

Summary
神話では、モデル生物出芽酵母で発現されるタンパク質間の過渡と安定な相互作用を高感度に検出することができます。それが正常にハイスループットな方法でそれらの相互作用パートナーを同定するために、外因性および酵母内在性膜タンパク質の研究に適用されています。
Abstract
内在性膜タンパク質の基本的な生物学的及び臨床的重要性は、完全長の膜貫通型タンパク質のタンパク質間相互作用(PPI)の高スループットを識別するための酵母ベースのシステムの開発を促した。この目的のために、私たちの研究室では、スプリットユビキチン系膜酵母ツーハイブリッド(神話)システムを開発した。この技術は使用して過渡安定したタンパク質相互作用の高感度な検出が可能
Protocol
1。背景情報
タンパク質 - タンパク質相互作用(PPIは)すべての細胞プロセスを支配するに関わる基本的なビルディングブロックです。その結果、この生物学的均衡の変化を一般的に病や癌の細胞形質転換の役割を果たしていると、すべての相互作用はしっかりと、細胞の恒常性を維持するために規制されていることが不可欠である。彼らは複雑なシグナル伝達カスケードを開始する、とに関連する問題として、医療の分野における最近の重要性がされている薬物を含むさまざまな分子が、、のインポートとエクスポートの両方を媒介することができるように膜関連タンパク質は、タンパク質の生物学的に最も重要なクラスの中です。薬剤耐性は、ますます一般的になってきている。このタンパク質のクラスの複雑さへの洞察を得ることはそれらの相互作用のパートナーの識別を必要とします。そのようなパートナーを発見すると、それぞれの膜結合タンパク質[1]のために最適化されている必要があります過酷な条件を必要とする多くの場合として、挑戦証明されています。
内在性膜タンパク質の基本的な生物学的及び臨床的重要性は、完全長の膜貫通タンパク質のためのPPIのハイスループット同定のための酵母ベースのシステムの開発を促した。この目的のために、我々は、分割ユビキチン系膜酵母ツーハイブリッド(神話)システム[2-4]を開発した。このツールは一時的かつ安定的なタンパク質相互作用の高感度な検出が可能になります。それが正常に[3-7]をモデル生物出芽酵母で発現して外因性と内因性のタンパク質を研究するために適用されています。神話は、ユビキチンが2つの部分に分割されるという観察を利用しています:。C末端側半分(C UB)とN -末端半分(NubI)in vivo試験のための彼らの高い親和性により、これらの部 分が自発的に再構成することが示されている一方、別の(Figure. 1A)。しかし、ユビキチンのN末端 側半分(N UB Gと呼ばれるフラグメントの生産)にグリシンの点突然変異にイソロイシン13を導入することはこの自発的な再アソシエーション[8](Figure. 1bを)防ぐことができます。
私たちは、神話のシステム(Figure. 1c及び2)にこの原理を使用してください。簡単に言えば、内在性膜baitタンパク質は、 大腸菌 DNA -結合タンパク質のLexAで構成される人工的な転写因子や単純ヘルペスウイルスからVP16の活性化ドメインにリンクされているC UBの部分に融合させる。餌はNubG部分へのcDNAまたはゲノムDNA由来のフラグメントの融合によって生成されます。酵母宿主の餌と獲物タンパク質間の相互作用は、細胞質脱ユビキチン化酵素(ダビング)と転写因子のタンパク質分解のリリースによって、その後の認識で、完全長の"擬似ユビキチン"分子の再構成につながる。転写因子は、細胞の核を入力し、餌と獲物の相互作用の指標となる選択培地上の酵母株の生育を可能にするレポーター遺伝子システムを(通常はHIS3、lacZおよびADE2遺伝子の発現を含む)、アクティブにすることができます[ 2-4]。
