



Overview
Source: Susannah C. Shissler1, Tonya J. Webb1
1 Département de microbiologie et d'immunologie, Université du Maryland, Baltimore, MD 21201
L'immunoprécipitation (IP, également connue sous le nom d'un «pull-down» d'essais) est une technique largement utilisée qui a des applications dans une variété de domaines. Conçu pour la première fois en 1984, il a été affiné en 1988 (1, 2). L'objectif fondamental de la propriété intellectuelle est la purification et l'isolement d'une protéine spécifique à l'aide d'un anticorps contre cette protéine. Le mot «immuno» fait référence à l'utilisation d'un anticorps tandis que le mot «précipitation» fait référence à l'arrêt d'une substance spécifique d'une solution. La protéine cible peut être endogène ou recombinante. La plupart des protéines recombinantes ont une étiquette d'épitope (c.-à-d. myc ou drapeau) attachée à elles pour simplifier la purification suivante. Typiquement, il est plus facile d'optimiser la protéine recombinante IP parce que les anticorps contre les étiquettes d'épitope recombinant sont très forts et efficaces. Les anticorps contre les protéines endogènes ont une efficacité extrêmement variable - ce qui rend beaucoup plus difficile d'optimiser ces adresses IP. Une étape nécessaire après l'immunoprécipitation est la vérification de la purification. La protéine isolée est résolue à l'aide de SDS-PAGE et par la suite sondée pour la pureté par les taches occidentales (figure 1). Un contrôle important est l'utilisation d'un anticorps différent pendant la tache occidentale pour vérifier tirer vers le bas de la protéine correcte. La combinaison de la propriété intellectuelle avec les techniques suivantes est un outil d'analyse puissant. L'objectif après la purification peut être la caractérisation de la protéine elle-même par la RMN, la spectrométrie de masse et les essais in vitro, ou l'analyse des partenaires interagissant de la protéine (c.-à-d. protéine, ADN, ARN) (3, 4, 5).
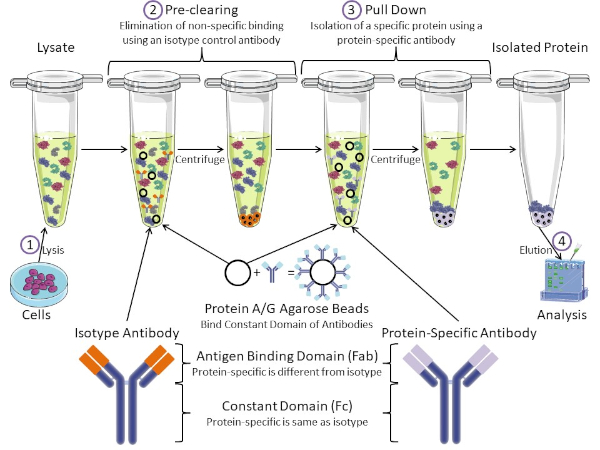
Figure 1 : Aperçu de la procédure d'immunoprécipitation. L'immunoprécipitation est l'isolement d'une protéine spécifique à l'aide d'un anticorps. Après la production de lysate à partir de cellules, il y a deux étapes principales- pré-dédouanement et tirer vers le bas. Pendant l'étape de pré-dédouanement, les lysates cellulaires sont pré-dédouanés des protéines qui se lient aux anticorps non spécifiquement à l'aide d'un anticorps anti-isotype. Dans l'étape de traction vers le bas, la protéine cible est tirée vers le bas à l'aide d'un anticorps protéique spécifique. La protéine isolée est ensuite analysée par Western blot. Les anticorps isotypes et les anticorps protéiques spécifiques ont le même domaine constant, mais différents domaines de liaison d'antigène. Un composant clé de ce protocole est protein A/G perles d'agarose qui lient le domaine constant des anticorps- permettant l'immunoprécipitation de la protéine cible. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Les anticorps sont la composante clé d'une immunoprécipitation qui la différencie des autres formes de purification des protéines (c.-à-d. la purification de la colonne d'affinité au nickel). Les anticorps sont des molécules fabriquées par des cellules B qui peuvent reconnaître des épitopes protéiques spécifiques. Les anticorps ont deux domaines : constante (Fc) et liaison antigène (Fab) (figure 1). Le domaine constant identifie le type d'anticorps et dicte la fonction in vivo. Habituellement, les domaines constants des anticorps utilisés pour la propriété intellectuelle sont la souris, le rat ou le lapin IgG. La partie de liaison d'antigène de l'anticorps reconnaît un épitope spécifique d'une protéine spécifique. Les anticorps peuvent reconnaître les épitopes sur les protéines pliées qui peuvent ne pas exister lorsque la protéine est dénaturée et vice versa. Par conséquent, la disponibilité de l'épitope dépend du pliage des protéines - identifier un facteur important à considérer lors du choix des anticorps et des conditions pour la propriété intellectuelle.
