



Overview
ソース: スザンナ C. シスラー1, トーニャ J. ウェッブ1
1メリーランド大学ボルチモア校微生物学・免疫学科、MD 21201
免疫沈殿(IP、また「プルダウンアッセイ」として知られている)は、様々な分野でアプリケーションを持っている広く使用されている技術です。1984年に初めて考案され、1988年(1、2)に精製されました。IPの基本的な目標は、そのタンパク質に対する抗体を用いた特定のタンパク質の精製と単離です。「免疫」という言葉は抗体の使用を指し、「沈殿」という言葉は溶液から特定の物質を引き下すことを指します。標的タンパク質は、内因性または組換えである可能性があります。ほとんどの組換えタンパク質は、その後の精製を簡素化するためにエピトープタグ(すなわちmycまたはフラグ)を付着しています。通常、組換えエピトープタグに対する抗体は非常に強力で効果的であるため、組換えタンパク質IPを最適化する方が簡単です。内因性タンパク質に対する抗体は非常に可変的な有効性を持ち、これらのIPの最適化がはるかに困難です。免疫沈殿後の必要なステップは、精製の検証です。単離されたタンパク質はSDS-PAGEを使用して解決され、その後、ウェスタンブロットによって純度を調べた(図1)。重要なコントロールは、正しいタンパク質のプルダウンを検証するために、ウェスタンブロット中に別の抗体を使用することです。IP と後続の手法の組み合わせは、強力な分析ツールです。精製後の目標は、NMR、質量分析、インビトロアッセイによるタンパク質自体の特徴付け、またはタンパク質の相互作用パートナー(タンパク質、DNA、RNA)の分析(3、4、5)であり得る。
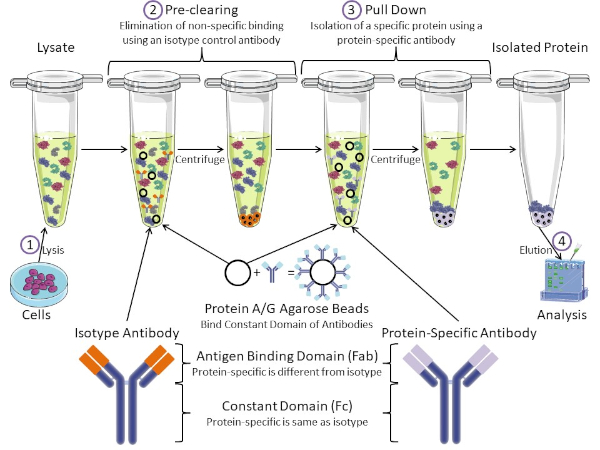
図1:免疫沈殿手順の概要免疫沈殿は、抗体を用いた特定のタンパク質の単離である。細胞からのリサートの生産後、2つの主要なステップがあります- 事前クリアとプルダウン。事前クリアステップの間、細胞分は、アイソタイプコントロール抗体を使用して非特異的に抗体に結合するタンパク質を事前に除去する。プルダウン工程では、標的タンパク質はタンパク質特異的抗体を用いてプルダウンされる。その後、単離されたタンパク質をウェスタンブロットで分析します。アイソタイプ抗体およびタンパク質特異的抗体は、同じ定常ドメインを持つが、異なる抗原結合ドメインを持つ。このプロトコルの重要な構成要素は、抗体の一定ドメインを結合するタンパク質A/Gアガロースビーズであり、標的タンパク質の免疫沈殿を可能にする。この図のより大きなバージョンを表示するには、ここをクリックしてください。
抗体は、他の形態のタンパク質精製(すなわちニッケル親和性カラム精製)と区別する免疫沈殿の重要な構成要素である。抗体は、特定のタンパク質エピトープを認識できるB細胞によって作られた分子です。抗体には、定数(Fc)と抗原結合(Fab)の2つのドメインがあります(図1)。定数ドメインは抗体の種類を識別し、生体内の機能を指示する。通常、IPに使用される抗体の一定ドメインは、マウス、ラット、またはウサギIgGである。抗体の抗原結合部は、特定のタンパク質の特異的なエピトープを認識する。抗体は、タンパク質が変性した場合に存在しない可能性のある折り畳まれたタンパク質上のエピトープを認識し、その逆も可能です。したがって、エピトープの利用可能性はタンパク質の折りたたみに依存し、IPの抗体および条件を選択する際に考慮すべき重要な要因を同定する。
原核生物系と真核生物系の両方が抗体結合タンパク質を持っています。真核生物系では、原核系では細菌からの免疫保護が目的であり、その目的は免疫系からの保護である。抗体結合タンパク質は、2つの方法でIP方法論に影響を与えます。まず、抗体を結合するタンパク質のライサートを取り除くために必要な事前清算工程(図1)があり、それによって最終製品における非特異的結合を減少させる。このステップでは、タンパク質特異的抗体とは異なる抗体結合ドメインと同じ定常ドメインを有するアイソタイプ抗体を使用します。細菌抗体結合タンパク質は、この方法の第2の重要な構成要素である。