



Overview
ソース: エワ・ブコフスカ・ファニバンド1, ティルデ・アンダーソン1, ロルフ・ルード1
1臨床科学ルンド, 感染医学の部門, ルンド大学生物医学センター, 221 00 ルンド, スウェーデン
惑星地球は何百万もの細菌種の生息地であり、それぞれが特定の特性を持っています。細菌種の同定は、感染した患者を診断するために環境試料および医療微生物学の生物多様性を決定するために微生物生態学で広く使用されている。細菌は、顕微鏡検査、特定の培法上の成長、生化学的および血清学的試験、抗生物質感受性アッセイなどの従来の微生物学的方法を使用して分類することができます。ここ数十年で、分子微生物学の方法は細菌の同定に革命を起こしました。一般的な方法は、16SリボソームRNA(rRNA)遺伝子シーケンシングです。この方法は、従来の方法よりも速く、より正確であるだけでなく、実験室の条件で成長することが困難な株の同定を可能にします。さらに、分子レベルでの株の分化は、典型的に同一の細菌(1-4)間の判別を可能にする。
16S rRNAは19個のタンパク質の複合体と結合し、細菌リボソーム(5)の30Sサブユニットを形成する。これは、リボソームアセンブリに不可欠な機能のために存在し、非常に保存されている16S rRNA遺伝子によってコードされています。ただし、特定の種の指紋として機能する可変領域も含まれています。これらの特徴により、16S rRNA遺伝子は、細菌の同定、比較、系統的分類に使用される理想的な遺伝的断片となっています(6)。
16S rRNA遺伝子シーケンシングは、ポリメラーゼ連鎖反応(PCR)(7-8)に基づいてDNAシーケンシング(9)を行う。PCRは、以下を含む一連のサイクルを通じてDNAの特定の断片を増幅するために使用される分子生物学的方法です。
i) 二本鎖DNAテンプレートの脱生
ii) テンプレートを補完するプライマー(短いオリゴヌクレオチド)のアニーリング
iii) 新しいDNA鎖を合成するDNAポリメラーゼ酵素によるプライマーの拡張
このメソッドの概略図を示します。
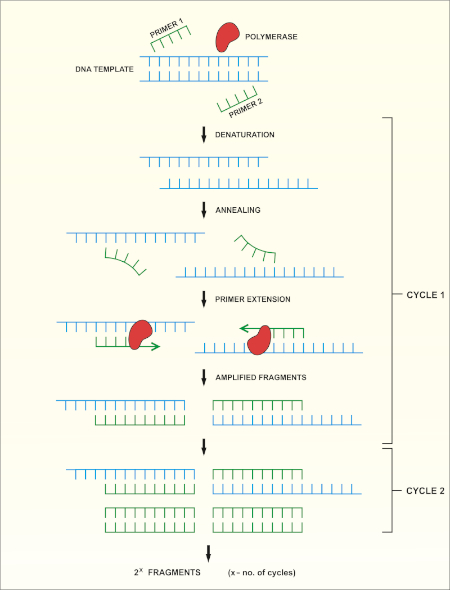
図 1:PCR反応の概略図。この図のより大きなバージョンを表示するには、ここをクリックしてください。
PCR反応を成功させるには重要ないくつかの要因があり、そのうちの1つはDNAテンプレートの品質です。細菌からの染色体DNAの単離は、標準プロトコルまたは市販キットを用いて行うことができる。PCR反応を阻害する汚染物質を含まないDNAを得るためには、特別な注意が必要です。
16S rRNA遺伝子の保存領域は、任意の細菌種の標的領域に結合し、増幅することができるユニバーサルプライマーペア(1つの前方および1逆)の設計を可能にする。ターゲット領域のサイズはさまざまです。プライマーペアの中には、16S rRNA遺伝子のほとんどを増幅できるものもあれば、その一部だけを増幅するものもあります。一般的に使用されるプライマーの例を表 1に示し、その結合部位を図 2 に示します。
| プライマー名 | シーケンス (5'→3') | フォワード/リバース | 参照 |
| 8F b) | アガグットガットックカッグカグ | 転送 | -1 |
| 27F | アガグットガットツクトカッグ | 転送 | -10 |
| 515F | GTGCCAGCMGGCGGTAA | 転送 | -11 |
| 911R | GCCCCCGTCAATTTTGA | 逆 | -12 |
| 1391R | ガグッググググGTRCA | 逆 | -11 |
| 1492R | グッタクットタクタクト | 逆 | -11 |
表 1:16S rRNA遺伝子a)の増幅に用いられる標準的なオリゴヌクレオチドの例。
a)異なるプライマーの組み合わせを使用して生成されたPCR製品の予想長さは、フォワードとリバースプライマーの結合部位間の距離を計算することによって推定できます(図2参照)。プライマーペア8F-1492Rを使用した製品は~1500bp、プライマーペア27F-911R~900bpです。
b) fD1 とも呼ばれます。

図 2:16S rRNA配列およびプライマー結合部位の代表的な図。保存された領域は灰色で色付けされ、可変領域は対角線で塗りつぶされます。最も高い分解能を可能にするために、プライマー8Fおよび1492R(rRNA配列上の位置に基づく名前)を使用して配列全体を増幅し、遺伝子のいくつかの可変領域のシーケンシングを可能にする。この図のより大きなバージョンを表示するには、ここをクリックしてください。
PCRのサイクリング条件(すなわち、DNAが変性し、プライマーでアニールされ、合成されるのに必要な温度と時間)は、使用されるポリメラーゼの種類およびプライマーの特性に依存する。特定のポリメラーゼの製造元のガイドラインに従うことをお勧めします。