Les systèmes procaryotes et eucaryotes ont des protéines liant les anticorps. Dans les systèmes eucaryotes, le but est la protection immunitaire contre les bactéries tandis que dans les systèmes procaryotes, le but est la protection contre le système immunitaire. Les protéines liant les anticorps affectent la méthodologie de la propriété intellectuelle de deux façons. Tout d'abord, il y a une étape nécessaire de pré-dédouanement (figure 1) pour débarrasser le lysate des protéines qui lient les anticorps - réduisant ainsi la liaison non spécifique dans le produit final. Cette étape utilise un anticorps isotype qui a le même domaine constant que, mais un domaine de liaison d'anticorps différent de votre anticorps spécifique aux protéines. Les protéines bactériennes liant les anticorps sont le deuxième composant clé de cette méthode. Après que l'anticorps protéique-spécifique lie la protéine cible, l'anticorps : complexe de protéine doit être tiré vers le bas (figure 1). Les protéines A, G et L sont des protéines bactériennes qui lient le domaine constant des anticorps. Alors que les bactéries l'utilisent pour subvertir le système immunitaire, les chercheurs ont coopté ce système pour la purification facile des anticorps, et il est utilisé à la fois pendant les étapes de pré-dédouanement et de traction. Ces protéines ont des affinités de liaison différentes pour différentes espèces et différents sous-types de domaine constants - un autre facteur à considérer lors du choix des conditions pour la propriété intellectuelle. De nombreuses entreprises vendent des perles d'agarose étiquetées Protéines A/G (figure 1), des colonnes de spin préfabriquées ou des résines pour fabriquer des colonnes. En général, les perles et les colonnes de spin sont utilisées pour de plus petites tailles d'échantillon tandis que les résines sont utilisées pour la purification en vrac.
Dans cet exercice de laboratoire, nous démontrons comment purifier la protéine endogène c-myc, des thymocytes murines primaires, utilisant la protéine A/G Plus agarose perles basées sur la technique d'immunoprécipitation de base. Le protocole commence à partir de la préparation du lysate cellulaire et se termine par la vérification de la protéine réussie tirer vers le bas en utilisant l'analyse de tache occidentale.
Procedure
1. Immunoprécipitation utilisant des perles d'agarose de protéine A/G PLUS
Préparation de lysate cellulaire
- Centrifuger 108 thymocytes dans un microcentrifugeà 13 000 tr/min pendant 3 min et retirer le supernatant.
Note: Le nombre de cellules varie en fonction des niveaux d'expression de la protéine désirée et du type de cellule choisi. - Re-suspendre les cellules dans 500 'L tampon de lyse RIPA avec PMSF.
- Perturber les cellules à l'aide de quelques impulsions rapides avec un vortex, puis aspirer le lysate à quelques reprises avec une aiguille de 25 G attachée à une seringue.
Note: Évitez de créer des bulles. Utilisez une aiguille plus grande comme une aiguille 21G pour les plus grands types de cellules. - Incuber le lysate cellulaire sur la glace pendant 10 min.
- Centrifuger le lysate à 13 000 tr/min pendant 15 min à 4 oC.
- Transférer le supernatant dans un tube microcentrifuge frais et étiqueté.
Pré-dédouanement
- Ajouter 20 perles d'agarose de protéines A/G PLUS et 1 g d'anticorps anti-isotype (ici, l'anticorps anti-isotype IgG1 de souris est utilisé), au lysate.