内在性膜タンパク質の相互作用の同定と特徴付けは、完全にその機能を理解するために役立つ情報を提供します。我々はより正確に理解し、内在性膜タンパク質と相互作用するタンパク質の役割を解剖するように、我々はこれらのタンパク質の調節に関与する動的な相互作用に関する洞察を得ることと治療の可能性を持っていることが新たな標的を発見することができる。
2。ベイトと適切な神話のシステムの選定
- 神話の分析を行う前に、あなたのタンパク質はそのN -および/またはC末端の細胞の細胞質ゾルになっていることを確認します。それは転写因子の放出に必要な脱ユビキチン化酵素が細胞質ゾルに配置されているので、C UB -のLexA - VP16タグは、このような末端のタンパク質に融合させることが不可欠である[4]。
- 次に、適している神話の二つの主要な亜種のかを決める。非ネイティブ酵母タンパク質、伝統的な神話(tMYTH)のために餌をプラスミドから異所性に過剰発現される、請求、使用することができます。ネイティブ酵母のタンパク質については、統合された神話(iMYTHは)選択の方法です。 iMYTH餌で内生的にその天然のプロモーターの制御下にそれらを残して、C UB -のLexA - VP16のタグが指定されています。これは餌の野生型発現レベルは、そのような偽陽性の数が増加[3]などのタンパク質の過剰発現に関連する問題を排除するのに役立つように、有利である。初期餌の構築と使用される選択培地を除いて、神話の両方の形式は本質的に同一の方法で実施されています。このバリアントは、原則として、事実上すべての生物体からの膜タンパク質と一緒に使用していることができるように分かりやすくするために、我々は、このレポートでtMYTHの使用に主に焦点を当てるしたがって、より広く適用できる。
3。餌の生成と検証
- 必要なメディアおよびソリューション
- 無菌のddH 2 Oは 121℃で、30分間15 PSIをオートクレーブして調製。
- 3 -アミノ-1,2,4 -トリアゾール(3 - AT)のddHで1Mストック溶液として調製した溶液2 O 0.2μmのフィルターを通すことによって滅菌する。
- 1パーセントw / vの酵母エキス、2%w / vのペプトン、2%(w / v)のグルコースおよびのddH 2 Oで調製した100μMのアデニンから成るYPADの成長メディア 121 ° C、30分15 psiでオートクレーブで滅菌。
- 10倍のアミノ酸/ヌクレオチド塩基ミックス:完全なミックスを1.0 mMのアデニン、1.8 mMのウラシル、1.0mMのアルギニン、1.0mMのヒスチジン、2.3 mMのイソロイシン、7.6 mMのロイシン、1.6 mMのリジン、メチオニン10.1 mMの、3.0 mMのフェニルアラニン、16.8 mMのが含まれているスレオニン、2.0 mMのトリプトファン、1.7 mMのチロシン、12.8 mMのバリンは、のddH 2 Oで調製した。ドロップアウトのために必要なアミノ酸(S)および/またはヌクレオチド塩基(s)を省略する。 121 ° C、30分15 psiでオートクレーブで滅菌。
- 0.67パーセントw / vの酵母窒素塩基(アミノ酸を含まないが硫安)、2%(w / v)のグルコース、2%w / vの寒天と1xアミノ酸/ヌクレオチドミックスから成る合成ドロップアウト(SD)増殖培地 、のddH 2 Oで調製した両方の液体と固体(2%寒天を含む)、SD -ロイシンのメディアを準備します。また、固体SD -トリプトファン-ロイシンとSD -トリプトファン-ロイシン-アデニン-ヒスチジンメディアを準備する。 121 ° C、30分15 psiでオートクレーブで滅菌。 100x15ミリメートルシャーレに固体培地を注ぐ。
- 3 - ATを含む合成ドロップアウト(SD)増殖培地。説明されているようにSD -トリプトファン-ロイシン-アデニン-ヒスチジンメディアを準備しますが、3 - AT 25、50、75および100 mMの濃度で含む。それはオートクレーブと冷却された後、メディアへの1M滅菌ストック溶液(まだ固化していない)から、ATの3の適切な量を追加。 100x15ミリメートルのペトリ皿に注ぐ。