PCRプログラムが完了すると、製品はアガロースゲル電気泳動によって分析されます。正常な PCR は、期待されるサイズの単一バンドを生成します。製品は、PCR反応に存在していた残留プライマー、デオキシリボヌクレオチド、ポリメラーゼ、およびバッファーを除去するために、シーケンシングの前に精製されなければならない。精製されたDNA断片は、通常、商用シーケンシングサービスにシーケンシングのために送られます。しかし、一部の機関は、独自のコア施設でDNAシーケンシングを行います。
DNA配列は、コンピュータによってDNAクロマトグラムから自動的に生成され、手動編集が必要な場合があるため、品質を慎重にチェックする必要があります。このステップに続いて、遺伝子配列は16S rRNAデータベースに沈着した配列と比較される。類似性の領域が識別され、最も類似したシーケンスが配信されます。
Procedure
1. 設定
- 微生物を取り扱いながら、良好な微生物の実践に従う必要がある。すべての微生物、特に未知のサンプルは、潜在的な病原体として扱われるべきです。無菌技術に従って、サンプル、研究者、または実験室の汚染を避けてください。細菌の取り扱い前後に手を洗い、手袋を着用し、防護服を着用してください。
- ゲノムDNA単離およびPCR産物精製のための実験プロトコルのリスク評価を行う。一部の試薬は有害である可能性があります!
- 純粋な培養は16S rRNAシーケンシングに不可欠です。ゲノムDNAの単離に進む前に、出発物質が完全に純粋であることを確認してください。これは、個々のコロニーを分離するためにストリークメッキによって行うことができます。これらは、必要に応じて、個別に、またはスープに、さらにストリークすることができます。
- 必要な実験装置:
- PCR用サーマルサイクラー。サーマルサイクラーの機能は、設定されたプログラムに応じて温度を上げたり下げたりすることです。プログラムの作成中に、すべてのPCRステップの温度と時間の値だけでなく、サイクルの合計数を入力するように求められます。
- アガロースゲル電気泳動システム。これは、そのサイズと電荷に基づいてDNA断片を分離するために使用されます。このプロトコルでは、アガロースゲル電気泳動を用いて、単離されたゲノムDNAおよびPCR産物の品質を可視化する。
2. プロトコル
注:実証されたプロトコルは、細菌の純粋な培養から16S rRNA遺伝子シーケンシングに適用されます。メタゲノム研究には適用されません。
-
ゲノムDNA(gDNA)の単離のための細菌を培養する。
- 適切な培地で微生物を育てなさい。液体および固体媒体の両方がこのステップで使用することができる。最高の成長をもたらす条件を選択します。実験を計画している間、成長が遅い細菌は、後期ログ/静止成長段階に到達するまでに数日を要する可能性があることに留意してください。このプロトコルでは、バチルス・サチリス168を200rpm、37°Cに設定した振盪インキュベーターで一晩リソジェニーブロス(LB)で増殖した。
-
gDNAの分離
- 細菌を固形培地で増殖させた場合、滅菌ループを用いて一部の細胞を削り取り、蒸留水の1mLで再懸濁する
- 細菌を液体培地で増殖させた場合は、一晩培養の約1.5mLを使用する。
- 遠心分離(1分、12,000~16,000×g)による細胞のペレットは、上清を除去し、市販キットまたは標準プロトコルを用いてgDNA単離のために細胞を使用する[例えばCTAB総DNA製剤(13)またはフェノールクロロホルム抽出(14)]。ここで、市販キットを用いて、B.subtilis 168の1.5mLからgDNAを一晩培養し、OD600=1.5を分離した。
注1:いくつかのグラム陰性細菌のために、このステップは省略し、沸騰することによって細胞からのDNAの簡単な放出に置き換えることができます。蒸留水で細菌ペレットを再中断し、100°Cに設定された加熱ブロックで10分間インキュベートします。
注2:グラム陽性細菌細胞は破壊しにくい。したがって、この細菌群からの単離に特化したgDNA単離方法またはキットを選択することをお勧めします。
-
gDNA品質チェック。
- アガロースゲル電気泳動による単離されたgDNAの品質を確認します。まず、単離したgDNAの5μLを装填色素(6倍)の1μLと混合し、DNA染色試料を含む0.8%のアガロースゲルに試料をロードします。
- 分子質量標準をロードし、色素フロントがゲルの底に達するまで電気泳動を実行します。
- 電気泳動が完了したら、適切なトランシルミレータ(UVまたは青色光)上のゲルを視覚化します。gDNAは厚い高分子バンド(10kb以上)として現れる。gDNA品質チェックの例を図 3 に示します。