Note: Le choix de l'anticorps isotype utilisé dépendra de l'anticorps protéique spécifique utilisé plus tard dans l'étape de traction. - Incuber le mélange de lysate sur un rotateur de centrifugeuse dans la chambre froide (4 oC) pendant 30 min.
- Centrifugelifuger l'échantillon à 3200 tr/min pour 30 s à 4oC.
- Transférer le supernatant prédéblant dans un tube microcentrifuge frais, étiqueté de 1,5 ml. Jeter la boulette.
Détermination de la concentration des protéines
- Déterminez la concentration protéique du lysate cellulaire en effectuant un résultat d'exécution de Bradford.
- Aliquot 1000 L Bradford Reagent en 7 tubes de microcentrifuge.
- Ajouter les quantités suivantes de protéine Standard BSA (2 mg/mL) dans 6 des tubes (tableau 1).
| Numéro de tube | Volume de BSA (L) (2 mg/mL) | Concentration de protéines (g/L) |
| 1 | 0 | 0 |
| 2 | 1 | 2 |
| 3 | 2 | 4 |
| 4 | 3 | 6 |
| 5 | 4 | 8 |
| 6 | 5 | 10 |
Tableau 1 : Quantités standard de protéines BSA
- Dans le tube de 7e, ajouter 1 l de lysate pré-éclairci.
Note: Pour s'assurer que la concentration de l'échantillon se situe dans la plage de détection d'analyse, préparez et analysez également une dilution de lysate de 1:2 ou 1:5. - Placer 200 l de chacun des 7 tubes dans des puits individuels d'une plaque à fond plat de 96 puits répétant chaque échantillon en triple.
- Lire la plaque sur un lecteur de plaque à 595 nm.
- Générez la courbe standard dans excel et calculez la concentration protéique du lysate pré-éclairci.
démolir
- Étiquetez deux tubes microcentrifugefrais de 1,5 ml, l'un comme « contrôle » et l'autre comme « test », qui dans cet exemple est c-myc.
- Placer 500 g de lysate pré-dédouané dans chacun de ces tubes.
Note: La quantité de protéines utilisée ici dépendra de la quantité de protéines désirées pour être purifiée. - Apportez le volume total pour chaque tube jusqu'à 500 L à l'aide de tampon de lyse.
- Ajouter 2 g d'anticorps anti-c-myc au tube du groupe d'essai et 2 anticorps anti-virus igG1 de souris à l'anticorps anti-myc.
Remarque : La quantité d'anticorps dépendra de l'efficacité des anticorps et de la quantité de la protéine cible. - Incuber les tubes sur un rotateur dans la chambre froide (4 oC) pendant 2 h.
- Ajouter 20 perles d'agarose de protéines A/G PLUS à chaque tube.
Note: Il est conseillé d'utiliser des pointes de pipette avec la coupe d'extrémité pour empêcher des dommages aux perles. - Incuber sur un rotateur dans la chambre froide (4 oC) pendant la nuit.
Note: Selon la protéine cible et l'efficacité des anticorps, cette étape peut varier de 1 h à la nuit. - Centrifuger les tubes à 3200 tr/min pour 30 s 4 oC pour tirer vers le bas les perles.
- Aspirez le supernatant de chaque tube.
Note: La protéine cible est maintenant liée aux perles. - Laver les perles deux fois à l'aide du PBS de 500 L 1X Dulbecco.
- Centrifuger les tubes à 3200 tr/min pour 30 s 4oC.
Note: Pour un lavage plus rigoureux, utilisez des tampons plus stricts, tels que RIPA. - Aspirez le tampon de chaque tube. À l'aide de pointes de gel, retirer les restes de tampon des perles et garder les perles sur la glace pour éliper la protéine.
Note: Dans cet exemple, la protéine est éludée dans SDS-PAGE en cours d'exécution tampon en faisant bouillir les perles, pour l'analyse de tache occidentale. Cette approche convient à la vérification des résultats de la propriété intellectuelle ou à l'examen des interactions protéines-protéines. Pour d'autres applications en aval, telles que la purification des protéines pour l'analyse structurelle ou enzymatique, des systèmes plus sophistiqués, tels que les étiquettes d'épitope (drapeau-tag ou myc-tag) sont utilisés pour éviter l'élution de l'anticorps avec la protéine d'intérêt.