- 40%の成るPEG /リチウムアセテートミックス W / V PEG - 3350、120mMの酢酸リチウムおよび167μgの/のddH 2 Oで調製されたmLのサケ精子DNA(タイプIIIのナトリウム塩)この混合物の無菌性は、滅菌水やソリューションからそれを準備するために、(すなわち、50%PEG - 3350をオートクレーブ滅菌のddH 2 Oで調製した1M酢酸リチウム、2 mg / mLのサケ精子DNAのタイプIIIのナトリウム塩をオートクレーブ)。
- 酵素とPCRを実施するための試薬 。
- 商業ミニプレップキット。
- ソーダライムガラスビーズ(0.5mm)の。
- プラスミド伝播 (例えばDH5α、XL10金)と細菌の増殖やプラスミドの選択に適した標準的なメディアに適した有能な大腸菌の細胞 。
- などのプロトコルで説明されている特定の酵母菌株、プラスミドとプライマー 。
- ギャップ修復によってtMYTH餌の世代
- 餌は、タグ付けと発現に適したベクター中にクローニングされている必要があります。 tMYTH種々のベクターは、餌の建設で現在使用可能です。このようなpCMBV4、pAMBV4とpTMBV4としてベクトルはそれぞれ、制御CYC1(弱い)、ADH1(強い)とTEF1(非常に強力な)プロモーター下でC末端タグ付きベイトの建設(BAIT - C UB -のLexA - VP16)を許可する。 N末端 にタグ付け餌(のLexA - VP16 - C UB - BAIT)がそれぞれTEF1とCYC1プロモーターの制御下で、そのようなpTLB - 1とpBT3 - Nのようなベクトルを用いて生成することができます。ベクトルの選択は、餌に依存し、経験的に決定する必要があります。他のケースでは餌の過剰発現が実際に偽陽性数の増加につながる、有害であるかもしれないが一定の場合に高い餌の式は、相互作用を検出するために必要です。
- 制約は、適切な制限部位(s)で選択されたプラスミドを消化。開裂は、C UB -のLexA - VP16のタグ(上流のC末端のタグ付けまたはN末端 のタグ付けのための下流のタグの)のすぐ近くにのみ発生する必要があります。例えば、pAMBV4ベクトルを使用する場合は、酵素SfiIは理想的な選択肢です。使用直前まで-20℃で消化したプラスミドを保管してください。
- 目的の遺伝子の増幅とクローニングのためにプライマーを設計します。エンド標的遺伝子の最初の18から20ヌクレオチドと一致していなければ5'3しながら、フォワードプライマーの端には、制限部位の上流に約35から40ヌクレオチドと一致する必要があります"が。 5終止コドンを省略する標的遺伝子(の最後の18から20ヌクレオチドの逆相補を対応するend"リバースプライマーの端は3で、制限部位の下流に約35から40ヌクレオチドの逆相補一致していなければなりません" C UB -のLexA - VP場合16タグをC末端に配置されている)。に応じてN -またはC末端のタグ付けが行われているかどうか、標的遺伝子がC UB -のLexA - VP16タグ付きフレームでクローニングされていること。このようなフォワードまたはリバースプライマーの35〜40ヌ クレオチドを選択します。
- 上記のプライマーを用いたPCRにより、目的の遺伝子を増幅する。 PCRのパラメータが使用される特定の酵素と特異的プライマーに依存するであろう。
- LEU2突然変異(例:BY4741)を担持する適切な酵母ラボ株に消化されたプラスミドと一緒にPCR産物を変換する。神話のレポーター株(例えばTHY.AP4またはL40)を使用できますが、必須ではありません、この時点で酵母の目的として、ギャップの修復は、相同組換えが発生する可能性のある環境として機能するように単純にすることができます。変換は以下のように実施する必要があります。
