

Summary
Dans cette vidéo, article, nous présentons une méthode pour l'isolement et la purification de
Abstract
L'
Protocol
Commentaires généraux sur le tri des billes magnétiques neurones périphériques drosophile (Timing Total pour l'achèvement du Protocole: 2.5-3 heures)
Procédures de laboratoire standard pour maintenir un environnement propre, RNAse environnement libre doivent être respectées en tout temps pour prévenir la dégradation de l'ARN.
Lorsque la cuticule des larves de drosophile est disséqué et placé dans le buffer dissociation cellulaire, les neurones périphériques sont l'une des dernières cellules de se détacher de la cuticule. Nous avons exploité cette propriété et conçu ce protocole pour éliminer la plupart des cellules non spécifiques de la cuticule comme le muscle et la graisse avant l'isolation des neurones DA.
Avec la pratique, le protocole de l'ensemble peut être complété avec succès au sein d'environ 2,5 heures. La préparation des billes revêtues d'anticorps doit être rempli avant l'expérience commence.
1. Préparation des billes magnétiques pour les piles à reliure:
Cette étape doit être terminée avant le début de l'expérience. Les billes préparées peuvent être préparés et conservés à 4 ° C jusqu'à ce que nécessaire.
- Lavez 100 ul de billes streptavidine Dynabeads M-280 recouvert de trois fois en PBS en le remettant en suspension dans 500 ul de PBS frais et de granulation dans un champ magnétique fort à chaque fois.
- Enfin remettre en suspension les perles directement dans 100 ul d'anticorps biotinylé dilué rat anti-souris-CD8a (concentration d'anticorps est de 100 pg / ml).
- Incuber le mélange pendant 1 heure sur la glace avec des vortex légers occasionnels à empêcher la sédimentation. [1 pl de Dynabeads M-280 peuvent se lier de 0.05 à 0,10 pg d'anticorps biotinylé].
- Laver le mélange billes-anticorps à trois reprises comme décrit dans l'étape 1.1 de supprimer les excès d'anticorps. Les billes magnétiques sont maintenant recouverts d'anticorps et prêt à être utilisé. Stocker le mélange de perles d'anticorps dans 100 ul de PBS 1X à 4 ° C jusqu'à utilisation.
2. Sélection et laver Larves: (10-15 minutes)
- Choisissez appariés pour l'âge de 30 à 50 larves troisième stade et les placer dans un tube de 1,6 ml avec 1 à 1,2 microcentrifugeuse ml de phosphate 1X saline tamponnée (PBS). (3-5 minutes)
- Fermer le tube et le vortex au réglage maximum trois fois pendant 1 seconde chacun.
- Utiliser un feu poli pipette Pasteur jeter le surnageant complètement. Répéter le lavage (2,1) et le vortex (2,2) étapes 3-4 fois jusqu'à ce que le surnageant est visiblement claire de toutes les particules de nourriture et de débris.
- Brièvement répéter le lavage avec 1 ml d'éthanol à 70% et jeter le surnageant.
- Laver les larves à deux reprises avec 1 ml de ddH2O et jeter le surnageant.
- Brièvement répéter les laver une fois avec 1 ml de RNase-AWAY et jeter le surnageant.
- Laver les larves à trois reprises dans 1 ml de ddH2O pour assurer l'élimination complète de RNase AWAY.
3. Dissection: (10-12 minutes)
- Placez 10-12 larves sur le centre d'une plaque recouverte Sylgard 35mm de Pétri. Placez-les légèrement à l'écart les uns des autres. [Etape critique: Jeter les larves qui ne semblent pas être au stade approprié de développement]
- Coupez l'extrémité antérieure de la larve à l'aide d'une paire de ciseaux de dissection fine pour toutes les larves.
