



Overview
Fuente: Natalia Martin1, Andrew J. Van Alst1, Rhiannon M. LeVeque1, y Victor J. DiRita1
1 Departamento de Microbiología y Genética Molecular de la Universidad Estatal de Michigan
Las bacterias tienen la capacidad de intercambiar material genético (ácido desoxirribonucleico, ADN) en un proceso conocido como transferencia de genes horizontal. La incorporación de ADN exógeno proporciona un mecanismo mediante el cual las bacterias pueden adquirir nuevos rasgos genéticos que les permiten adaptarse a las condiciones ambientales cambiantes, como la presencia de antibióticos o anticuerpos (1) o moléculas que se encuentran en hábitats naturales (2). Existen tres mecanismos de transferencia génica horizontal: transformación, transducción y conjugación (3). Aquí nos centraremos en la transformación, la capacidad de las bacterias para tomar ADN libre del medio ambiente. En el laboratorio, el proceso de transformación consta de cuatro pasos generales: 1) Preparación de células competentes, 2) Incubación de células competentes con ADN, 3) Recuperación de células y 4) Placado de las células para el crecimiento de los transformadores (Figura 1).
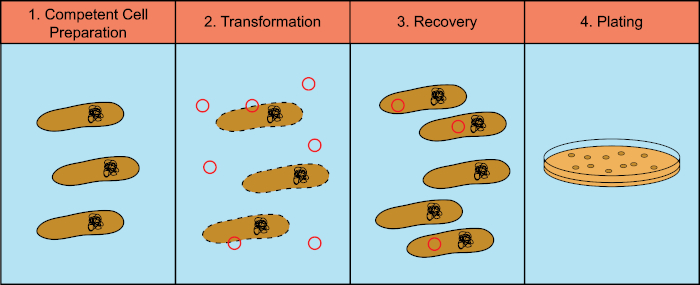
Figura 1: Pasos generales del proceso de transformación. El proceso de transformación tiene cuatro pasos generales: 1) Preparación de células competentes, 2) Incubación con ADN, 3) Recuperación de las células y 4) Células de chapado para el crecimiento de los transformadores.
Para que se produzca la transformación, las bacterias receptoras deben estar en un estado conocido como competencia. Algunas bacterias tienen la capacidad de llegar a ser naturalmente competentes en respuesta a ciertas condiciones ambientales. Sin embargo, muchas otras bacterias no se vuelven competentes naturalmente, o las condiciones para este proceso son aún desconocidas. La capacidad de introducir ADN en bacterias tiene una gama de aplicaciones de investigación: generar múltiples copias de una molécula de ADN de interés, para expresar una gran cantidad de proteínas, como un componente en los procedimientos de clonación, y otros. Debido al valor de la transformación a la biología molecular, existen varios protocolos destinados a hacer que las células sean artificialmente competentes cuando se desconocen las condiciones de competencia natural. Se utilizan dos métodos principales para preparar células artificialmente competentes: 1) a través del tratamiento químico de las células y 2) la exposición de las células a pulsos eléctricos (electroporación). El primero utiliza diferentes productos químicos dependiendo del procedimiento para crear atracción entre el ADN y la superficie celular, mientras que el segundo utiliza campos eléctricos para generar poros en la membrana celular bacteriana a través de la cual pueden entrar las moléculas de ADN. El enfoque más eficiente para la competencia química es la incubación con cationes divalentes, sobre todo calcio (Ca2+) (4,5) Competencia inducida por calcio es el procedimiento que se describirá aquí (6). Este método se utiliza principalmente para la transformación de bacterias Gram-negativas, y ese será el foco de este protocolo.
El procedimiento de transformación química implica una serie de pasos en los que las células están expuestas a cationes para inducir la competencia química. Estos pasos son seguidos posteriormente por un cambio de temperatura - choque de calor - que favorece la absorción de ADN extraño por la célula competente (7). Los sobres de las células bacterianas se cargan negativamente. En bacterias Gram-negativas como Escherichia coli, la membrana externa se carga negativamente debido a la presencia de lipopolisacárido (LPS) (8). Esto resulta en la repulsión de las moléculas de ADN cargadas negativamente de manera similar. En la inducción de competencia química, los iones de calcio cargados positivamente neutralizan esta repulsión de carga permitiendo la absorción del ADN en la superficie celular (9). El tratamiento del calcio y la incubación con ADN se realizan sobre hielo. Posteriormente, se realiza una incubación a temperaturas más altas (42oC), el choque de calor. Este desequilibrio de temperatura favorece aún más la aceptación del ADN. Las células bacterianas deben estar en la fase de crecimiento exponencial medio para soportar el tratamiento de choque térmico; en otras etapas de crecimiento las células bacterianas son demasiado sensibles al calor, lo que resulta en una pérdida de viabilidad que disminuye significativamente la eficiencia de transformación.