- gDNAが品質管理を通過した場合(すなわち、高分子バンドが存在し、gDNAの塗りつぶしがほとんどない場合)、最初に3つのマイクロ遠心管を次のようにラベル付けしてgDNAを連続的に希釈します:「10x」、「100x」、「1000x」。
- 3つの管のそれぞれに無菌蒸留水のピペット90 μL。
- gDNA溶液の10 μLを取り、「10x」とマークされたチューブに追加します。
- 溶液が均一に混合されていることを確認するために、全体の体積(すなわち100 μL)を徹底的に上下にピプテします。次に、このチューブから溶液の10 μLを取り、「100x」とマークされたチューブに移します。
- 前述のように混合し、チューブ「100x」からチューブ「1000x」に溶液の10 μLを転送することにより、同じ手順を繰り返します。これらの希釈物は、PCR反応においてテンプレートとして使用される。

図 3:バチルス・サチリスから単離されたgDNAのアガロースゲル電気泳動.レーン 1: M - 分子量マーカー (上から下へ: 10000 bp, 8000 bp, 6000 bp, 5000 bp, 4000 bp, 3500 bp, 3000 bp, 2500 bp, 2000 bp, 1500 bp, 1000 bp)レーン2: gDNA -バチルス・サチリスから単離されたゲノムDNA.この図のより大きなバージョンを表示するには、ここをクリックしてください。
-
PCRによる16S rRNA遺伝子の増幅
注:以下のPCRプロトコルは、特定のDNAポリメラーゼおよびプライマーペア8F~1492Rに対して最適化されています(表1参照)。各ポリメラーゼおよびプライマーペアに対してプロトコルの最適化が必要である。- 氷上のすべての試薬を解凍します。
- 表 2 に示すように、PCR マスター ミックスを準備します。DNAポリメラーゼは室温で活性であるため、反応セットアップは氷上で行う必要があり、すなわちPCRチューブおよび反応成分は常に氷上に保持されるべきである。gDNAサンプルごとに1つの反応を調作し、1つの反応を陰性対照に調用する。ネガティブコントロールは、gDNAテンプレートのないPCRミックスであり、反応の他の成分が汚染されていないことを保証するために使用されます。
注:複数のサンプルの場合、マスターミックスが一般的に調製されます。マスターミックスは、テンプレートを除く全ての反応成分を含む溶液である。反復的なピペットを省略し、ピペットエラーを回避し、サンプル間の高い一貫性を確保するのに役立ちます。マスターミックスを調作成するには、各成分の体積(DNAテンプレートを除く)に、テストされたサンプル数を掛けます。マイクロ遠心管内のすべてのコンポーネントを混ぜ、ボリューム全体を数回上下にピペットします。 - 個々のPCRチューブにマスターミックスのアリコート49 μL。
- マスターミックスでチューブに1 μLテンプレートを追加します。陰性制御のために無菌水の1 μLを追加します。成分がよく混合されていることを確認するには、30-50μLに設定されたピペットでミックスを約10回ゆっくりとピペットします。
- 表 3 に示すプログラムを使用して PCR マシンを設定します。
- チューブをPCRマシンに入れ、プログラムを起動します。
- プログラムが完了したら、アガロースゲル電気泳動によってPCR製品の品質を調べます。
- 8F-1492Rプライマーペアを用いてPCR反応が成功すると、約1.5kbの単一バンドが得られます(図4)。他のバンド(すなわち、非特異的な製品)が存在する場合は、アニーリング温度を調整してPCRプログラムを最適化します。期待されるサイズのバンドが 1 つある場合は、次の手順に進みます。ここでは、100倍希釈されたgDNAテンプレートを使用したPCR反応は、期待されるサイズの鋭いバンドを有し、非特異的な製品を欠いていたため、最良の製品を生み出した。したがって、それは浄化され、シーケンスのために送信されるように選択されました。
- シーケンシングの前に、PCR反応に存在していた残留プライマー、デオキシリボヌクレオチド、ポリメラーゼ、およびバッファーから製品をクリーンアップする必要があります。PCR製品は、市販のPCR精製キットを用いて単離することができる。PCR反応は、DNA結合マトリックスを含むカラムにロードされます。PCR 製品は列にバインドされ、他のコンポーネントは列を通過します。その後、カラムは洗浄バッファーを使用して洗浄され、最後にDNAが選択したバッファーでELE化されます。キットで補完される溶出バッファーがシーケンスと互換性があることを確認します。