- En utilisant une paire de ternes Dumont n ° 5 pinces à inverser la larve à l'envers. Insérez une pince à l'intérieur de la cuticule des larves tout le chemin à l'extrémité postérieure. Pincez le bout de la pince ensemble pour attraper l'extrémité postérieure de la cuticule (figure 1a) (essayez d'appuyer sur la cuticule sur la surface Sylgard pour le rendre plus facile). Utilisation de la deuxième paire de pinces, poussez la cuticule des larves intérieur. [Etape critique: Essayez de pratiquer cette méthode à quelques reprises avant de tenter l'expérience d'isolement cellulaire. Essayez d'obtenir les larves complètement renversé, pour s'assurer que tous les tissus mous sont exposées à la solution pour faciliter la dissociation.]
- Après dissection des larves 3-4, les transférer immédiatement à frais, PBS glacé (placer le tube sur la glace) dans un tube à centrifuger 1,6 ml.
- Répétez les étapes 3.1 à 3.4 jusqu'à ce que toutes les larves exigées sont recueillies (30-40 larves de ce protocole). [Etape critique:. Anticiper 10-20% de perte lors de la dissection et la dissociation, et planifier en conséquence]
4. Retrait de faiblement adhérentes non spécifiques des cellules: (2-3 minutes)
[Cette étape aides de compensation des faiblement adhérentes non spécifiques des tissus tels que les corps gras et du SNC.]
- Prenez le tube de microcentrifugation 1,6 ml contenant les cuticules larvaires inversé et remplacer le surnageant avec environ 700 à 800 l de frais PBS glacé.
- Pulse-vortex du tube de centrifugeuse 5 fois (3 secondes par impulsion) à pleine vitesse.
- Jeter le surnageant et le remplacer par quelque 700 à 800 ul frais, PBS glacé.
- Répétez les étapes 4.2 et 4.3 à trois reprises.
- Resuspend les cuticules des larves dans 400 ul de frais, PBS glacé
5. Dissociation du tissu dans une suspension cellulaire unique: (18-20 minutes)
[Etape critique: Plus-dissociation peut entraîner la perte du marqueur de surface cellulaire menant à rendement cellulaire pauvres et la viabilité cellulaire faible. Le tissu larvaire peut être dissociée ni par dissociation mécanique (sonication, douncing), dissociation enzymatique (trypsine, la collagénase etc) ou une combinaison des deux. Comme ces tissus larvaires sont difficiles à dissocier, nous avons constaté que la combinaison des deux mécaniques et dissociation enzymatique donné les meilleurs résultats.]
- Ajouter 1,5 l de Libérase 1X Blendzyme 3 (28 W unités nsch / flacon) à la cuticule des larves en suspension dans 400 ul de PBS.
- Vortex la solution 2-3 fois pendant 1 seconde au réglage maximum (Cela devrait éliminer les cellules faiblement adhérentes de la cuticule dans la solution).
- Incuber la solution à température ambiante (22-25 ° C) pendant 5 minutes. [Etape critique: temps d'incubation affecte grandement l'efficacité de cellules finales de tri. Le temps d'incubation recommandée devrait être suffisante pour desserrer le tissu. Ne pas dépasser 15 minutes.]
- Pulse-vortex le tube 20-30 fois au maximum de paramètres pour 2 secondes par impulsion à pleine vitesse. Cela devrait libère les muscles et autres tissus dans la solution. Inspectez un petit échantillon de la solution sous une lampe fluorescente stéréomicroscope à chaque étape. (Avec l'expérience on devrait être capable de déterminer le niveau de dissociation en observant le tube de micro directement sous une fluorescence activée stéréomicroscope)
- Lavez les cuticules larvaires 2-3 fois avec frais, PBS glacé et enfin de les remettre en suspension dans 500 ul de frais, PBS glacé contenant 1% de BSA.
- Pour éviter la cuticule des larves coller à la surface de verre de la broyeur de tissus Kontes 2 ml et un pilon grand dégagement, pré-enduire le broyeur de tissus et d'un pilon avec un BSA à 1% dans une solution PBS et après un bref rinçage jeter la solution de BSA. Par la suite, à l'aide d'une pipette Pasteur polie au feu, transférer les cuticules à l'étape 5.5 de la BSA broyeur de tissus enduits. [Etape critique: Pré-refroidir le broyeur de tissus / pilon en le plaçant sur la glace pendant quelques minutes afin de prévenir les dommages cellulaires / lyse].