Diferentes fuentes de ADN se pueden utilizar para la transformación. Típicamente, plásmidos, pequeñas moléculas circulares de ADN de doble cadena, se utilizan para la transformación en la mayoría de los procedimientos de laboratorio en E. coli. Para que los plásmidos se mantengan en la célula bacteriana después de la transformación, deben contener un origen de replicación. Esto permite que se repliquen en la célula bacteriana independientemente del cromosoma bacteriano. No todas las células bacterianas se transforman durante el procedimiento de transformación. Por lo tanto, la transformación produce una mezcla de células transformadas y células no transformadas. Para distinguir entre estas dos poblaciones, se utiliza un método de selección para identificar las celdas que han adquirido el plásmido. Los plásmidos suelen contener marcadores seleccionables, que son genes que codifican un rasgo que confiere una ventaja para el crecimiento (es decir, resistencia a un antibiótico o químico o rescate de una auxotrofia de crecimiento). Después de la transformación, las células bacterianas se chapan en medios selectivos, lo que sólo permite el crecimiento de las células transformadas. En el caso de las células transformadas con un plásmido que confiere resistencia a un antibiótico dado, los medios selectivos serán medios de crecimiento que contengan ese antibiótico. Se pueden utilizar varios métodos diferentes para confirmar que las colonias cultivadas en los medios selectivos son transformadores (es decir, han incorporado el plásmido). Por ejemplo, los plásmidos pueden recuperarse de estas células utilizando métodos de preparación de plásmidos (10) y digeridos para confirmar el tamaño del plásmido. Alternativamente, la PCR de colonia se puede utilizar para confirmar la presencia del plásmido de interés (11).
El objetivo de este experimento es preparar las células químicamente competentes de E. coli DH5, utilizando una adaptación del procedimiento de cloruro de calcio (12), y transformarlas con el plásmido pUC19 para determinar la eficiencia de transformación. La cepa de E. coli DH5 es una cepa comúnmente utilizada en aplicaciones de biología molecular. Debido a su genotipo, específicamente el recA1 y endA1, esta cepa permite una mayor estabilidad de la plaquita y mejorar la calidad del ADN plásmido en preparaciones posteriores. Dado que la eficiencia de transformación disminuye con el aumento del tamaño del ADN, el plásmido pUC19 se utilizó en este protocolo debido a su pequeño tamaño (2686 bp) (ver https://www.mobitec.com/cms/products/bio/04_vector_sys/standard_cloning_vectors.html para un vectores). pUC19 confiere resistencia a la ampicilina y, por lo tanto, este fue el antibiótico utilizado para la selección.
Procedure
Este protocolo describe la preparación y transformación de E. coli DH5 competente utilizando una adaptación del procedimiento de cloruro de calcio (12).
1. Configuración
-
Equipo
- Espectrofotómetro
- Centrífuga Sorval (o equivalente)
- Centrífuga de sobremesa
- Bloque de calor o baño de agua
- Agitador orbital
- Incubadora estacionaria
- Bandeja de fundición de gel
- Bueno peines
- Fuente de tensión
- Caja de gel
- Fuente de luz UV
- Microondas
-
Soluciones y reactivos
- Caldo Luria-Bertani (LB) (10 g de hidrolizado enzimático de caseína, extracto de levadura de 5 g y 5 g de cloruro sódico en 1000 ml de H2O)
- Caldo super óptimo con represión catabolita (SOC): (2% (p.v) triptona, 0,5% (p/v) extracto de levadura, 10 mM de NaCl, 2,5 mM de KCl, 10 mM MgCl2,10 mM MgSO4y 20 mM de glucosa)
- CaCl2-MgCl2 (80 mM MgCl2, 20 mM CaCl2) solución.
- M CaCl2 solución (si las células se transformarán inmediatamente) o 0.1M CaCl2 solución que contiene 10% (v/ v) glicerol (si las células se congelarán para uso futuro).