タンパク質特異的抗体が標的タンパク質と結合した後、抗体:タンパク質複合体をプルダウンする必要があります(図1)。タンパク質A、G、およびLは、抗体の一定ドメインに結合する細菌タンパク質である。細菌はこれを使用して免疫系を破壊しますが、研究者は抗体精製を容易にするためにこのシステムを共同で選択し、事前クリアとプルダウンの両方のステップで使用されます。これらのタンパク質は、異なる種と異なる定数ドメインサブタイプに対して異なる結合親和性を持っています - IPの条件を選択する際に考慮すべきもう一つの要因。多くの企業では、タンパク質A/Gラベルのアガロースビーズ(図1)、あらかじめ作られたスピンカラム、またはカラムを作る樹脂を販売しています。一般に、ビーズとスピンカラムはサンプルサイズが小さく、樹脂はバルク精製に使用されます。
このラボでは、タンパク質A/Gプラスアガロースビーズをベースにした基本的な免疫沈殿技術を用いて、原発性マウス甲状腺細胞から内因性タンパク質c-mycを精製する方法を示す。プロトコルは細胞リサート調製から始まり、ウェスタンブロット分析を用いて成功したタンパク質プルダウンの検証で終わります。
Procedure
1. タンパク質A/G PLUSアガロースビーズを用した免疫沈殿
細胞リサート製剤
- 遠心分離機108チモシグネタを13,000rpmのマイクロ遠心分離機で3分間、上清を除去する。
注:細胞数は、所望のタンパク質の発現レベルと選択した細胞タイプによって異なります。 - PMSFを使用して500 μLリシスバッファRIPAで細胞を再中断します。
- 渦でいくつかのクイックパルスを使用して細胞を破壊し、注射器に取り付けられた25G針で数回ライサートを吸引します。
注:バブルの作成は避けてください。より大きなセルタイプには、21G針などの大きな針を使用します。 - 細胞を氷上で10分間インキュベートします。
- 4°Cで15分間13,000rpmでリザートを遠心分離します。
- 上清を新鮮な標識マイクロ遠心管に移します。
事前清算
- 20 μLタンパク質A/G PLUSアガロースビーズと1μgのアイソタイプコントロール抗体(ここでは、マウスIgG1アイソタイプコントロール抗体が使用される)をライサートに添加する。
注:使用されるアイソタイプ抗体の選択は、プルダウン工程の後半で使用されるタンパク質特異的抗体に依存する。 - 冷たい部屋(4°C)の遠心回転子でリザートミックスを30分間インキュベートします。
- 4°Cで30sのための3200のrpmでサンプルを遠心分離する。
- クリア済みの上清を、ラベル付きの1.5mLマイクロ遠心管に移します。ペレットを捨てます。
タンパク質濃度決定
- ブラッドフォードアッセイを行うことにより、細胞のタンパク質濃度を決定します。
- アリコート1000 μLブラッドフォード試薬を7マイクロ遠心管に入れます。
- 次の量の BSA タンパク質標準 (2 mg/mL) をチューブの 6 本に加えます (表 1)。
| チューブ番号 | BSA 体積 (μL) (2 mg/mL) | タンパク質濃度(μg/μL) |
| 1 | 0 | 0 |
| 2 | 1 | 2 |
| 3 | 2 | 4 |
| 4 | 3 | 6 |
| 5 | 4 | 8 |
| 6 | 5 | 10 |
表 1:BSAタンパク質標準量
- 第7管に、予クリアされたライサトの1μLを加える。
注:サンプル濃度がアッセイ検出範囲内にあることを確認するには、1:2 または 1:5 のリサート希釈も準備して分析します。 - 7つのチューブのそれぞれから200 μLを平底の個々のウェルに入れ、各サンプルを三つ編みで繰り返す96ウェルプレートに入れます。
- 595 nmのプレートリーダーのプレートを読み取ります。
- Excelで標準曲線を生成し、クリア済みリサートのタンパク質濃度を計算します。
プルダウン
- 2つの新鮮な1.5 mLマイクロ遠心管チューブに「コントロール」、もう1本を「テスト」とラベル付けします。
- これらのチューブのそれぞれに500 μgの事前にクリアされたライサートを置きます。