- Dounce le tissu avec des coups lente et régulière, en évitant moussage (environ 20-30 coups). [Etape critique:. Dounce lentement et régulièrement, sinon les cellules peuvent lyser]
- Pour évaluer les niveaux de dissociation cellulaire, essuyez la paroi extérieure du broyeur de tissus avec un tissu Kimwipe propre, et l'inspecter sous une lampe fluorescente stéréomicroscope. Les neurones doivent avoir détaché de la cuticule, et peut être vu dans la solution. Si cela s'avère difficile, sinon la pipette sur un petit échantillon de la solution, et l'observer sous une lampe fluorescente stéréomicroscope. Une bonne indication de la dissociation cellulaire est l'absence de neurones à partir de la cuticule larvaire. Toutefois, si l'on observe encore des neurones attaché à la cuticule, ou observe des cellules incomplètement dissocié, Dounce encore jusqu'à ce que la cellules obtenir une suspension cellulaire unique.
- Triturer la solution 5 fois avec une pipette Pasteur polie au feu réduit à environ 50% du diamètre de la pointe standard, suivie par 10 fois avec une pipette Pasteur polie au feu réduit à environ 25% du diamètre de la pointe standard. [Etape critique: la trituration énergique peut endommager les cellules. Surveiller les cellules entre les étapes, et d'ajuster la procédure en conséquence].
- Filtrer la solution à travers un filtre 30 um cellulaire et recueillir le filtrat de cellules dans un tube à centrifuger 1,6 ml. La solution obtenue doit consister en une suspension cellulaire unique et est maintenant prêt pour le tri magnétique de cellules.
6. Magnétique tri cellulaire perle: (45 - 75 minutes, dépendant de la durée d'incubation d'anticorps)
- Ajouter 15 ul de billes magnétiques recouvertes d'anticorps à 500 pl de suspension cellulaire (étape 5.10). Les autres anticorps conjugués billes magnétiques peuvent être stockées jusqu'à ce que nécessaire pour les isolements cellulaires ultérieures.
- Incuber les cellules avec des billes magnétiques recouvertes d'anticorps pendant 30-60 minutes sur la glace avec parfois la main de mélange. [Etape critique: Incubation à une température plus élevée ou plus de temps peut entraîner la liaison d'anticorps non spécifiques.]
- Placez le tube de centrifugeuse dans un fort champ magnétique pendant 2 minutes. Toutes les cellules sélectionnés positivement le long avec des perles non liée seront séparés sur le côté du tube.
- Lentement la pipette le surnageant en veillant à ne pas perturber le culot cellulaire.
- Laver les cellules 3-4 fois en eau douce, PBS glacé pour enlever tout reste non spécifique des cellules.
- Resuspendre les cellules dans 30 ul de frais, PBS glacé.
- Pour estimer la pureté et le rendement des cellules, des pipettes de 5 pl de la cellule de suspension sur la surface polie d'un hémocytomètre et compter toutes les cellules visibles fluorescentes sous une lampe fluorescente stéréomicroscope. Vérifiez également la quantité de cellules non fluorescentes et de tout signe d'impuretés. Typiquement, l'échantillon sera hautement enrichi pour les cellules fluorescentes.
7. Isolement d'ARN à partir de billes magnétiques cellules triées: (60 - 75 minutes)
- Après le dépouillement, un culot cellulaire dans un champ magnétique, éliminer le surnageant et ajouter 20 ul de tampon d'extraction de l'ARN PicoPure ™ Kit d'isolation (Molecular Devices). Selon le nombre de cellules on peut avoir besoin d'ajouter un volume plus élevé de tampon d'extraction.