- 滅菌YPAD媒体5mlに、選択した酵母株の単一コロニーを接種し、30℃一晩インキュベート℃で一定に振盪しながら(200rpm)し。
- 〜0.15のOD600に新鮮なYPADメディア50mlに一晩培養を希釈し、30℃でインキュベートCを(200rpm)し振とうしながら。 〜0.6のOD600に達するまで約3-4時間のために成長する。
- 5分間700xgで細胞を遠心し上清を取り除く。
- 5分間700xgで25 mLの滅菌のddH 2 O及び遠心分離で細胞ペレットを再懸濁します。
- 上清を除去し、1mLの滅菌のddH 2 Oで細胞ペレットを再懸濁します。
- マイクロ遠心チューブに細胞を100μL、300μLPEG /リチウムアセテートの混合、および消化したプラスミド(50 fmol)をし、PCR産物を(250〜500 fmol)を追加。
- 30分間30℃でインキュベートする。
- 1時間42℃での熱ショック。
- 5分間3000xgで遠心分離し、上清を取り除く。
- 200μLの滅菌のddH 2 Oとプレート固体、SD -ロイシン選択培地上に、ボリューム全体の細胞ペレットを再懸濁します。 2-4日間、30℃で成長する。
- 30℃で一晩で5 mLのSD -ロイシン液体培地で形質転換株の単一コロニーを生育させます。
- 5分間700xgで細胞を遠心し上清を取り除く。
- いかなる商業的なミニプレップキットを使用して細胞ペレットから餌プラスミドDNAを分離します。少し変わりますが標準プロトコルに従ってください。十分な酵母細胞の溶解を確実にするために、5分間激しく初期再懸濁とボルテックスの後ペレットに0.5mmのソーダライムガラスビーズの小さなボリュームを追加します。その後、通常のような商用プロトコルに進みます。
- コンピテント大腸菌に分離された酵母DNAを変換大腸菌は、少なくとも1 × 10 7細胞/μgのDNAの形質転換効率を持つプラスミド伝播(例えばDH5α、XL10金)に適した菌株。ベイトプラスミドはカナマイシンを使用するために選択できることに注意してください。
- 形質転換したE.から収穫プラスミドDNA標準的なDNAの単離法や市販のキットを用いて大腸菌 。
- シークエンシングによるプラスミド餌の適切な構造を確認してください。
- 適切な神話のレポーター株(例えばTHY.AP4、L40)に検証された餌の構造を変換する。先ほど説明した酵母の形質転換プロトコールは、消化されたプラスミドとPCR産物の代わりに餌プラスミドDNAに置き換えて、使用することができます。
- 餌の検証 - 適切なローカライゼーション
- 使用前に、餌の菌株は、それらが適切に酵母の細胞膜に局在していることを保証するために分析されます。 iMYTHを使用する場合は、この局在は、タグ付けされた餌の特性に特に左右されます。 tMYTHの場合は、餌のプラスミドは一般に原形質膜に発現されたタンパク質を誘導するシグナル配列(例えばMATα)が含まれます。局在を蛍光顕微鏡を用いて決定されます。餌タグ配列のYFP分子の取り込み(すなわちC UB - YFP -のLexA - VP16)が生きている細胞の直接可視化を可能にする、最も単純で最も直接的なアプローチであり、そして一般的にiMYTHで使用されています。また、タグののLexAまたはVP16成分に対する抗体を用いて標準的な免疫蛍光アプローチを使用することができます。
- 餌の検証- N UB G / Iコントロールテスト
- 一度餌の適切なローカライズが確立されて、それは餌が単独または相互作用しない餌(餌は、自己活性化ではないこと、すなわちを確認する)の存在下でレポーター系を活性化しないことを保証するために必要です。これは、餌が相互作用(正)と相互作用しない(陰性)コントロール餌で形質転換され、そして成長は選択培地で評価されるN UB G / Iテストを、使用して行われます。餌は、選択培地で成長する必要があります私n個の陽性対照の存在は、神話での使用に適したものにするために、陰性対照の存在下で成長しません。
