予想サイズを有することを示した。
Subscription Required. Please recommend JoVE to your librarian.
Results
TEは多くの要因に依存しているが、このような非商業的な有能な細胞製剤は、通常、プラスミドのマイクログラム当たり106〜10 7形質転換体を得る。したがって、この調製物は、TE= 2.46 x 108 cfu/μgを有し、TEが予想範囲をはるかに超えた。特定のアプリケーションに対して高い変換効率が必要な場合に、超有能なセルを作成するための追加のプロトコルが利用可能です (13) .
形質転換細胞から回収されたプラスミドDNAの消化の分析は、このプラスミドがpUC19 DNA(2686 bp)の期待サイズを有することを示した。
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
形質転換は、研究室の多くの分子生物学アプリケーションの鍵となる細菌細胞に外因性DNAを導入するための強力な方法です。さらに、細菌細胞が遺伝性の変化を増加させ、幅広い条件下で生存のための異なる有益な形質の獲得を可能にする遺伝物質を交換できるようにすることで、自然界で大きな役割を果たしています。多くの細菌株は、自然な能力に必要な遺伝子をコードします。しかし、これらの遺伝子が誘導される条件はまだ不明である。これらの条件を決定するためにさらなる研究が必要です。.
Subscription Required. Please recommend JoVE to your librarian.
References
- Croucher, N. J. et al. Rapid pneumococcal evolution in response to clinical interventions. Science. 331 (6016):430-434. (2011)
- Borgeaud, S. et al. The type VI secretion system of Vibrio cholerae fosters horizontal gene transfer. Science. 347(6217):63-67. (2015)
- Burmeister, A. R. Horizontal Gene Transfer. Evol Med Public Health. 2015 (1):193-194. (2015)
- Weston A, Brown MG, Perkins HR, Saunders JR, Humphreys GO. Transformation of Escherichia coli with plasmid deoxyribonucleic acid: calcium-induced binding of deoxyribonucleic acid to whole cells and to isolated membrane fractions. J Bacteriol. 145 (2):780-7. (1981)
- Dagert M, Ehrlich SD. Prolonged incubation in calcium chloride improves the competence of Escherichia coli cells. Gene. 6 (1):23-8. (1979)
- Asif A, Mohsin H, Tanvir R, and Rehman Y. Revisiting the Mechanisms Involved in Calcium Chloride Induced Bacterial Transformation. Front Microbiol. 8:2169. (2017)
- Panja S, Aich P, Jana B, Basu T. How does plasmid DNA penetrate cell membranes in artificial transformation process of Escherichia coli? Mol Membr Biol. 25 (5):411-22. (2008)
- Silhavy, TJ, Kahne D, Walker S. The Bacterial Cell Envelope. Cold Spring Harb Perspect Biol. 2 (5): a000414. (2010)
- Panja S, Aich P, Jana B, Basu T. (2008) Plasmid DNA binds to the core oligosaccharide domain of LPS molecules of E. coli cell surface in the CaCl2-mediated transformation process. Biomacromolecules. 9 (9):2501-9.
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Plasmid Purification. JoVE, Cambridge, MA. (2018)
- Bergkessel M and Guthrie C. Colony PCR. Methods in Enzymology. 529: 299-309. (2013)
- Sambrook J and Russell DW. Molecular Cloning A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.Protocol 25 (1.116-118). (2001)
- Wirth R, Friesenegger A, Fiedler S. Transformation of various species of gram-negative bacteria belonging to 11 different genera by electroporation. Molecular & General Genetics. 216 (1): 175-7. (1989)