2. Vérification de la propriété intellectuelle par l'analyse western Blot
SDS-PAGE Electrophorésis:
- Suspendre à nouveau les perles dans le colorant de chargement SDS-PAGE de 20 L contenant le mercapto-ethonol.
- Faire bouillir les échantillons à 95 oC pendant 5 min.
- Centrifuger les perles à 13 000 tr/min pour 10 s à température ambiante.
- À l'aide de pointes de chargement de gel, épileverts soigneusement les échantillons obtenus à partir des perles et les charger dans des puits de 4-15% gradient SDS-PAGE gel.
- En plus des échantillons, chargez une voie avec une échelle protéique ainsi qu'une voie avec le lysate pré-dédouané pour servir de contrôle de chargement.
- Exécuter à 100 V jusqu'à ce que l'avant de teinture atteigne le fond du gel (1h).
Analyse Western Blot:
- Faire sandwich western blot, en veillant à ce que la membrane PVDF est entre le gel et la cathode rouge.
- Transfert pour 1 h à 100 V.
- Placez la membrane dans un tampon de blocage de 5 ml à température ambiante pendant 1 h sur un rocher à un réglage bas, pour bloquer les sites de liaison protéique non spécifiques.
Remarque : Les volumes de tampon de blocage, d'anticorps primaires, d'anticorps secondaires et de lavages peuvent devoir être augmentés pourles taches de plus grande taille. - Incuber la tache avec 5 mL d'anticorps anti-c-myc dans le tampon de blocage pendant la nuit à 4 oC sur le rocker à un réglage bas.
Remarque : L'anticorps utilisé ici doit être différent de celui utilisé dans l'étapede traction . - Laver la tache 3-6 fois à l'aide de 5 mL de TTL avec chaque lavage étant de 5 min à température ambiante sur un rocker à un réglage bas.
- Incuber la tache avec HRP-marqué anti-lapin anti-lapin chaîne secondaire chaîne de lumière dans le tampon de blocage, pour 1 h à température ambiante sur le rocker à un réglage bas.
Note: Le choix de l'anticorps secondaire dépendra de l'anticorps primaire utilisé pour la tache occidentale. En outre, une chaîne légère secondaire spécifique est utilisé dans le protocole parce que la protéine cible est proche en poids moléculaire de la chaîne lourde de l'anticorps. Si la protéine cible est proche de 50kDa, utilisez une chaîne de lumière secondaire spécifique. Si la protéine cible est proche de 25kDa, l'utilisation etla chaîne lourde spécifique secondaire . - Laver la tache 3-6 fois à l'aide de 5 mL de TTL avec chaque lavage étant de 5 min à température ambiante sur un rocker à un réglage bas.
- Retirer le liquide de la tache et tamponner le bord de la tache sur les lingettes de laboratoire pour enlever l'excès de liquide.
- Couvrir la tache avec 1x réagent de détection chemiluminescente et couver pendant 1 min.
Note: Les étapes suivantes doivent être effectuées en succession rapide car le réactif de détection est léger et sensible au temps. - Dab bord de tache sur les lingettes de laboratoire pour enlever le réactif de détection excédentaire.
- Placez la tache sur la surface d'imagerie du plateau Imager.
Note: Les taches chemiluminescentes peuvent également être visualisées à l'aidede film . - Image utilisant le «Programme Chemiluminescent» pour capturer plusieurs points de temps de 10 s à 5 min.
Remarque : Le temps optimal peut changer en fonction de la quantité de protéines et de la qualité des anticorps. - Choisissez une image avec une visibilité optimale de la bande, puis exportez cette image.
- Avant de déplacer la tache, prenez une photo de la tache à l'aide de l'imageur, pour capturer l'emplacement de l'échelle. Ensuite, exportez cette image aussi.
- À l'aide d'un logiciel de préparation de diapositives (comme PowerPoint), alignez les bandes et les images de l'échelle pour former une seule image.