- Placas de agar LB
- Placas selectivas de agar LB (para este experimento, ya que el plásmido utilizado confiere resistencia a la ampicilina, se utilizaron placas de agar LB que contienen ampicilina 100 g/ml)
- Cepa de E. coli DH5
- ADN plásmido pUC19 (100 pg/l)
- Kit de miniprep de giro QIAprep (Qiagen)
- Hind III enzima de restricción
- 1 kb más escalera de ADN
- Bajo punto de fusión Agarose
- 1X tacolcha TAE (base Tris de 40 mM, ácido acético de 20 mM y EDTA de 1 mM)
- Bromuro de etidio (10mg/ml)
-
Notas generales de seguridad
E. coli DH5 está clasificado como Nivel de Bioseguridad 1 (BSL1). Los microbios de esta categoría representan poca o ninguna amenaza de infección en adultos sanos. Sin embargo, se requiere una manipulación cuidadosa del microorganismo.
IMPORTANTE todos los pasos de este protocolo deben llevarse a cabo utilizando técnicas asépticas y sobre temperaturas de hielo o de 4oC a menos que se indique.
2. Protocolo
- A partir de un stock congelado de E. coli DH5 (congelado en 20% glicerol de un cultivo nocturno cultivado en LB) raya las bacterias para el aislamiento en una placa de agar LB. Incubar a 37oC durante la noche (16-20 horas).
- Inocular una sola colonia en 3 ml de caldo LB en un tubo. Crecer agitando a 210 rpm a 37oC durante la noche (16-20 horas).
- Mida el OD600 de la cultura nocturna. Utilice el cultivo nocturno para inocular 100 ml de caldo LB en un matraz de 1 litro a un OD600-0.01. Incubar el cultivo agitando vigorosamente (210 rpm) a 37oC monitoreando OD600 en el espectrofotómetro cada 15-20 min, hasta que el cultivo alcance OD600-0,35 (aproximadamente 3 horas).
NOTA: Para que la transformación sea eficiente, las células bacterianas deben estar en la fase de crecimiento exponencial medio. El número máximo de células debe ser de 108 células/ml, que para la mayoría de las cepas de E. coli corresponde a OD600-0.4. El uso del espectrofotómetro permite medir el OD600, lo que permite determinar que las células se encuentran en la etapa de crecimiento adecuada. Si este protocolo se utilizará para otras cepas de bacterias, la calibración para determinar el número de colonias que forman unidades en valores OD600 específicos será necesaria para determinar esta correlación. - Transfiera los 50 ml del cultivo a cada una de las 2 botellas de centrífuga de polipropileno helada. Coloque las botellas sobre hielo durante 20 minutos para enfriarlas.
- Recuperar las células por centrifugación a 2700g (4100 rpm en un rotor Sorval GSA) durante 10 min a 4oC.
- Retire el sobrenadante. Escurra los últimos rastros de medios colocando la botella boca abajo sobre una almohadilla o una toalla de papel.
- Resuspenda cada pellet bacteriano en 30 ml de una solución helada CaCl2-MgCl2 (80 mM MgCl2, 20 mM CaCl2). Primero agregue 5 ml de la solución, gire cuidadosamente hasta que el pellet se haya disuelto por completo y luego agregue los 25 ml restantes de solución.
- Repita el paso 2.4.
- Repita el paso 2.5.
- Si las células competentes van a ser transformadas directamente, resuspende cada pellet bacteriano en 2 ml de una solución helada CaCl2 (0.1 M) girando los tubos cuidadosamente. Si el pellet no se resuspende con este método, resuspende pipeteando suavemente hacia arriba y hacia abajo (evitando la formación de burbujas).
Alternativamente, las células competentes se pueden congelar y almacenar para su uso posterior. Para preparar las existencias congeladas de células competentes, resuspenda el pellet en 2 ml de una solución caCl2 de 0,1M que contenga 10% (v/v) de glicerol. Esta solución debe estar helada. Suspensión de células alícuotas en tubos de polipropileno de 1,5 ml de hielo (160 ml por tubo). Congele las células competentes inmediatamente en un baño de hielo seco/etanol. Transfiera los tubos a un congelador de -70oC. - Para transformar las células tratadas con CaCl2,transfiera 50 ml de células competentes a cada uno de los 2 tubos de polipropileno de 1,5 ml. Añadir los 1 l (100 pg) de ADN plásmido pUC19 a uno de los tubos y dejar el segundo tubo sin ADN (control negativo). Mezclar suavemente (evitar la formación de burbujas). Incubar durante 30 minutos sobre hielo.