- Vortexer le tube à la vitesse maximale pour permettre le mélange de culot cellulaire avec un tampon d'extraction.
- Incuber le tube à 42 ° C pendant 30 minutes.
- Pour assurer l'enlèvement des billes magnétiques avant purification sur colonne de l'ARN (voir étape 7.5), le tube est centrifugé à 2000 brièvement (x) g pendant 2 minutes pour sédimenter les billes magnétiques. Le tube est ensuite placé dans un champ magnétique fort pour conserver le culot et le surnageant est transféré à un tube de centrifugeuse nouvelle.
- Extraire et purifier la colonne de l'ARN conformément aux instructions du fabricant de kit d'extraction d'ARN PicoPure est. Traitement à la DNase est facultative et peut être effectuée sur une colonne lors de la purification d'ARN selon l'exigence d'analyse. Enfin, éluer l'ARN lié au total dans un petit volume (11-30 pi) de tampon d'élution et conserver à -80 ° C jusqu'à utilisation. Si vous le souhaitez une aliquote de 1 ul peuvent être utilisés pour évaluer la qualité totale ARN sur un bioanalyseur 2100 (Agilent Technologies, Inc.)
Les résultats représentatifs:
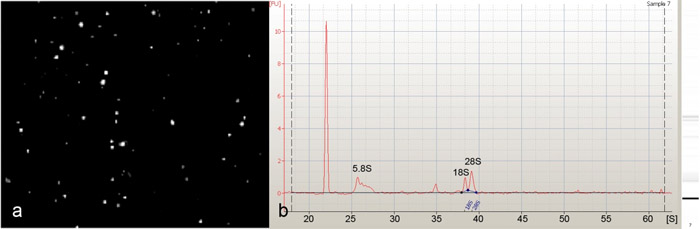
Magnétique de tri perle a été utilisée pour isoler des drosophile da neurones (figure 1). Les ARN purifiés à partir de ces neurones isolés da (figure 2a) a été jugée d'excellente qualité, comme indiqué par la présence de 5.8S forte, 18S et 28S du ribosome pics ARN lorsqu'il est analysé sur une bioanalyseur Agilent 2100 (Agilent Technologies, Inc) ( Figure 2b). Début avec 30-40 larves du troisième que nous étions capables d'isoler, en moyenne 300 à 500 neurones da classe IV en utilisant le pilote PPK-GAL4, et 1500-2000 neurones DA (classe I, II, III et IV) en utilisant le pan- da spécifique des neurones GAL4 21-7 conducteur. Pour évaluer l'enrichissement neuronaux spécifiques de nos cellules isolées nous avons effectué quantitative reverse transcription PCR (qRT-PCR) en utilisant deux gènes spécifiques neuronaux marqueurs (elav et futsch). Ces analyses ont révélé un enrichissement significatif pli de gènes marqueurs indiquant à la fois un enrichissement très spécifiques pour les neurones da par rapport à circuler à travers l'aide de notre protocole (Figure 3). Enfin, l'ARN isolé à la fois pan-da neurones et de classe-IV neurones DA a été utilisé pour effectuer des profils d'expression transcriptionnelle sur Agilent Drosophila melanogaster microarrays oligo génome entier (4 x 44K) (figure 4). Ces analyses identifié de nombreux régulateurs préalablement impliqués de da neurone dendrite morphogenèse en plus à un large éventail de molécules non définies auparavant et putatifs voies de transduction de signal qui pourrait jouer des rôles fonctionnels importants dans les neurones da développement. Des études visant à évaluer le rôle potentiel (s) de ces molécules non définies auparavant dans la médiation da neurone développement, et de la morphogenèse spécifiquement dendrite, sont actuellement en cours.