L'immunoprécipitation, ou IP, est une technique largement utilisée pour isoler une protéine d'intérêt d'un lysate cellulaire ou tissulaire ou d'un liquide corporel pour la caractérisation des protéines ou pour étudier les interactions protéines-protéines.
Le processus commence par un anticorps, qui a une forte affinité et spécificité pour la protéine cible. Cet anticorps est mélangé à l'échantillon, ce qui permet aux complexes anticorps-cibles de se former. Toute protéine liée à la protéine cible est également indirectement attaché à l'anticorps dans le processus. Ensuite, la solution est incubée avec des perles d'agarose, conjuguées à une protéine bactérienne, qui a une forte affinité pour la région constante des anticorps. La protéine bactérienne se lie à l'anticorps et relie les complexes anticorps cibles aux perles. Ensuite, la solution est centrifugepour précipiter les perles, extrayant ainsi l'ensemble du complexe contenant l'anticorps liant, la protéine cible et toutes les protéines en interaction. Enfin, les protéines liées sont extraites des perles et libérées les unes des autres et sont utilisées pour une analyse plus approfondie par des techniques telles que le ballonnement occidental.
Plusieurs variantes de différentes parties de cette technique sont couramment utilisées, comme le pré-dédouanement, l'utilisation d'étiquettes peptidiques ou de perles magnétiques, ou l'analyse d'autres partenaires de liaison non protéiques. La propriété intellectuelle peut être précédée par une étape de pré-compensation, afin d'éliminer les protéines non spécifiques liant les anticorps dans l'échantillon et de minimiser l'arrière-plan. Il s'agit d'abord d'incuber l'échantillon avec des anticorps anti-isotypes, ce qui leur permet de se lier à ces protéines, puis d'utiliser des perles d'agarose pour précipiter les complexes. L'échantillon est alors prêt à passer à la propriété intellectuelle réelle.
Les balises Peptide sont utiles si un anticorps spécifique n'est pas disponible pour la propriété intellectuelle. Ici, la protéine cible peut être génétiquement modifiée pour contenir une étiquette d'épitope peptidique et un anticorps contre l'étiquette est capable de retirer la protéine d'intérêt. Les perles magnétiques sont souvent utilisées au lieu d'agarose pour précipiter la cible. Après s'être reliuté au complexe anticorps-cible, le tube d'échantillon est placé dans un champ magnétique fort, qui extrait les perles de la solution. Cela élimine le besoin de centrifugation et améliore la vitesse et la commodité.
L'immunoprécipitation est également utilisée pour l'étude de l'ADN ou des protéines liant esquimables par l'ARN et sont connues sous le nom d'immunoprécipitation de chromatine et d'immunoprécipitation d'ARN, respectivement. Ces variations sont utiles pour le dépannage et l'adaptation de la méthode pour différentes applications expérimentales. Dans cette vidéo, vous observerez comment pré-effacer un lysate cellulaire et effectuer l'immunoprécipitation pour extraire une protéine d'intérêt, suivie de l'analyse de tache occidentale pour valider l'expérience.
Pour commencer, placez les cellules pré-collectées dans un microcentrifugeet et tournez à 13 mille tr/min pendant trois minutes. Après le spin, retirez le supernatant, puis resuspendles les cellules en 500 microlitres de tampon de lyse RIPA avec PMSF. Maintenant, perturber les cellules en utilisant quelques impulsions rapides avec un vortex, puis aspirer le lysate à quelques reprises avec une aiguille de 25 jauges attachée à une seringue, en prenant soin d'éviter de créer des bulles. Placer les cellules sur la glace pendant 15 minutes. Après avoir incubé les échantillons sur la glace, centrifuger la lysation pendant 15 minutes à quatre degrés Celsius.