NOTA: No se deben utilizar más de 50 ng de ADN en un volumen de 10 ml o menos en la transformación. - Transfiera los tubos al bloque de calor e incubar a 42oC durante 45 s exactamente.
NOTA: El choque por calor es un paso crítico. No exceda la temperatura ni el tiempo de incubación. - Transfiera fácilmente los tubos al hielo. Incubar durante 2 min.
- Añadir 950 l de medios SOC e incubar los tubos durante 1 hora a 37 oC para permitir que las bacterias se recuperen y expresen el marcador resistente a los antibióticos codificado en el plásmido.
- Diluir 10 ml de la suspensión celular en 1000 ml en SOC (1/100 de dilución) y 100 l de la suspensión celular en 1000 ml en SOC (1/10 de dilución). Placa 100 l de las diluciones, así como el control, en placas selectivas, y se extiende nifique utilizando una espátula. Por lo general, el chapado de 100 ml de una dilución de 1/100 y 1/10 producirá un número suficiente de unidades formadoras de colonias (cfu) por placa. Idealmente, este número debe oscilar entre 30-300 cfu para que haya suficientes colonias pero separadas entre sí. Sin embargo, el número de cfu dependerá de la eficiencia de la transformación (consulte la Sección análisis de datos y resultados).
- Incubar las placas a 37oC. Las colonias transformadas deben aparecer en 12-16 horas (este rango dependerá de la cepa celular y el método de selección). Ninguna colonia debe crecer en el control negativo.
- Contar el cfu/placa obtenido para la transformación (Tabla 1).
- Para verificar que los transformadores albergan el plásmido pUC19, se realizará una preparación plásmido y posterior digestión. Para ello, inocular una sola colonia en 3 ml de caldo LB en un tubo. Crecer agitando a 210 rpm a 37oC durante la noche (16-20 horas).
- Prepare una preparación de plásmido utilizando el kit miniprep QIAprep Spin, de acuerdo con las instrucciones del fabricante.
- Digerir el 1 g de pUC19 purificado con la enzima de restricción HindIII a 37oC durante 1 hora.
NOTA: Cualquier enzima que corte en el sitio de clonación múltiple pUC19 se puede utilizar para este paso.
| Componente | Cantidad |
| 10X Búfer de resumen de restricción | 2,5 l |
| Plásmido pUC19 | 1 g |
| Hind Ⅲ | 1 l |
| H2O | 20,5 l (a 25 l) |
- Ejecuta una escalera de peso molecular, ADN pUC19 digerido y la misma cantidad de ADN pUC19 no digerido en un gel de agarosa del 1% que contiene bromuro de etidio de 1 g/ml durante 1 hora a 95 V.
NOTA: el tiempo y el voltaje variarán dependiendo del equipo utilizado. - Visualice el gel bajo luz UV. Compare el tamaño del ADN pUC19 digerido y no digerido(Figura 2) (consulte la Sección Análisis de datos y resultados).
Continúe con los pasos necesarios necesarios para verificar la transformación de acuerdo con el objetivo de cada experimento de transformación en particular.

Figura 2: Digestión del ADN plásmido recuperado de células transformadas DH5. El ADN del plásmido se recuperó de las células transformadas de DH5, digerido con HindIII, ejecutado en un gel de agarosa del 1% y visualizado con una fuente UV (pasos 2.19 a 2.22).
3. Análisis de datos y resultados
Para calcular la eficiencia de la transformación, es necesario contar un indicador de lo bien que las células tomaron el ADN extracelular, las colonias obtenidas en la transformación:
| Dilución | Ufc |
| 1/100 | 34 |
| 1/10 | 246 |
Tabla 1: Unidades formadoras de colonias (cfu) contadas a partir del experimento de transformación.