- DNAシーケンシング用の精製PCR製品を送付します。選択したシーケンシング施設でのシーケンスサンプルの提出に関するガイドラインに従ってください。最適なシーケンス カバレッジを行うには、PCR 増幅プライマー(セクション 2.4.1 で使用されているのと同じ)をシーケンス プライマーとして使用します。ここで、プライマー8F及び1492Rを用いてPCR製品のシーケンシングを行った。
| コンポーネント | 最終濃度 | 反応あたりの体積 | x反応あたりの体積(マスターミックス) |
| 5x反応バッファー | 1x | 10 μL | 10 μL × x |
| 10 mM dNTP | 200 μM | 1 μL | 1 μL × x |
| 10 μM プライマー 8F | 0.5 μM | 2.5 μL | 2.5 μL × x |
| 10 μM プライマー 1492R | 0.5 μM | 2.5 μL | 2.5 μL × x |
| フシオンポリメラーゼ | 1ユニット | 0.5 μL | 0.5 μL × x |
| テンプレート DNA * | - | 1 μL | - |
| ddH2O | - | 32.5 μL | 32.5 μL × x |
| 総ボリューム | 50 μL | 49 μL × x |
表 2:PCR反応成分。*ステップ2.3から10x、100xまたは1000x希釈されたgDNAを使用してください。
| ステップ | 温度 | 時間 | サイクル |
| 初期の非核化 | 98°C | 30秒 | |
| 変性 | 98°C | 10秒 | 25-30 |
| 焼鈍 | 60°C | 30秒 | |
| 拡張子 | 72°C | 45秒 | |
| 最終拡張子 | 72°C | 7 分 | |
| 保持 | 4°C | ∞ |
表 3:16S rRNA遺伝子の増幅のためのPCRプログラム。

図 4:プライマー8F及び1492R及びgDNAをテンプレートとして増幅したPCR製品のアガロースゲル電気泳動。B.subtilis由来のgDNAサンプル(図3参照)を、最良の結果をテストするために10、100および1000回希釈した。レーン1:M - 分子量マーカー(上から下へ:10000 bp、8000 bp、6000 bp、5000 bp、4000 bp、3500 bp、3000 bp、2500 bp、2000 bp、1500 bp、1000bp、750bp、500bp)。レーン2:10倍希釈テンプレートを使用したPCR反応。レーン 3:100倍希釈テンプレートを使用したPCR反応。レーン4:1000x希釈テンプレートを使用したPCR反応。レーン5:(C-)-陰性制御(DNAテンプレートを使用まない反応)。この図のより大きなバージョンを表示するには、ここをクリックしてください。
3. データ分析と結果
注:PCRプロダクトは、フォワード(ここでは8F)と逆(ここでは1492R)プライマーを使用してシーケンス化されます。したがって、2 組のデータ シーケンスが生成され、1 つはフォワード用、もう 1 つはリバース プライマー用です。各配列について少なくとも2種類のファイルが生成される:i)DNA配列およびii)DNAクロマトグラムは、シーケンス実行の品質を示す。
- フォワードプライマーの場合は、クロマトグラムを開き、シーケンスを注意深く調べます。品質シーケンスの理想的なクロマトグラムは、均等に間隔をあけたピークを持ち、バックグラウンド信号をほとんどまたは全く持っていない必要があります(図5A)。
- クロマトグラムが高品質でない場合は、シーケンスを破棄するか、シーケンステキスト ファイルを次のように変更する必要があります。
- クロマトグラム全体の二重ピークの存在は、複数のDNAテンプレートの存在を示す。これは、細菌培養が純粋ではなかった場合に当てはまる可能性があります。このようなシーケンスは破棄する必要があります (図 5B)。
- あいまいなクロマトグラムは、同じ場所に異なる色のピークの存在から生じる可能性があります。最も一般的なエラーの 1 つは、同じ位置に 2 つの異なる色のピークが存在し、シーケンス ソフトウェアによるベースの不適切な割り当てです (図 5C)。誤って割り当てられたヌクレオチドを手動で修正し、テキストファイルで編集します。
- 低分解能クロマトグラムは、これらの領域におけるヌクレオチドの誤カウントを引き起こす「広いピーク」をもたらす可能性があります(図5D)。このエラーは修正が難しいため、それ以降の位置合わせステップでミスマッチが発生する可能性があるため、信頼性の高い方法として扱う必要はありません。