Étiquetez un nouveau tube microcentrifuge de 1,5 millilitre. Après la rotation, transférer le supernatant dans le tube fraîchement étiqueté et jeter la pastille. Ensuite, pré-effacer le lysate des contaminants qui se lient non spécifiquement aux perles d'agarose ou à l'anticorps primaire en ajoutant 20 microlitres des perles de protéine A/G PLUS-agarose et un microgramme d'un anticorps de contrôle d'isotype au lysate, qui dans ce par exemple est une souris IgG1 isotype de contrôle. Incuber le tube sur un rotateur dans une chambre froide pendant 30 minutes. Après avoir fait tourner le lysate dans la chambre froide pendant 30 minutes, centrifuger l'échantillon à 3200 tr/min pendant 30 secondes à quatre degrés Celsius. Retirez le tube de la centrifugeuse et transférez le supernatant pré-autorisé dans un tube microcentrifuge de 1,5 millilitre étiqueté frais. Jeter la boulette.
Maintenant, déterminez la concentration en protéines du lysate cellulaire en effectuant un résultat d'exécution bradford. Étiquette sept 1. Tubes microcentrifugede de 5 millilitres un à six et échantillon et aliquot 1000 microlitres du réactif Bradford dans chaque tube. Six des tubes seront utilisés pour faire une courbe standard en ajoutant diverses quantités de quantités connues de BSA à chaque tube. Les montants à ajouter sont énumérés dans ce tableau. Dans le septième tube d'échantillon, ajouter un microlitre du lysate pré-éclairci. Placer 200 microlitres de chacun des sept tubes dans des puits individuels d'une plaque plate de 96 puits, en répétant chaque échantillon en triplette afin qu'il y ait trois colonnes de sept échantillons. Lisez la plaque sur un lecteur de plaque, à l'aide d'une longueur d'onde de 595 nanomètres. Après avoir créé une courbe standard dans Excel, calculer la concentration en protéines du lysate pré-éclairci.
Ensuite, étiquetez deux tubes microcentrifugede de 1,5 millilitre, l'un comme contrôle et l'autre comme test, qui dans cet exemple, sera l'anticorps c-myc. Placez 500 microgrammes du lysate pré-dédouané dans chacun de ces tubes, puis apportez le volume total pour chaque tube jusqu'à 500 microlitres à l'aide de tampon de lyse. Ensuite, ajoutez deux microgrammes de l'anticorps anti-c-myc au tube du groupe d'essai. Pour le contrôle, ajouter deux microgrammes de la souris IgG1 anticorps anti-isotype. Une fois que les anticorps sont ajoutés aux tubes, placez les échantillons sur un rotateur dans une chambre froide et incuber pendant deux heures. Maintenant, ajoutez les perles d'agarose. Pour ce faire, il est recommandé de couper l'extrémité d'une pointe de pipette, puis, à l'aide de cette pointe modifiée, ajouter 200 microlitres de la protéine A/ G PLUS-agarose perles à chaque tube. Incuber les tubes sur un rotateur dans la chambre froide pendant la nuit.
Après l'incubation, retirer les tubes du rotateur et faire tourner les lysates dans le microcentrifuge pour tirer vers le bas les perles. Une fois la rotation terminée, retirer les tubes de la centrifugeuse et aspirer le supernatant de chaque tube. Ensuite, lavez les perles à l'aide de 500 microlitres de 1X Dulbecco PBS. Placer les tubes dans un microcentrifugeet et faire tourner pendant 30 secondes à quatre degrés Celsius. Après cela, retirez le supernatant. Répétez le lavage et la centrifugeuse marche une fois de plus pour un total de deux fois. Retirez les tubes de la microcentrifuge et aspirez le tampon de chaque tube. À l'aide de pointes de chargement de gel, retirer les restes de tampon des perles, en gardant les perles sur la glace pour éliper la protéine liée.
Dans cet exemple, la protéine est éludée dans le tampon de fonctionnement SDS-PAGE en faisant bouillir pour l'analyse de tache occidentale. Pour ce faire, resuspendre les perles dans 20 microlitres de colorant de chargement SDS-PAGE contenant du bêta-mercaptoéthanol, ou BME. Faire bouillir les échantillons à 95 degrés Celsius pendant cinq minutes pour dissocier les immunocomplexes des perles. Ensuite, centrifuger les perles à vitesse maximale pendant 10 secondes à température ambiante. Retirer les tubes de la microcentrifugeuse et les tenir dans une grille à température ambiante. À l'aide de pointes de chargement de gel, pipette soigneusement les échantillons des perles et les charger dans des puits d'un gel SDS-PAGE de gradient de 4 à 15 %. En plus des échantillons, chargez une voie avec une échelle protéique ainsi qu'une voie avec le lysate pré-dédouané pour servir de contrôle de chargement. Une fois le gel chargé, faire fonctionner le gel à 100 volts.