La eficiencia de transformación (TE) es una medida del número de cfu resultante de la transformación de 1 g de plásmido en un volumen determinado de células competentes. Muchos parámetros afectan a la eficiencia de la transformación: tamaño plásmido, genotipo celular, etapa de crecimiento durante la preparación de la competencia, métodos de transformación, etc.). Al calcular el TE es importante considerar qué dilución (si la hay) se realizó antes del enchapado e incorporarla en el cálculo del número total de cfu. La eficiencia de transformación (TE) se calcula con la siguiente ecuación:

En primer lugar, divida el cfu por el g de ADN, en este ejemplo 0.0001 g. A continuación, divida el resultado por el factor de dilución. En este ejemplo, se utilizó una dilución de 1/10 y se celenó 100 ml de una solución de 1 ml (dilución: 1/10 a 100 l /1000 l a 0,01).

Las bacterias son notablemente adaptables y un mecanismo que facilita esta adaptación es su capacidad para tomar moléculas externas de ADN. Un tipo de ADN que las bacterias pueden tomar se llama plásmido, una pieza circular de ADN que con frecuencia contiene información útil, como genes de resistencia a antibióticos. El proceso de modificación de bacterias por nueva información genética incorporada de una fuente externa se conoce como transformación. La transformación se puede realizar fácilmente en el laboratorio utilizando Escherichia coli, o E. coli.
Para ser transformados, las células de E. coli deben ser obligadas primero, lo que significa ser capaces de tomar moléculas de ADN de su entorno. El protocolo para lograr esto es sorprendentemente simple, una incubación corta de las células en una solución de cloruro de calcio. Esta incubación hace que las células se vuelvan permeables a las moléculas de ADN. Después de que las células son peletadas por centrifugación, se extrae el sobrenadante. El ADN plásmido ahora se añade a las células competentes. Después de incubar las células con ADN, la mezcla se calienta brevemente a 42 grados Celsius, seguido de un enfriamiento rápido en el hielo. Este choque térmico hace que el ADN se transfiera a través de la pared y las membranas de la célula. Las células se incuban en medios frescos. Luego, las bacterias se colocan a 37 grados para permitirles volver a sellar sus membranas y expresar proteínas resistentes.
Esas células que han tomado en los plásmidos copiarán fielmente el ADN y lo transmitirán a su progenie y expresarán cualquier proteína que pueda ser codificada por él, incluidos los mediadores de resistencia a los antibióticos. Esos genes de resistencia se pueden utilizar como marcadores seleccionables para identificar bacterias que se han transformado con éxito porque las células que no han tomado el plásmido no expresarán el producto del gen de resistencia. Esto significa que cuando las células están chapadas en un medio sólido que contiene el antibiótico apropiado, sólo las células que han tomado el plásmido crecerán. La transformación de las células en una colonia en crecimiento se puede confirmar aún más mediante el cultivo de esas células en medios líquidos durante la noche para aumentar el rendimiento antes de extraer el ADN de la muestra. Una vez que el ADN está aislado, se puede llevar a cabo una enzima de restricción diagnóstica. Debido a que las enzimas de restricción cortan el ADN en lugares predecibles, ejecutar estos digeridos en un gel debe mostrar un patrón predecible si el plásmido deseado se transformó con éxito. Por ejemplo, si pUC19 está preparado y cortado con la enzima de restricción HindIII, se debe ver una sola banda de 2686 nucleótidos en el gel.
En este laboratorio, transformará la cepa de E. coli DH-5 Alpha con pUC19, y luego confirmará la transformación exitosa por electroforesis de gel de ADN.
Antes de iniciar el procedimiento, ponte el equipo de protección personal adecuado, incluyendo una capa de laboratorio y guantes. A continuación, esterilizar el espacio de trabajo con 70% de etanol.
Ahora, prepare células químicamente competentes depositando un bucle lleno de bacterias en una placa de agar LB estéril y rayando las bacterias con un nuevo lazo. Luego, incubar la placa a 37 grados centígrados durante la noche. Al día siguiente, esterilizar la parte superior del banco con 70% de etanol de nuevo, y retirar la placa de la incubadora.
Inocular una sola colonia bien aislada en 3 mililitros de caldo LB en un tubo con un lazo estéril. Luego, hacer crecer el cultivo a 37 grados centígrados durante la noche, con temblores a 210 RPM. Al día siguiente, medir la densidad óptica del cultivo nocturno con un espectrofotómetro. Luego, agregue 100 mililitros de caldo LB a un matraz de un litro, e inocularlo con el cultivo nocturno a una densidad óptica de 0. 01. Ahora, incubar la cultura a 37 grados centígrados con temblores, y comprobar el OD600 cada 15 a 20 minutos hasta que el cultivo alcance la fase de crecimiento medio exponencial.