- コントロールプラスミドの獲物100〜200 ngでテストされる餌の菌株を形質転換することから始めます。前述の酵母の形質転換プロトコールはYPADの代わりにSD -ロイシンメディアを代入し、最後のめっき工程用にSD -トリプトファン、ロイシンメディアを使用して、使用することができます。次のコントロール餌の構造は一般的に使用されています:
- POST1 - N UB I(ポジティブコントロール)
- POST1 - N UB G(ネガティブコントロール)
- pFUR4 - N UB I(ポジティブコントロール)
- pFUR4 - N UB G(ネガティブコントロール)
- OST1はoligosaccharyl複合体の構成要素であり、小胞体膜[9]にローカライズされていますFUR4はウラシルのパーミアーゼであり、形質膜に局在している間に[10]。これらのタンパク質は一般に非相互作用の餌として使用するために私たちの最初の選択肢ですが、その適合性はケースバイケースで異なります。あなたの餌が純粋にこれらのコントロールの両方と相互作用すると予測されていること万一、代替餌は、選択する必要があります。 N UB私はユビキチンのN末端 の野生型形態である、と自発的にC ubとN UBが融合しているタンパク質間の相互作用のC UB独立と相互作用することを思い出してください。 N UB G捕食が(N ubとC UBの自発的な関連付けを防止するグリシンの突然変異にイソロイシン13を運ぶ)ネガティブコントロールとして機能しながらこのようにN UB私の餌は、ポジティブコントロールを構成する。
- 100μLの滅菌のddH 2 Oにそれぞれ変換された餌の単一コロニーを懸濁します
- シリアル1 / 10、1 / 100、1 / 1000の希釈液を生成するために無菌のddH 2 Oに再懸濁した細胞を希釈する。
- 濃度の範囲での3 - ATがある場合とない場合のスポットSD -トリプトファン、ロイシンおよびSD -トリプトファン-ロイシン-アデニン-ヒスチジンメディアに原液と希釈細胞の5μLボリューム。 3 - AT HIS3レポーター遺伝子の競合阻害剤として作用し、選択プロセスの厳密性を高めるのに役立ちます。それは弱く緩やかに自己活性化ベイトの阻害非特異的成長のためにいくつかのケースで役立ちます。
- スポットを乾燥してから2-4日間、30℃でプレートをインキュベートすることができます。
- すべての形質転換体は、それらが正常にプラスミド獲物で形質転換されたことを示す、SD -トリプトファン、ロイシンプレート上で成長する必要があります。自己活性化は、N UBで形質転換した場合にのみ、SD -トリプトファン-ロイシン-アデニン-ヒスチジンメディアに成長するとしていない餌私が構造を捕食ではなく、N UB G捕食と。 (注)これはスクリーニング中に使用する必要があるとして3 - ATのどの濃度は(もしあれば)メディアで必要とされる。
4。スクリーニング
- 必要なメディアおよびソリューション
- 無菌のddH 2 Oは 121℃で、30分間15 PSIをオートクレーブして調製。
- 0.9%NaCl溶液でのddH 2 Oで調製し121℃オートクレーブ滅菌° C、30分間15 PSI。
- 493 mMのリン酸ナトリウム塩基とのddH 2 O中の250mMリン酸ナトリウム一塩基から構成されるリン酸ナトリウム溶液 121 ° C、30分15 psiでオートクレーブで滅菌。
- X - Galを(5 -ブロモ-4 -クロロ-3 -インドリル-β- D -ガラクトピラノシド)N、N -ジメチルホルムアミドの100 mg / mLの原液として調製した溶液 。