Figure 1:. Schéma de tri bille magnétique des neurones DA drosophile (a) appariés pour l'âge des larves du troisième stade portant da spécifique des neurones GAL4, journaliste UAS-GFP-mCD8 transgène sont disséqués en inversant la cuticule des larves dedans-dehors, pour exposer les SNP à la dissociation de tampon et stocké dans PBS glacé. (b) la dissociation enzymatique est effectuée en ajoutant Libérase Blendzyme 3 à la solution contenant la cuticule larvaire. (c) Les tissus larvaires sont encore dissociés par une combinaison de vortex, la trituration et douncing pour enlever les tissus non spécifiquement étiquetés comme les graisses des organismes, des intestins et du SNC. (d, e) Les cellules sont alors filtrés en utilisant un filtre de 30 um cellule. La solution contient une suspension cellulaire unique de différents types cellulaires, y compris les épithéliums, les muscles et les neurones. (F) anti-souris CD8a-anticorps revêtement Dynabeads M-280 sont ajoutés à la suspension de cellules et incubé sur glace pendant 30-60 minutes. ( g) Les billes magnétiques se lie à des neurones qui expriment da une souris CD8 marqués protéine de fusion GFP. (h, i) La bille magnétique cellules enrobées sont séparés en plaçant la solution dans un fort champ magnétique. Le surnageant est jeté et les cellules sont lavées trois fois pour éliminer tout résidu non-spécifique des cellules, ce qui entraîne (j) highly purifiée populations de neurones DA. S'il vous plaît cliquez ici pour voir une version agrandie de la figure 1.

Figure 2: Image représentant (a) de la sélection positive des neurones, la GFP fluorescente da classe IV isolés par dissociation cellulaire et magnétiques de tri de billes. La population résultant de neurones a été jugée hautement enrichi pour les neurones da classe IV avec peu ou pas d'impuretés de cellules contaminantes. (B) Une bioanalyseur Agilent 2100 (Agilent Technologies, Inc) électrophérogramme de l'ARN total isolé à partir bille magnétique neurones da triés , montrant une excellente qualité d'ARN total, comme indiqué par la présence de 5.8S, 18S, 28S et ARNr. S'il vous plaît cliquez ici pour voir une version agrandie de la figure 2.

Figure 3: qRT-PCR de l'expression du gène marqueur de neurones isolés dans les neurones da (GAL421-7, SAMU-mCD8-GFP) et le débit à travers fraction a été réalisée en trois exemplaires. Les niveaux d'expression des deux gènes marqueurs neuronaux spécifiques (elav et futsch) ont été évalués par qRT-PCR. Les valeurs obtenues à partir de ces analyses ont été normalisées pour le contrôle endogène (rp49), et les niveaux par rapport à ceux observés dans traversent fraction ont été calculés en utilisant la méthode ΔΔCτ 6. Les deux elav et futsch ont été considérablement enrichie dans la population isolée da neurone par rapport à l'écoulement à travers fraction.

Figure 4: Représentant de classe IV da spécifique des neurones marqués Cy3 fichier image de biopuces. On voit ici une Agilent Drosophila melanogaster génome entier oligo biopuces (4 x 44K) hybridées avec Cy3 labeld ARN total isolé à partir de neurones da classe IV purifié par le tri bille magnétique. S'il vous plaît cliquez ici pour voir une version agrandie de la figure 4.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Le protocole présenté ici est optimisé pour l'isolement et la purification des neurones périphériques qui adhèrent fermement à la surface interne de la cuticule drosophile troisième stade larvaire en utilisant une stratégie de billes magnétiques tri cellulaire. Alors que nous avons utilisé ce protocole pour isoler spécifiquement drosophile neurones DA, les applications de ce protocole à l'isolement des autres types de cellules qui adhèrent à la cuticule larvaire ou pupal de développement (par exemple l'épithélium, le muscle, d'autres neurones périphériques) peuvent être adaptés par variant de quelques paramètres et l'utilisation de GAL4 distinctes, journaliste de transgènes UAS-GFP-mCD8 dont l'étiquette du type de cellule ou de types d'intérêts. En outre, ce protocole peut être utilisé à la fois dans la perte de fonction et le gain de fonction des approches où un gène d'intérêt peut être cloné dans un transgène UAS-GFP-mCD8 qui peut être couplé avec un transgène pour diriger GAL4 soit gènes spécifiques de perte de fonction (par exemple SAMU-ARNi) ou gain de fonction d'un type cellulaire d'intérêt. Par exemple, dans le cas d'un facteur de transcription on peut souhaiter identifier des gènes potentiellement haut ou le bas-réglementé en cas de perte de fonction ou gain de fonction d'expression dans un type cellulaire d'intérêt. En isolant l'ARN total à partir du type cellulaire d'intérêt purifiée via ce protocole et l'utilisation de cet ARN pour effectuer profilage de l'expression puces, il est possible d'identifier les gènes différentiellement réglementés qui peuvent représenter des cibles en aval de la régulation transcriptionnelle qui jouent un rôle dans la médiation de changements phénotypiques au sein de la cellule .