- クロマトグラムの読み取り品質が悪く、複数のピークの存在は、一般的にシーケンスの5'と3'の終わりに見られます。シーケンサーソフトウェアの中には、これらの低品質フラグメントが自動的に除去され(図5E)、ヌクレオチドはテキストファイルに含まれません。シーケンスが自動的に切り捨てられなかった場合は、端部にある低品質のフラグメント(弱い信号、重なり合うピーク、解像度の損失など)を特定し、テキストファイルからそれぞれのベースを削除します。

図 5:DNA シーケンシングのトラブルシューティングの例。A) 高品質のクロマトグラム配列の例(均等間隔、明確なピーク)。B)通常クロマトグラムの先頭に発生する品質の悪いシーケンス。グレーゾーン領域は低品質と見なされ、シーケンスソフトウェアによって自動的に削除されます。より多くの塩基は手動でトリミングすることができます。C) ダブルピークの存在(矢印で示す)。赤い矢印で示されるヌクレオチドはシーケンサーで「T」(赤いピーク)と読み取られたが、青いピークが強く、「C」とも解釈できる。D)重なり合うピークはDNA汚染(すなわち複数のテンプレート)を示す。E)解像度の損失と、信頼性の高いベースコールを防ぐいわゆる「広いピーク」(長方形でマーク)。この図のより大きなバージョンを表示するには、ここをクリックしてください。
- リバースプライマーの場合は、3.1 と 3.2 を繰り返します。
- 最後に、前方シーケンスと逆シーケンスを 1 つの連続したシーケンスに組み立てます。良好なシーケンス実行では、最大 1100 bp のシーケンスが生成されます。
- DNA配列アセンブリプログラムを使用して2つの配列をマージし、例えばCAP3(http://doua.prabi.fr/software/cap3)(15)のような自由なツールを使用してマージする。
- 指定されたボックスに FASTA 形式の 2 つのシーケンスを挿入します。[送信] ボタンをクリックし、結果が返ってくるのを待ちます。
- 組み立てられたシーケンスを表示するには、結果タブで「Contigs」を押します。アライメントの詳細を表示するには、「アセンブリの詳細」を押します。
注1:CAP3 ソフトウェアをコンティグ アセンブリに使用する場合、逆プライマー シーケンスを逆補完に変換する必要はありません。ただし、別のプログラムを使用する場合は、この手順が必要になる場合があります。
注2:FASTA形式は、ヌクレオチド配列を表すテキストベースのフォーマットである。FASTA ファイルの最初の行 (説明行) は、シンボル ">" で始まり、その後にシーケンスの名前または一意の識別子が続きます。説明行に続いて、ヌクレオチド配列である。シーケンスを次の形式で貼り付けします。
>シーケンス_frw_プライマー
ここにテキストファイルからシーケンスを貼り付けます
>シーケンス_rvs_プライマー
ここにテキストファイルからシーケンスを貼り付けます
- 基本的なローカルアライメント検索ツール(BLAST;https://blast.ncbi.nlm.nih.gov/Blast.cgi)のウェブサイトにアクセスして、データベース検索を行います。
- シーケンスをデータベースと比較するには、「ヌクレオチド BLAST」ツールを選択します。
- シーケンス(3.5で組み立てられたコンティグ)を「クエリシーケンス」テキストボックスに入力し、スクロールダウンメニューのデータベース「16S rRNAシーケンス(細菌とArchea)」を選択します。
- ページ下部の「BLAST」ボタンを押します。最も類似したシーケンスが返されます。BLAST の結果の例を図 6 に示します。提示された実験では、トップヒットはB.subtilis株168であり、BLASTデータベースで利用可能な配列を持つ100%のアイデンティティを示す。
- トップヒットに 100% の ID が表示されない場合は、位置合わせに移動し、不一致がないかどうかを確認します。トップヒットをクリックすると、アライメントの詳細が表示されます。整列したヌクレオチドは短い垂直線で結合され、不一致のヌクレオチドはそれらの間にギャップを持つ。シーケンシング会社から受け取ったクロマトグラムに戻り、不一致の領域に焦点を当ててシーケンスをもう一度修正します。追加のエラーが見つかった場合は、シーケンスを修正します。修正されたシーケンスを使用して、もう一度 BLAST を実行します。

図 6:ヌクレオチドBLAST結果の例。B.subtilis 168の純粋培養からの16S rRNA遺伝子配列を照会配列として用いられた。トップヒットは、期待どおりにB.サチリス株168に100%アイデンティティ(下線付き)を示す。この図のより大きなバージョンを表示するには、ここをクリックしてください。
地球には何百万もの細菌種が生息しており、それぞれがユニークな特徴を持っています。これらの種を同定することは、環境サンプルを評価する上で重要です。医師はまた、感染した患者を診断するために異なる細菌種を区別する必要があります。