- の2%w / vの酵母エキス、DDH 2 Oで調製した4%(w / v)のペプトン、4%(w / v)のグルコースおよび100μMのアデニンを含む2xYPADの増殖培地 121 ° C、30分15 psiでオートクレーブで滅菌。
- 以前に説明したように調製した合成ドロップアウト(SD)増殖培地、。液体SD -ロイシンと固体SD -トリプトファン、ロイシンを準備します。両方100x15 mmのシャーレに固体培地を注ぐ。 3 - AT、必要に応じて、N UB G / I制御の試験から決定された濃度で含む150ミリメートルラウンドプレート、各画面用16プレート、の固体SD -トリプトファン-ロイシン-アデニン-ヒスチジンメディアを準備します。
- 合成ドロップアウト(SD)メディア+ 5 -ブロモ-4 -クロロ-3 - Indoyl -β- D -ガラクトピラノシド(X - GAL)。前述のように寒天を含むSD -トリプトファン-ロイシン-アデニン-ヒスチジンメディアを準備します。オートクレーブした後、10分の1の体積Oが続く(必要に応じて)3 - ATを追加、冷却することができますF滅菌リン酸ナトリウム溶液。次に、80μg/ mLの最終濃度にX - gal溶液を追加してください。完全に混和および150mm円形プレートに注ぐ。
- PEG /酢酸リチウム溶液IIは、40%PEG - 3350、100 mMの酢酸リチウム、1mMのEDTAと10mMのトリスpH7.5を含む。無菌のddH 2 Oとソリューション(例えば、オートクレーブを50%PEG - 3350、1 M酢酸リチウム、100mMのトリスpH7.5、500 mMのEDTA pH 8.0)を用いてこの溶液を調製します。
- 酢酸リチウム/ 110 mMの酢酸リチウム、11 mMのトリスpH7.5お よび1.1 mM EDTAを含むトリスEDTA溶液 。無菌のddH 2 Oとソリューション(例えばオートクレーブ1M酢酸リチウム、100mMのトリスpH7.5、500 mMのEDTA pH 8.0)を使用して、この溶液を調製します。
- 100mMトリスpH7.5お よび10mMのEDTAから成る10倍トリスEDTA溶液ではのddH 2 Oで調製した121 ° C、30分15 psiでオートクレーブで滅菌。
- 無菌のddH 2 Oで調製した2 mg / mLのサケ精子DNAタイプIIIのナトリウム塩を含む一本鎖キャリアーDNA(ssDNAの)ソリューション
- 商業ミニプレップキット。
- ソーダライムガラスビーズ(0.5mm)の。
- プラスミド伝播 (例えばDH5α、XL10金)と細菌の増殖やプラスミドの選択に適した標準的なメディアに適した有能な大腸菌の細胞 。
- などのプロトコルで説明されている特定の酵母菌株とプラスミド
- 大規模なスケール変換
- SD -ロイシンのメディアを5 mLにあなたの餌を含む神話レポーター株のシングルコロニーを植菌し、30℃一晩インキュベート° C(200 rpm)で振盪しながら。
- 30℃でOD600 = 0.15とインキュベートする200mLのSD -ロイシンのメディアに一晩培養を希釈°(200rpm)し振とうしながらC。 0.7(約4-5時間) - OD600 = 0.6まで成長する。
- ターゲットのOD600に到達する直前に、ssDNAの溶液のアリコートを解凍。 100℃沸騰° C氷上で冷却して5分のため。一回繰り返します。
- ターゲットOD600に到達したとき、(4x50 mlのスクリューキャップ付き遠心管の間に200 mLの培養液を分周)、5分間700xgで遠心分離を介して細胞を採取する。
- 30 mLの滅菌のddH 2 Oと簡単にボルテックスサンプルでそれぞれのペレットを洗浄します。 5分間700xgで遠心分離します。