注:ここで使用されるタンパク質の量は、精製を希望するタンパク質の量に依存する。 - ライシスバッファーを使用して、各チューブの合計体積を最大 500 μL まで持ち上げます。
- 試験群チューブに2μgの抗c-myc抗体を添加し、2μgマウスIgG1アイソタイプコントロール抗体を対照群に添加する。
注:抗体の量は、抗体の有効性と標的タンパク質の量に依存します。 - 冷たい部屋(4°C)の回転子の管を2時間インキュベートする。
- 各チューブに20μLタンパク質A/G PLUSアガロースビーズを追加します。
注:ビーズの損傷を防ぐために、エンドカットでピペットの先端を使用することをお勧めします。 - 冷たい部屋(4°C)で一晩回転子でインキュベートします。
注:標的タンパク質および抗体の有効性に応じて、このステップは1時間から一晩に変化しうる。 - 30 s 4°Cでチューブを遠心分離し、ビーズを引き下げてください。
- 各チューブから上清を吸引します。
注:標的タンパク質がビーズに結合するようになりました。 - 500 μL 1X ダルベッコのPBSを使用してビーズを2回洗います。
- 30 s 4°Cで3200のrpmで管を遠心分離する。
注:より厳格な洗浄を行うには、RIPA などのより厳格なバッファを使用します。 - 各チューブからバッファを吸引します。ゲルローディングの先端を使用して、ビーズから残りのバッファーを取り除き、ビーズを氷の上に保ち、タンパク質を溶出します。
注:この例では、タンパク質は、ウェスタンブロット分析用にビーズを沸騰させることにより、SDS-PAGE実行バッファーにタンパク質をタンパク質にタンパク質でタンパク質にタンパク質でタンパク質にタンパク質でタンパク質を再生します。このアプローチは、IP結果を検証したり、タンパク質とタンパク質の相互作用を調べたりするのに適しています。構造または酵素解析のためのタンパク質の精製などの他の下流アプリケーションでは、エピトープタグ(フラグタグまたはmyc-tag)などのより洗練されたシステムが目的のタンパク質を有する抗体の溶出を避けるために使用される。
2. ウェスタンブロット解析を用いるIP検証
SDS-PAGE 電気泳動:
- β-メルカプト-エトノールを含む20 μL SDS-PAGE負荷染料でビーズを再停止します。
- サンプルを95°Cで5分間沸騰させます。
- ビーズを室温で10sの13,000 rpmで遠心分離します。
- ゲルローディングの先端を使用して、ビーズから得られたサンプルを慎重に配管し、4-15%の勾配SDS-PAGEゲルの井戸にロードします。
- サンプルに加えて、タンパク質のはしごを持つレーンと、事前にクリアされたライサテを持つレーンをロードして、ローディングコントロールとして機能します。
- 染料の前部がゲルの底に達するまで100Vで走る(~1h)。
ウェスタンブロット分析:
- 西洋のブロットサンドイッチを作り、PVDF膜がゲルと赤陰極の間であることを確認します。
- 100Vで1時間の転送。
- 低設定のロッカー上で1時間の室温で5mLブロッキングバッファーに膜を置き、非特異的タンパク質結合部位を遮断する。
注:ブロッキングバッファー、一次抗体、二次抗体、および洗後の体積は、より大きなサイズのブロットに対して増加する必要がある場合があります。 - 低い設定でロッカーで4°Cで一晩ブロッキングバッファーに5 mL抗c-myc抗体でブロットをインキュベートします。
注:ここで使用する抗体は、プルダウンステップで使用される抗体とは異なる必要があります。 - 5 mL TBST を使用してブロットを 3~6 回洗浄し、各洗浄は低い設定でロッカーの室温で 5 分です。
- HRPタグ付き抗ウサギ軽鎖二次抗体をブロッキングバッファーに入れ、低設定でロッカーの室温で1時間インキュベートします。
注:二次抗体の選択は、ウェスタンブロットに使用される一次抗体に依存します。さらに、標的タンパク質が抗体の重鎖に分子量に近いため、軽鎖特異的二次二次がプロトコルで使用される。標的タンパク質が50kDaに近い場合は、軽鎖特異的二次を使用する。標的タンパク質が25kDaに近い場合は、重鎖特異的二次を使用する。 - 5 mL TBST を使用してブロットを 3~6 回洗浄し、各洗浄は低い設定でロッカーの室温で 5 分です。