細菌を同定するために、コロニーの形態を観察するために、特定の媒体上の形態の顕微鏡観察や成長を含む様々な技術を採用することができます。遺伝子解析は、細菌を同定するための別の技術が近年人気が高まっており、一部は16SリボソームRNA遺伝子シーケンシングに起因する。
細菌リボソームは、2つのサブユニットからなるタンパク質RNA複合体である。これら2つのサブユニットのうち小さい30Sサブユニットには、ゲノムDNAに含まれる16S rRNA遺伝子によってコードされる16S rRNAが含まれています。16S rRNAの特定の領域は、リボソームアセンブリに不可欠な機能のために、高度に保存されています。他の領域は、機能に対してそれほど重要ではないが、細菌種によって異なる場合がある。16S rRNAの可変領域は、細菌種に対するユニークな分子指紋として機能することができ、表現価が一般的に同一の株を区別することができます。
gDNAの品質サンプルを得た後、16S rRNAコード遺伝子のPCRを開始することができる。PCRは、二本鎖DNAテンプレートの変性のサイクル、遺伝子の保存状態の高い領域を増幅するユニバーサルプライマーペアのアニーリング、およびDNAポリメラーゼによるプライマーの拡張からなる、一般的に使用される分子生物学法です。プライマーの中には、16S rRNAコード遺伝子のほとんどを増幅するプライマーもあれば、その断片のみを増幅するものもあります。PCR後、製品はアガロースゲル電気泳動を介して分析することができる。増幅が成功した場合、ゲルは、使用されるプライマーペアに応じて、16S rRNA遺伝子のおおよその長さである、期待されるサイズの単一バンドを含む必要があります。
精製およびシーケンシングの後、得られた配列をBLASTデータベースに入力し、参照16S rRNA配列と比較することができます。このデータベースは、最も高い類似性に基づいて一致を返すので、これは目的の細菌の同一性を確認することができます。このビデオでは、PCR、DNA配列解析および編集、配列アセンブリ、データベース検索を含む16S rRNA遺伝子シーケンシングを観察します。
微生物を取り扱う場合、無菌技術の使用や適切な個人用保護具の着用など、良好な微生物生物学的実践に従うことが不可欠です。対象の微生物または環境試料に対して適切なリスク評価を行った後、試験培養を得る。この例では、バチルス・サビリスの純粋な培養が用いられる。
まず、適切な条件で適切な培地で微生物を増育します。この例では、バチルス・サチリス168は、37°Cで200rpmに設定された揺動インキュベーターで一晩LBブロスで成長する。次に、市販のキットを使用して、B.サチリス一晩培養の1.5ミリリットルからゲノムDNAまたはgDNAを分離します。
単離されたDNAの品質を確認するには、まず、単離されたgDNAの5マイクロリットルを1マイクロリットルのDNAゲルロード色素と混合します。次に、サンプルを0.8%のアガロースゲルにロードし、SYBRセーフまたはEtBrなどのDNA染色試薬を含む。この後、ゲルに1キロベースの分子質量標準をロードし、前色素がゲルの底から約0.5センチメートルになるまで電気泳動を実行します。ゲル電気泳動が完了したら、青色光トランジユリニエータ上のゲルを視覚化します。gDNAは、サイズが10キロベースを超える厚いバンドとして表示され、塗りつぶしが最小限に抑えられます。
この後、gDNAのシリアル希釈を作成するために、3つのマイクロ遠心分離管に10X、100X、および1000Xとしてラベルを付けます。次に、ピペットを使用して、90マイクロリットルの無菌蒸留水を各チューブに分配します。次に、10XチューブにgDNA溶液の10マイクロリットルを追加します。溶液が完全に混合されていることを確認するために、ボリューム全体を上下にピペット。次に、10Xチューブから溶液の10マイクロリットルを取り出し、これを100Xチューブに移します。前述のように溶液を混ぜます。最後に、100Xチューブ内の溶液の10マイクロリットルを、1000Xチューブに移す。
PCRプロトコルを開始するには、氷上で必要な試薬を解凍します。次に、PCRマスターミックスを準備します。DNAポリメラーゼは室温で活性であるため、氷上で起こる反応が必要です。各PCRチューブにマスターミックスのアリコット49マイクロリットル。次に、各実験管に1マイクロリットルのテンプレートを加え、1マイクロリットルの滅菌水を負の制御管に加え、上下に配管して混合する。この後、表に記載されているプログラムに従ってPCRマシンを設定する。チューブをサーモサイクラーに入れ、プログラムを開始します。