Después de aproximadamente tres horas, transfiera 50 mililitros del cultivo a dos botellas de polipropileno heladas. Luego, vuelva a colocar las botellas en hielo durante 20 minutos para enfriarlas. A continuación, recupere las células a través de la centrifugación. Deseche los supernatantes y coloque las botellas boca abajo sobre una toalla de papel. A continuación, resuspenda el pellet bacteriano en cinco mililitros de solución de cloruro de magnesio de cloruro de calcio en frío y gire cuidadosamente hasta que el pellet se haya disuelto por completo. A continuación, agregue otros 25 mililitros de la solución al pellet bacteriano disuelto. Resuspenda el otro pellet bacteriano como se ha demostrado anteriormente. Después de esto, repita la centrifugación y retire los sobrenatantes.
Si las células competentes van a ser transformadas directamente, resuspenda cada pellet bacteriano en dos mililitros de una solución de cloruro de calcio de 0,1 molar es 0,1 hielo girando los tubos cuidadosamente. Para comenzar el procedimiento de transformación, transfiera 50 microlitros de células competentes a dos tubos de polipropileno de 1,5 mililitros etiquetados. Luego, agregue un microlitro de ADN plásmido pUC19 a uno de los tubos. Mezclar suavemente, evitando la formación de burbujas, e incubar ambos tubos durante 30 minutos sobre hielo. Después de la incubación, transfiera los tubos a un bloque de calor e incubar a 42 grados centígrados durante 45 segundos. Transfiera inmediatamente los tubos al hielo e incubar durante dos minutos. Ahora, agregue 950 microlitros de medios SOC a cada tubo e incubarlos durante una hora a 37 grados Celsius para permitir que las bacterias se recuperen, y exprese el marcador resistente a los antibióticos codificado en el plásmido.
Para hacer una dilución de 1 a 100, agregue 990 microlitros de medios SOC y 10 microlitros de suspensión celular a un tubo de 1,5 mililitros. Luego, hacer una dilución de 1 a 10 mediante la adición de 900 microlitros de medios SOC y 100 microlitros de suspensión celular a un tubo de 1.5 mililitros. A continuación, placa 100 microlitros de las suspensiones celulares diluidas y 100 microlitros del control negativo, en placas selectivas separadas que contienen ampicilina utilizando un esparcidor e incubar las placas a 37 grados centígrados durante 12 a 16 horas. Después de la incubación, cuente las unidades formadoras de colonias, o UFC, por placa, obtenidas a través de la transformación, y registre estos datos. Para verificar que los transformadores tienen el plásmido pUC19, elija una sola colonia bien aislada de una placa con un lazo estéril, e introdúzcalo en un tubo que contenga 3 mililitros de caldo LB. Luego, incubar la cultura a 37 grados centígrados con temblores, de la noche a la mañana. Al día siguiente, utilice un mini kit de preparación de ADN para aislar el ADN de 3 mililitros del cultivo, de acuerdo con las instrucciones del fabricante. Después de completar la mini preparación del ADN, digiere el 1 microgramo de pUC19 purificado con una enzima de restricción a 37 grados Celsius durante 1 hora. Ahora, cargue 20 microlitros de una escalera de peso molecular, 1 microgramo de ADN de plásmido digerido y 1 microgramo de ADN plásmido no digerido en pozos consecutivos de un gel de agarosa del 1% que contiene 1 bromuro de etidio de microgramo por mililitro. A continuación, ejecute el gel durante 1 hora a 95 voltios. Por último, visualice el gel con un iluminador UV.