Pour le tri cellulaire réussie, il est essentiel de donner une attention particulière aux étapes critiques mis en évidence dans le protocole ci-dessus. Exemples de domaines problème commun qui peut exiger une certaine dépannage et d'optimisation, en fonction du type cellulaire, notamment (1) rendement cellulaire faible et (2) des cellules d'isolement pendant l'agglutination de billes magnétiques. Dans le premier cas, on peut essayer de réduire la concentration de Libérase Blendzyme 3, et de compenser en augmentant dissociation mécanique via douncing. Dans le second cas, on peut essayer de réduire l'intensité du champ magnétique par l'application d'un seul ou plusieurs couches de ruban adhésif sur le laboratoire de l'aimant.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Nous remercions les Drs. Yuh-Nung Jan et Wes Grueber pour fournir des stocks volantes utilisées dans cette étude. Les auteurs remercient F. Thomas et Kate Miller Jeffress Memorial Trust pour le soutien de cette recherche (DNC) et le Bureau du l'Université George Mason Provost (EPRI).
Materials
| Name | Company | Catalog Number | Comments |
| 10X Phosphate Buffered Saline (PBS) | MP Biomedicals | PBS10X02 | Diluted to 1X working solution |
| 10X Liberase Blendzyme 3 | Roche Group | 11814176001 | Diluted to 1X working solution (28 Wünsch units/vial) |
| RNase-AWAY | Sigma-Aldrich | 83931 | |
| Biotinylated Rat anti-Mouse-CD8a antibody | Invitrogen | MCD0815 | 100 μg/ml stock concentration |
| BSA (Bovine Serum Albumin), Fraction V | GIBCO, by Life Technologies | 11018-017 | Prepare a 1% BSA solution in PBS |
| Dynabeads M-280 Streptavidin | Invitrogen | 11205D | 1 μl can bind 0.05-0.10 μg of biotinylated antibody |
| PicoPure RNA Isolation Kit | Molecular Devices | KIT0204 | Follow manufacturer’s instructions |
Equipment
|
|||
References
- Corty, M. M., Matthews, B. J., Grueber, W. B. Molecules and mechanisms of dendrite development in Drosophila. Development. 136, 1049-1061 (2009).
- Parrish, J. Z., Emoto, K., Kim,, Jan, Y. N. Mechanisms that regulate establishment, maintenance, and remodeling of dendritic fields. Ann. Rev. Neurosci. 30, 399-423 (2007).
- Lee, T., Luo, L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 22, 451-461 (1999).
- Grueber, W. B. Projections of Drosophila multidendritic neurons in the central nervous system: links with peripheral dendrite morphology. Development. 134, 55-64 (2007).
- Song, W., Onishi, M., Jan, L. Y., Jan, Y. N. Peripheral multidendritic sensory neurons are necessary for rhythmic locomotion behavior in Drosophila larvae. Proc Natl Acad Sci USA. 104, 5199-5204 (2007).
- Livak, K. J., Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) methods. Methods. 25, 402-408 (2001).

{kind=link}
{kind=link}
{kind=link}