プログラムが完了したら、前に示したように、アガロースゲル電気泳動を介して製品の品質を調べます。記載されたプロトコルを使用して成功した反応は、約1.5キロベースの単一バンドを生成する必要があります。この例では、100X希釈gDNAを含むサンプルは、最高品質の製品を得た。次に、市販のキットで、最高のPCR製品(この場合は100X gDNA)を精製します。これで、PCR 製品をシーケンス用に送信できるようになりました。
この例では、PCR 製品はフォワードプライマーとリバース プライマーを使用してシーケンスされます。したがって、DNA配列とDNAクロマトグラムを含む2つのデータセットが生成されます。まず、各プライマーから生成されたクロマトグラムを調べます。理想的なクロマトグラムは、バックグラウンド信号をほとんどまたは全く持たない均等なピークを持つ必要があります。
クロマトグラムが二重ピークを示す場合、PCR製品に複数のDNAテンプレートが存在している可能性があり、配列を破棄する必要があります。クロマトグラムに同じ場所に異なる色のピークが含まれている場合、シーケンシングソフトウェアはヌクレオチドと誤呼び出された可能性があります。このエラーは、テキスト ファイルで手動で識別および修正できます。クロマトグラムの広範なピークの存在は、関連する領域におけるヌクレオチドの誤カウントを引き起こす分解能の喪失を示す。このエラーは修正が困難であり、後続の手順のいずれかで不一致は信頼できないものとして扱う必要があります。クロマトグラム読み取り品質が悪く、複数のピークの存在によって示され、通常、配列の5つの素数および3つの素数端で生じる。一部のシーケンシング プログラムでは、これらの低品質のセクションが自動的に削除されます。シーケンスが自動的に切り捨てられなかった場合は、低品質のフラグメントを識別し、テキスト ファイルからそれぞれのベースを削除します。
DNA アセンブリ プログラムを使用して、2 つのプライマー 配列を 1 つの連続配列に組み立てます。フォワードプライマーとリバースプライマーを使用して取得したシーケンスは、部分的に重複する必要があります。DNA 組立プログラムで、FASTA 形式の 2 つのシーケンスを適切なボックスに挿入します。次に、送信ボタンをクリックし、プログラムが結果を返すのを待ちます。
組み立てられたシーケンスを表示するには、[結果] タブで [Contigs] をクリックします。次に、線形の詳細を表示するには、アセンブリの詳細を選択します。基本的なローカルアライメント検索ツール(BLAST)のウェブサイトに移動し、ヌクレオチドBLASTツールを選択してシーケンスをデータベースと比較します。クエリ シーケンステキスト ボックスにシーケンスを入力し、下のスクロール メニューで適切なデータベースを選択します。最後に、ページの下部にある BLAST ボタンをクリックし、ツールがデータベースから最も類似したシーケンスを返すのを待ちます。
この例では、トップヒットはB.subtilis株168であり、BLASTデータベース内のシーケンスとの100%のアイデンティティを示す。トップヒットが期待される種やひずみに100%のアイデンティティを示さない場合は、クエリに最も近いシーケンスをクリックして、線形の詳細を確認します。整列されたヌクレオチドは短い垂直線で結合され、不一致のヌクレオチドはそれらの間にギャップを持つことになります。識別された不一致の領域に焦点を当てて、シーケンスを修正し、必要に応じて BLAST 検索を繰り返します。
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
細菌種の特定は、異なる研究者にとっても、ヘルスケアの研究者にとっても重要です。16S rRNAシーケンシングは、最初に細菌間の系統的関係を決定するために研究者によって使用されました。やがて、潜在的な病原体を同定する方法として、環境試料の生物多様性を決定するメタゲノム研究や臨床検査室で実施されてきた。それは臨床サンプルに存在する細菌の速く、正確な同一証明を可能にし、患者の早期診断およびより速い処置を促進する。
Subscription Required. Please recommend JoVE to your librarian.
References
- Weisburg, W.G., Barns, S.M., Pelletier, D.A. and Lane D.J. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 173 (2): 697-703. (1991)
- Drancourt, M., Bollet, C., Carlioz, A., Martelin, R., Gayral, J.P., Raoult D. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol. 38 (10):3623-3630. (2000)