En este experimento, E. coli DH5 Alpha se prepararon células químicamente competentes utilizando una adaptación del procedimiento de cloruro de calcio, y luego se transformaron con el plásmido pUC19 para determinar la eficiencia de transformación. Para calcular la eficiencia de transformación, utilice los recuentos de UFC registrados para las diluciones 1 de cada 100 y 1 en 10, y cualquier otra dilución con CFU cuenta entre 30 y 300. En primer lugar, el recuento de UFC registrado, 246 en este ejemplo, se divide por la cantidad de ADN, .0001 microgramos aquí, que fue chapado. A continuación, este número se divide por el factor de dilución utilizado para dar la eficiencia de transformación en UFC por microgramo. En este ejemplo, se utilizó una dilución de 1 a 10 y se chaparon 100 microlitros de una solución de 1 mililitro, lo que le da un factor de dilución final de 0,01. En el carril plásmido no digerido, el ADN circular puede aparecer como dos o tres bandas diferentes de brillo variable. Esto se debe a que el ADN circular sin cortar puede existir en varios estados de conformación diferentes, como supercoiled, círculo abierto, o más lineal, y cada uno de estos se mueve nalto a través del gel a diferentes velocidades. El análisis de la digestión del ADN plásmido recuperado indicó que el plásmido utilizado tiene un tamaño esperado de ADN pUC19, 2.686 pares de bases.
Subscription Required. Please recommend JoVE to your librarian.
Results
Aunque TE depende de muchos factores, las preparaciones celulares competentes no comerciales, como esta, normalmente producen de 106 a 107 transformadores por microgramo de plásmido. Por lo tanto, esta preparación, con un TE de 2,46 x 108 cfu/g, produjo un TE mucho más allá del rango esperado. Existen protocolos adicionales para fabricar celdas supercompetentes cuando se requieren mayores eficiencias de transformación para una aplicación determinada (13).
El análisis de la digestión del ADN plásmido recuperado de las células transformadas indicó que este plásmido tiene el tamaño esperado del ADN pUC19 (2686 bp).
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
La transformación es un método potente para introducir ADN exógeno en células bacterianas que es clave para muchas aplicaciones de biología molecular en el laboratorio. Además, desempeña un papel importante en la naturaleza al permitir que las células bacterianas intercambien material genético que podría resultar en una mayor variación genética y permitir la adquisición de diferentes rasgos beneficiosos para la supervivencia bajo una amplia gama de condiciones. Muchas cepas bacterianas codifican los genes necesarios para la competencia natural. Sin embargo, todavía se desconocen las condiciones en las que se inducen estos genes. Se requiere más investigación para determinar estas condiciones.
Subscription Required. Please recommend JoVE to your librarian.
References
- Croucher, N. J. et al. Rapid pneumococcal evolution in response to clinical interventions. Science. 331 (6016):430-434. (2011)
- Borgeaud, S. et al. The type VI secretion system of Vibrio cholerae fosters horizontal gene transfer. Science. 347(6217):63-67. (2015)
- Burmeister, A. R. Horizontal Gene Transfer. Evol Med Public Health. 2015 (1):193-194. (2015)
- Weston A, Brown MG, Perkins HR, Saunders JR, Humphreys GO. Transformation of Escherichia coli with plasmid deoxyribonucleic acid: calcium-induced binding of deoxyribonucleic acid to whole cells and to isolated membrane fractions. J Bacteriol. 145 (2):780-7. (1981)
- Dagert M, Ehrlich SD. Prolonged incubation in calcium chloride improves the competence of Escherichia coli cells. Gene. 6 (1):23-8. (1979)
- Asif A, Mohsin H, Tanvir R, and Rehman Y. Revisiting the Mechanisms Involved in Calcium Chloride Induced Bacterial Transformation. Front Microbiol. 8:2169. (2017)
- Panja S, Aich P, Jana B, Basu T. How does plasmid DNA penetrate cell membranes in artificial transformation process of Escherichia coli? Mol Membr Biol. 25 (5):411-22. (2008)
- Silhavy, TJ, Kahne D, Walker S. The Bacterial Cell Envelope. Cold Spring Harb Perspect Biol. 2 (5): a000414. (2010)
- Panja S, Aich P, Jana B, Basu T. (2008) Plasmid DNA binds to the core oligosaccharide domain of LPS molecules of E. coli cell surface in the CaCl2-mediated transformation process. Biomacromolecules. 9 (9):2501-9.
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Plasmid Purification. JoVE, Cambridge, MA. (2018)
- Bergkessel M and Guthrie C. Colony PCR. Methods in Enzymology. 529: 299-309. (2013)
- Sambrook J and Russell DW. Molecular Cloning A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.Protocol 25 (1.116-118). (2001)
- Wirth R, Friesenegger A, Fiedler S. Transformation of various species of gram-negative bacteria belonging to 11 different genera by electroporation. Molecular & General Genetics. 216 (1): 175-7. (1989)