- 余分な液体を除去するために実験室のワイプのブロットおよびダブエッジから液体を取り除く。
- 1xケミルミネッセンス検出試薬でブロットをカバーし、1分間インキュベートします。
注:検出試薬は軽く、時間に敏感であるので、次のステップは迅速に連続して行われるべきである。 - 余分な検出試薬を除去するために実験室のワイプのブロットのダブ端。
- イメージャートレイのイメージング面にブロットを配置します。
注:ケミルミネッセンスブロットは、フィルムを使用して視覚化することもできます。 - 「ケミルミネッセンスプログラム」を使用して、10~5分の複数のタイムポイントをキャプチャする画像。
注:最適な時間は、タンパク質の量と抗体の質に基づいて変化する可能性があります。 - 最適なバンドの可視性を持つ画像を選択し、そのイメージを書き出します。
- ブロットを移動する前に、イメージャーを使用してブロットの写真を撮り、はしごの位置をキャプチャする。次に、そのイメージもエクスポートします。
- スライド準備ソフトウェア (PowerPoint など) を使用して、バンドとラダー イメージを位置合わせして単一のイメージを形成します。
免疫沈殿、またはIPは、細胞または組織のリサートまたはタンパク質特性を有する体液から目的のタンパク質を分離したり、タンパク質とタンパク質の相互作用を調べたりするために広く使用されている技術です。
このプロセスは、標的タンパク質に対する高い親和性および特異性を有する抗体から始まる。この抗体は試料と混合され、抗体標的複合体が形成される。標的タンパク質に結合したタンパク質も、その過程で抗体に間接的に付着します。次に、溶液をアガロースビーズでインキュベートし、抗体の一定領域に対して強い親和性を有する細菌タンパク質に結合する。細菌タンパク質は抗体に結合し、抗体標的複合体をビーズに接続します。次いで、溶液を遠心分離してビーズを沈殿させ、それによって結合抗体、標的タンパク質、および任意の相互作用タンパク質を含む複合体全体を抽出する。最後に、結合タンパク質をビーズから抽出し、互いに放出し、ウェスタンブロッティングなどの技術によるさらなる分析に使用されます。
この技術のいくつかのバリエーションは、事前クリア、ペプチドタグまたは磁気ビーズの使用、または他の非タンパク質結合パートナーの分析など、一般的に使用されます。IPは、事前クリアステップによって前接することができ、試料中の非特異的抗体結合タンパク質を除去し、背景を最小限に抑える。これは、最初にアイソタイプコントロール抗体でサンプルをインキュベートし、これらのタンパク質に結合させ、次にアガロースビーズを使用して複合体を沈殿させることを含みます。次に、サンプルは実際の IP に進む準備が整いました。
ペプチドタグは、特定の抗体がIPに使用できない場合に有用である。ここで、標的タンパク質は、ペプチドエピトープタグとタグに対する抗体を含有するように遺伝子改変することができ、目的とするタンパク質を引き出すことができる。磁気ビーズは、多くの場合、ターゲットを沈殿させるためにアガロースの代わりに使用されます。抗体標的複合体に結合した後、試料チューブは強い磁場に置かれ、溶液からビーズを抽出する。これは遠心分離のための必要性を除去し、速度および便利を改善する。
免疫沈殿は、DNAまたはRNA結合タンパク質の研究にも使用され、クロマチン免疫沈殿およびRNA免疫沈殿としてそれぞれ知られています。これらのバリエーションは、さまざまな実験アプリケーションのトラブルシューティングと方法の適応に役立ちます。このビデオでは、細胞リサートを事前にクリアし、目的のタンパク質を抽出するために免疫沈殿を行う方法を観察し、続いてウェスタンブロット分析を行って実験を検証します。