- Woo, P.C., Lau, S.K., Teng, J.L., Tse, H., Yuen, K.Y. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect. 14 (10):908-934. (2008)
- Tang, Y.W., Ellis, N.M., Hopkins, M.K., Smith, D.H., Dodge, D.E., Persing, D.H. Comparison of phenotypic and genotypic techniques for identification of unusual aerobic pathogenic gram-negative bacilli. J Clin Microbiol. 36 (12):3674-3679. (1998)
- Tsiboli, P., Herfurth, E., Choli, T. Purification and characterization of the 30S ribosomal proteins from the bacterium Thermus thermophilus. Eur J Biochem. 226 (1):169-177. (1994)
- Woese, C.R. Bacterial evolution. Microbiol Rev. 51 (2):221-271. (1987)
- Bartlett, J.M., Stirling, D. A short history of the polymerase chain reaction. Methods Mol Biol. 226:3-6. (2003)
- Wilson, K.H., Blitchington, R.B., Greene, R.C. Amplification of bacterial 16S ribosomal DNA with polymerase chain reaction. J Clin Microbiol. 28 (9):1942-1946. (1990)
- Shendure, J., Balasubramanian, S., Church, G.M., Gilbert, W., Rogers, J., Schloss, J.A., Waterston, R.H. (2017) DNA sequencing at 40: past, present and future. Nature. 550:345-353.
- Lane, D.J. 16S/23S rRNA sequencing. (1991) In Nucleic acid techniques in bacterial systematics. (Goodfellow, M. and Stackebrandt, E., eds.) p.115-175. Wiley and Sons, Chichester, United Kingdom.
- Turner, S., Pryer, K.M., Miao, V.P., Palmer, J.D. (1999) Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryot Microbiol. 46:327-338.
- Fredricks, D.N., Relman, D.A. (1998) Improved amplification of microbial DNA from blood cultures by removal of the PCR inhibitor sodium polyanetholesulfonate. J Clin Microbiol. 36:2810-2816.
- Wilson, K. Preparation of genomic DNA from bacteria. (2001) Curr Protoc Mol Biol. Chapter 2:Unit 2.4.
- Wright, M. H., Adelskov, J., Greene, A.C. (2017) Bacterial DNA extraction using individual enzymes and phenol/chloroform separation. J Microbiol Biol Educ. 18:18.2.48.
- Huang, X., Madan, A. (1999). CAP3: A DNA sequence assembly program. Genome Res. 9:868-877.