Après que l'avant de teinture a atteint le fond du gel, qui devrait prendre approximativement une heure, arrêter le gel et faire un sandwich de tache occidentale, s'assurant que la membrane de PVDF est entre le gel et la cathode. Placer le sandwich western dans l'appareil de transfert et transférer les protéines sur le gel à la membrane pendant une heure à 100 volts. Une fois le transfert terminé, placez la membrane en cinq millilitres de bloc pour empêcher les anticorps de se lier non spécifiquement à la membrane. Rock à un réglage bas pendant une heure à température ambiante. Lorsque la minuterie retentit, retirez le tampon de blocage. Ajouter cinq millilitres du tampon de blocage avec l'anticorps de détection à la membrane. Ici, un anticorps anti-c-myc, qui est différent de celui utilisé pour la traction vers le bas, est utilisé.
Incuber la tache pendant la nuit, à quatre degrés Celsius sur un rocker à un réglage bas. Après l'incubation, retirer l'anticorps et bloquer le tampon. Laver la tache, en utilisant cinq millilitres de TBST pendant cinq minutes à température ambiante, sur un rocker à un réglage bas. Cette étape de lavage doit être répétée deux à cinq fois pour un total de trois à six lavages, en utilisant du TBST frais pour chaque lavage. Ajouter cinq millilitres d'un à 1000 anticorps secondaires et bloquer tampon à la tache. Dans ce cas, l'anticorps secondaire est HRP-étiqueté anti-lapin chaîne de lumière. Incuber la tache sur un rocker à un réglage bas pour un notre à température ambiante. Ensuite, retirez le tampon et lavez la tache avec cinq millilitres de TBST. Incuber ce lavage sur un rocker à un réglage bas pendant cinq minutes à température ambiante. Répétez ce lavage pour un total de six à 12 lavages, chacun avec un frais de cinq millilitres de TBST. Retirer le lavage final en versant d'abord le liquide hors de la tache. Ensuite, à l'aide d'une pince à épiler, tamponner le bord de la tache sur une lingette de laboratoire pour enlever tout excès de liquide, puis placer la tache dans un récipient frais. Ensuite, couvrir la tache avec 1X Chemiluminescent Detection Reagent et couver pendant une minute.
En travaillant rapidement, tamponnez le bord de la tache sur une lingette de laboratoire pour enlever tout réactif de détection excédentaire, puis placez la tache sur la surface d'imagerie du plateau imageur. Image utilisant le programme Chemiluminescent pour capturer plusieurs points de temps de 10 à 30 secondes. Une fois la tache représentée, choisissez une image avec une visibilité optimale de la bande, puis exportez cette image. Avant de déplacer la tache, utilisez l'Imager pour prendre une photo de la tache pour capturer l'emplacement de l'échelle. Ensuite, exportez cette image aussi. Enfin, à l'aide d'un logiciel de préparation de diapositives, comme PowerPoint, alignez les bandes et les images de l'échelle pour former une image.
Cette image montre le résultat de tache occidentale pour l'immunoprécipitation du c-myc de protéine des cellules de thymocyte. De gauche à droite, les voies représentent le contrôle de l'isotype, l'IP c-myc et l'entrée de lysate pré-dédouanée. La voie à l'extrême droite est une image fusionnée de l'échelle de poids moléculaire. La bande forte, à environ 25 kilodaltons est de la chaîne de lumière et celui à 50 kilodaltons est de la chaîne lourde de l'anticorps de liaison et ne sont pas spécifiques à la propriété intellectuelle ou les échantillons. C-myc court environ 67 kilodaltons sur les taches occidentales et est généralement visible juste en dessous de la bande d'échelle de 75 kilodalton. Dans cette tache, la bande de c-myc est visible dans la deuxième voie mais absente dans la première voie, indiquant que l'anticorps IP a réussi à tirer vers le bas c-myc. Il n'y a pas de bande visible dans la voie de lysate pré-dédouanée, ce qui suggère que cette protéine a de faibles niveaux d'expression endogènes.