まず、あらかじめ収集した細胞をマイクロ遠心分離機に入れ、13,000rpmで3分間回転させます。スピンに続いて、上清を取り除き、PMSFで500マイクロリットルのリシスバッファーRIPAで細胞を再中断します。さて、渦でいくつかのクイックパルスを使用して細胞を破壊し、その後、気泡を作成しないように注意して、注射器に取り付けられた25ゲージの針で数回リサートを吸引します。細胞を氷の上に15分間置きます。氷上でサンプルをインキュベートした後、4°Cで15分間リサートを遠心分離します。
新しい1.5ミリリットルのマイクロ遠心管にラベルを付けます。スピンに続いて、上清を新たに標識されたチューブに移し、ペレットを廃棄します。次に、タンパク質A/G PLUS-アガロースビーズの20マイクロリットルと同種コントロール抗体の1マイクログラムをリザートに添加することにより、非特異的にアガロースビーズまたは一次抗体に結合する汚染物質のライサートを事前にクリアします。例は、マウス IgG1 アイソタイプコントロールです。冷たい部屋で30分間回転子の管をインキュベートする。冷たい部屋で30分間リザートを回転させた後、サンプルを3200rpmで30秒間遠心分離し、摂氏4度で30秒間遠心分離する。遠心分離機からチューブを取り出し、クリア済みの上清を1.5ミリリットルのマイクロ遠心分離管に移します。ペレットを捨てます。
さて、ブラッドフォードアッセイを行うことにより、細胞のタンパク質濃度を決定する。ラベル 7 1.5ミリリットルのマイクロ遠心チューブ1~6本、サンプルとアリコート1000マイクロリットルのブラッドフォード試薬を各チューブに入れます。6つのチューブは、各チューブにBSAの様々な量の既知の量を追加することにより、標準的な曲線を作るために使用されます。追加する金額は、この表に一覧表示されます。第7のサンプルチューブに、予クリアされたライサートの1マイクロリットルを加える。7つのチューブのそれぞれから200マイクロリットルを平底96ウェルプレートの個々のウェルに入れ、各サンプルを三つ三つ三つ返しにして、7つのサンプルの3つの列があるようにします。595ナノメートルの波長を使用して、プレートリーダー上のプレートを読みます。Excel で標準曲線を作成した後、クリア済みリサートのタンパク質濃度を計算します。
次に、2つの1.5ミリリットルマイクロ遠心分離管に標識を付け、1つは対照管として、もう1つは試験として、この例ではc-myc抗体となる。これらのチューブのそれぞれに500マイクログラムのクリアされたライサートを配置し、その後、ライシスバッファを使用して、各チューブの総容積を500マイクロリットルまで持って来ます。次に、抗c-myc抗体の2マイクログラムを試験群チューブに添加する。対照のために、マウスIgG1アイソタイプ対照抗体の2マイクログラムを添加する。抗体をチューブに加えたら、サンプルを冷たい部屋の回転子に置き、2時間インキュベートします。次に、アガロゼビーズを追加します。これを行うには、ピペット先端の端を切り取り、この修正された先端を使用して、各チューブにプロテインA/Gプラスアガロースビーズの200マイクロリットルを追加することをお勧めします。一晩冷たい部屋の回転子の管をインキュベートする。
インキュベーションの後、回転子からチューブを取り出し、マイクロ遠心分離機でライサテを回転させ、ビーズを引き下げる。スピンが完了したら、遠心分離機からチューブを取り外し、各チューブから上清を吸引します。次に、1XダルベッコのPBSの500マイクロリットルを使用してビーズを洗浄します。チューブをマイクロ遠心分離機に入れ、4°Cで30秒間スピンダウンします。この後、上清を取り除く。洗浄と遠心分離機のステップを合計2回繰り返します。マイクロ遠心分離機からチューブを取り外し、各チューブからバッファを吸引します。ゲルローディングの先端を使用して、ビーズから残ったバッファーを取り除き、ビーズを氷の上に保ち、結合したタンパク質を溶出させる。