- 上清を捨て、1 mLの酢酸リチウム/トリスEDTA溶液で各ペレットを再懸濁します。 5分間700xgで滅菌済み1.5mlマイクロチューブや遠沈管に移す。
- 上清を捨て、酢酸リチウム/トリスEDTA溶液600μLで各ペレットを再懸濁します。
- 4x15 mlのスクリューキャップ付き遠心管に以下を追加します。
- 2.5mLのPEG /酢酸リチウム溶液II
- 600μL懸濁している細胞
- 100μLのssDNAのソリューション
- 獲物ライブラリーDNAの7μgの
- N UB GとN -またはC -末端のいずれかでタグ付けされた、とのcDNAまたはゲノム、さまざまなソースから調製した餌を含むライブラリは、(市販されているwww.dualsystems.com )。使用される特定のライブラリが選択された餌や実験目的に依存し、ケースバイケースで決定する必要があります。
- ボルテックスで1分間チューブを45分間30℃の水浴中でインキュベートして完全に混合して確保する。 15分ごとに簡単に混ぜる。
- 各チューブに160μLのジメチルスルホキシド(DMSO)を追加し、チューブを反転により、すぐに混ぜる。
- 20分間42℃の水浴中でインキュベートする。
- 5分間700xgで遠心分離によって形質転換体を収集する。
- 上清を捨てる。 3 mLの2xYPADの各ペレットの再懸濁によって形質転換体を回復する。シングル50mLのスクリューキャップ付き遠心管に一緒にプールのすべてのサンプルを。
- 細胞の回復のための90分間30℃でインキュベートする。
- 5分間700xgで遠心分離し、上清を捨てる。
- 4.9 mLの滅菌0.9%NaCl中で細胞ペレットを再懸濁します。
- 再懸濁した細胞を100μLを使用すると、10倍から1000倍まで無菌の0.9%NaClで10倍段階希釈液を準備。
- 30でSD -トリプトファン-ロイシンメディアインキュベート上に100倍と1000倍希釈のプレート100μL° 2〜3日のためのC。これらのプレートは、コントロールとして使用し、形質転換の効率を計算するために使用されています。
- 同様に、大(150mm)をSD -トリプトファン-ロイシン-アデニン-ヒスチジンプレート上に懸濁している細胞とプレートの残りの4.8 mLを分けるとしてN UB G / I試験で決定した、と3〜4日間、30℃でインキュベート3 - ATの必要な量を含む。
- 上に0.9%のNaClとプレート5μLアリコートを100μLに、単一のコロニーを(それぞれ表す細胞が潜在的な相互作用餌 - 捕食のペアを含む)再懸濁し、SD -トリプトファン-ロイシン-アデニン-ヒスチジン+ X - Galをメディア(および場合3 - ATを含む必要)。 2〜4日間に成長することができます。このステップでは、選択的スクリーニングの第二ラウンドとなっており、最初のラウンドで得られた偽陽性の除去に役立ちます。力強い成長と青の色を表示するだけコロニーはさらなる分析のために選択されています。
- 獲物のDNAの単離とシーケンシング
- 前述の変更でミニプレップのプロトコルを使用して青色の酵母のコロニーからプラスミドDNAを単離する。獲物の保持ではなく、餌を選択するには、SD -トリプトファンのメディアのみで細胞を育つようにしてください、プラスミド。ヒットの非常に大きな数を生成する画面の場合は、市販の高スループットのミニプレップキットには、この時点では有利かもしれない。
- コンピテント大腸菌に分離された酵母プラスミドDNAを変換大腸菌は、少なくとも1 × 10 7細胞/μgのDNAの形質転換効率を持つプラスミド伝播(例えばDH5α、XL10金)に適した菌株。獲物プラスミドは、アンピシリンを使用するために選択することができることに注意してください。