Subscription Required. Please recommend JoVE to your librarian.
Results
Les résultats de la procédure décrite ci-dessus sont indiqués à la figure 2. De gauche à droite, les voies contiennent le groupe témoin (isotype), le groupe d'essai (c-myc), le lysate pré-dédouané (lysate) et l'échelle de poids moléculaire (échelle). Les bandes d'échelle de 25 et 75 kDa sont marquées. Les deux bandes proéminentes à 25 kDa et 50 kDa sont la chaîne légère et lourde de l'anticorps de liaison, respectivement et ne sont pas spécifiques à la propriété intellectuelle ou aux échantillons. protéine c-myc qui fonctionne autour de 67kDa sur les taches occidentales et est généralement visible juste en dessous de la bande d'échelle de 75 kDa. Dans cette tache, la bande de c-myc est visible dans la deuxième voie, mais absente dans la première voie, indiquant que l'anticorps IP a réussi à tirer vers le bas c-myc. Il n'y a pas de bande visible dans la voie de lysate pré-dédouanée, ce qui suggère que cette protéine a de faibles niveaux d'expression endogènes.

Figure 2 : Résultats d'une analyse de blot occidental, utilisée pour évaluer la purification du c-myc par immunoprécipitation. Une bande à 67 kDa, correspondant au c-myc, est visible dans la voie anti-c-myc, mais pas dans la voie de contrôle de l'isotype. Notez que les niveaux de c-myc n'étaient pas assez élevés pour être visualisés dans la voie de lysate. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
En bref, l'immunoprécipitation est l'isolement d'une protéine spécifique à l'aide d'un anticorps. Dans cet exemple, les résultats de l'immunoprécipitation ont été analysés par Western blot pour évaluer la pureté. La protéine isolée pourrait être utilisée dans un certain nombre d'applications par la suite, y compris: RMN pour la structure des protéines, Spectrométrie de masse pour la séquence d'acides aminés, ou des essais in vitro pour la caractérisation enzymatique. Les adresses IP peuvent également caractériser les partenaires en interaction des protéines. Par exemple, après l'isolement, l'ADN ou l'ARN pourrait être isolé pour le séquençage. Les co-immunoprécipitations évaluent les interactions protéines-protéines. Lorsque la protéine cible est tirée vers le bas au cours d'une propriété intellectuelle, les protéines en interaction peuvent également être tirées vers le bas. Ces partenaires en interaction peuvent être évalués par spectrométrie de masse et tache occidentale. L'immunoprécipitation est une technique puissante pour étudier la biologie des protéines.
Subscription Required. Please recommend JoVE to your librarian.
References
- Olliver, C. L. and Boyd, C. D. (1984). Immunoprecipitation of In Vitro Translation Products with Protein A Bound to Sepharose. In J. M. Walker (eds), Nucleic Acids. Methods in Molecular Biology (pp. 157-160). New Jersey: Humana Press.
- Thurston, C. F. and Henley, L. F. (1988). Direct Immunoprecipitation of Protein. In J. M. Walker (eds), New Protein Techniques. Methods in Molecular Biology (pp. 149-158). New Jersey: Humana Press.
- Anderson, N. G. (1998). Co-immunoprecipitation: Identification of Interacting Proteins. In R. A. Clegg (eds), Protein Targeting Protocols.Methods in Molecular Biology (pp. 35-45). New Jersey: Humana Press.
- Jackson, D. I. and Dickson, C. (1999). Protein Techniques: Immunoprecipitation, In Vitro Kinase Assays, and Western Blotting. In P.T. Sharpe and I. Mason (eds), Molecular Embryology. Methods in Molecular Biology (pp. 699-708). New Jersey: Humana Press.
- Trieu, E. P. and Targoff, I. N. (2015). Immunoprecipitation: Western Blot for Proteins of Low Abundance. In B. Kurien and R. Scofield (eds), Western Blotting. Methods in Molecular Biology (pp. 327-342). New York, NY: Humana Press.