この例では、タンパク質は、ウェスタンブロット分析のために沸騰することにより、SDS-PAGE実行バッファーにタンパク質をタンパク質にタンパク質で再生します。これを行うには、ベータメルカプトエタノール、またはBMEを含むSDS-PAGE負荷染料の20マイクロリットルでビーズを再中断します。サンプルを95°Cで5分間沸騰させ、ビーズから免疫錯体を解離します。次に、ビーズを室温で10秒間最高速度で遠心分離する。マイクロ遠心分離機からチューブを取り外し、室温でラックに保持します。ゲルローディングの先端を使用して、慎重にビーズからサンプルをピペットし、4〜15%の勾配SDS-PAGEゲルの井戸にそれらをロードします。サンプルに加えて、タンパク質のはしごを持つレーンと、事前にクリアされたライサテを持つレーンをロードして、ローディングコントロールとして機能します。ゲルがロードされたら、100ボルトでゲルを実行します。
染料の前面がゲルの底に達した後、約1時間かかります, ゲルを停止し、西洋のブロットサンドイッチを作ります, PVDF膜がゲルと陰極の間にあることを確認.移送装置にウェスタンブロットサンドイッチを置き、ゲル上のタンパク質を100ボルトで1時間膜に移します。転移が完了したら、抗体が膜に非特異的に結合するのを防ぐために、膜を5ミリリットルのブロックに入れます。室温で1時間の低い設定でロックします。タイマーが鳴ったら、ブロッキング バッファを削除します。検出抗体を膜に5ミリリットルのブロッキングバッファーを添加する。ここでは、プルダウンに使用されるものとは異なる抗c-myc抗体が使用される。
低い設定でロッカーに摂氏4度で、一晩ブロットをインキュベートします。インキュベーションに続いて、抗体および遮断バッファーを除去する。室温で5ミリリットルのTBSTを使用して、低い設定でロッカーでブロットを洗います。この洗浄工程は、各洗浄のために新鮮なTBSTを使用して、合計3〜6回の洗浄のために2〜5回繰り返す必要があります。1~1000の二次抗体の5ミリリットルを追加し、ブロットにブロックバッファーを追加します。この場合、二次抗体はHRPタグ付き抗ウサギ軽鎖である。室温で低い設定でロッカーのブロットをインキュベートします。次に、バッファを取り外し、TBSTの5ミリリットルでブロットを洗浄します。この洗浄を室温で5分間低い設定でロッカーにインキュベートします。この洗浄を6~12回繰り返し、それぞれに新鮮な5ミリリットルのTBSTを入れ直します。最初にブロットから液体を注ぐことによって、最終的な洗浄を削除します。次に、ピンセットを使用して、余分な液体を除去し、新鮮な容器にブロットを置くために実験室のワイプにブロットの端を塗ります。次に、1Xケミルミネッセンス検出試薬でブロットを覆い、1分間インキュベートします。
迅速に作業し、実験室のワイプでブロットの端を塗り、余分な検出試薬を取り除き、Imager トレイのイメージングサートの画像面にブロットを配置します。ケミルミネッセンスプログラムを使用して10~30秒の複数の時間ポイントをキャプチャする画像。ブロットをイメージした後、最適なバンドの可視性を持つ画像を選択し、そのイメージを書き出します。ブロットを移動する前に、Imager を使用してブロットの写真を撮り、はしごの位置をキャプチャします。次に、そのイメージもエクスポートします。最後に、PowerPoint などのスライド準備ソフトウェアを使用して、バンドとラダー イメージを位置合わせして 1 つのイメージを形成します。
この画像は、胸腺細胞からのタンパク質c-mycの免疫沈殿に対するウェスタンブロット結果を示す。左から右に、レーンはアイソタイプコントロール、c-myc IP、および事前にクリアされたリサート入力を表します。右上の車線は、分子量はしごのマージされた画像です。強いバンドは、約25キロダルトンで軽鎖からであり、50キロダルトンの1つは結合抗体の重鎖からのものであり、IPまたはサンプルに非特異的である。C-mycは西部のブロットで約67キロダルトンを走り、通常は75キロダルトンのはしごバンドのすぐ下に見えます。このブロットでは、c-mycバンドは第2車線に見えますが、第1レーンに存在せず、IP抗体が正常にc-mycを引き下がっていることを示します。事前にクリアされたリサートレーンには目に見えるバンドがなく、このタンパク質の内因性発現レベルが低いことが示唆されています。
Subscription Required. Please recommend JoVE to your librarian.
Results
上記の手順の結果を図 2 に示します。左から右に、レーンは対照群(アイソタイプ)、試験群(c-myc)、事前にクリアされたリサート(リサート)、および分子量はしご(はしご)を含む。25 kDa のはしごバンドには、次のマークが付いています。約25 kDaおよび50 kDaの2つの顕著なバンドは、それぞれ結合抗体の軽くて重い鎖であり、IPまたはサンプルに非特異的である。c-mycタンパク質は、西洋のブロット上で67kDaの周りを実行し、通常75 kDaラダーバンドのすぐ下に表示されます。このブロットでは、c-mycバンドは第2車線に見えますが、第1レーンに存在せず、IP抗体が正常にc-mycを引き下がっていることを示します。事前にクリアされたリサートレーンには目に見えるバンドがなく、このタンパク質の内因性発現レベルが低いことが示唆されています。

図2:ウェスタンブロット分析の結果は、免疫沈殿によるc-mycの精製を評価するために用いられる。c-mycに対応する67kDaのバンドは、アンチc-mycレーンに見えますが、アイソタイプコントロールレーンには表示されません。c-myc レベルは、ライサテレーンで視覚化するのに十分な高さではなかったことに注意してください。この図のより大きなバージョンを表示するには、ここをクリックしてください。
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
要するに、免疫沈殿は、抗体を用いた特定のタンパク質の単離である。この例では、免疫沈殿の結果をウェスタンブロットによって分析し、純度を評価した。単離されたタンパク質は、タンパク質構造のためのNMR、アミノ酸配列のための質量分析、または酵素特性を特徴付けるインビトロアッセイを含む多くのアプリケーションで使用することができます。また、AP は、タンパク質の相互作用するパートナーを特徴付けることもできます。例えば、次の単離では、DNAまたはRNAをシーケンシングのために単離することができる。共免疫沈殿は、タンパク質とタンパク質の相互作用を評価します。IP中に標的タンパク質をプルダウンすると、相互作用するタンパク質もプルダウンできます。これらの相互作用するパートナーは、質量分析とウェスタンブロットによって評価することができます。免疫沈殿は、タンパク質生物学を研究するための強力な技術です。
Subscription Required. Please recommend JoVE to your librarian.
References
- Olliver, C. L. and Boyd, C. D. (1984). Immunoprecipitation of In Vitro Translation Products with Protein A Bound to Sepharose. In J. M. Walker (eds), Nucleic Acids. Methods in Molecular Biology (pp. 157-160). New Jersey: Humana Press.
- Thurston, C. F. and Henley, L. F. (1988). Direct Immunoprecipitation of Protein. In J. M. Walker (eds), New Protein Techniques. Methods in Molecular Biology (pp. 149-158). New Jersey: Humana Press.
- Anderson, N. G. (1998). Co-immunoprecipitation: Identification of Interacting Proteins. In R. A. Clegg (eds), Protein Targeting Protocols.Methods in Molecular Biology (pp. 35-45). New Jersey: Humana Press.
- Jackson, D. I. and Dickson, C. (1999). Protein Techniques: Immunoprecipitation, In Vitro Kinase Assays, and Western Blotting. In P.T. Sharpe and I. Mason (eds), Molecular Embryology. Methods in Molecular Biology (pp. 699-708). New Jersey: Humana Press.
- Trieu, E. P. and Targoff, I. N. (2015). Immunoprecipitation: Western Blot for Proteins of Low Abundance. In B. Kurien and R. Scofield (eds), Western Blotting. Methods in Molecular Biology (pp. 327-342). New York, NY: Humana Press.