- 形質転換したE.からプラスミドDNAを単離標準的なDNAの単離法や市販のキットを用いて大腸菌 。サンプル数が多い場合、再度、高スループットのミニプレップキットは役に立つかもしれません。 E.におけるDNAの増幅大腸菌は非常にプラスミドの収量を増加させ、DNAの十分な量の塩基配列決定とさらなる分析の両方に存在することを確認。
- N UB G.内で配列に相補的なプライマーを用いて単離されたプラスミドシーケンス
- インターアクターのあなたの予備的なリストを組み立てるために、すべての配列データをコンパイルし、分析する。これは、手動で、または適切なソフトウェアを使用して自動化された方法で行われることがあります。
- 餌の依存関係のテスト
- インターアクターの予備的なリストの組み立て後に、それは相互作用を再確認し、相互作用/餌のアイデンティティに依存しない方法でレポーターシステムをアクティブに無差別捕食を排除することが重要です。これは、ベイトの依存関係のテストを使用して行われます。このテストでは、特定されたインターアクターのすべてがオリジナルの餌のひずみだけでなく、C UB -のLexA - VP16タグに融合した、単一の膜貫通ドメインで構成されるコントロールの人工餌を抱いて株に戻って変換されます。トランスフォーメーションは、標準プロトコルに従って行われ、最終めっき工程のためにYPADとSD -トリプトファン、ロイシン固形培地の代わりにSD -ロイシンメディアを使用して、以前に説明した。
- SD -トリプトファン-ロイシン-アデニン-ヒスチジン+ X - Galをメディア(および必要に応じて3 - ATを含む)の上に無菌のddH 2 Oとスポット5μLボリュームの100μLで上記の変換から単一のコロニーを懸濁します。その後、プレートを30℃2〜4日℃でインキュベートする。理想的には複数の形質転換体は、それぞれの獲物に合わせて選択してください、そしてオリジナルの餌と人工餌の両方を同じプレート上にスポットしてください。
- レポーターシステム(すなわち成長と青色)の原因の活性化が無差別と見なされ、その特定の獲物は、インターアクターのリストから削除されていることを人工的な餌と獲物を運ぶ酵母。
- 餌オブ関心を持って酵母の成長と青色の着色の原因となる餌ではなく、人工的な餌は、この特定の相互作用を確認する。しかし、酵母は獲物を保有して餌の利子が成長していない場合、この獲物は、インターアクターのリストから削除されます。
- 残り餌は、神話のスクリーニングで同定されたインターアクターの完全なリストを構成する。
5。さらなる研究
神話のスクリーニングが完了したら、さらなる分析が検出された相互作用の生物学的意義を検証し、決定するために実行する必要があります。実行する特定の研究では、ケースバイケースでそれぞれ異なり、個々の研究者によって決定される必要があります。フォローアップ作業の一般的な例としては、ネイティブの生物における共免疫沈降実験と削除の研究が含まれています。さらに、得られたデータのコンピューター解析では、パターンを検出し、さまざまな相互作用が果たす可能性がある潜在的な関連性と役割を識別するのに役立ちますことができます。このように、神話の技術は、膜タンパク質の重要な機能的相互作用の識別と理解に向けて強力な第一段階"としての役割を果たします。詳細なフォローアップ試験および他の最近の先進国および新興TEと結合chnologies、それは細胞の謎の鍵を開けるための貴重なツールとなるでしょう。

図1。スプリットユビキチン。aの原則 C末端側半分(C UB)とN -末端半分(N UB I):ユビキチンは、2つの部分に分けることができる。一方、別の用のため、その高親和性の自発的に再構成するこれらの部分。 B.イソロイシンからグリシンへの位置13におけるUB私の点突然変異(N UB G)は、この自発的な再アソシエーションを防ぎます。 C.神話のシステムでは、C UBは、餌の利子(B)に融合され、獲物はN UB G(A)に融合させる。 ABのタンパク質相互作用は、擬似ユビキチン再構成します。
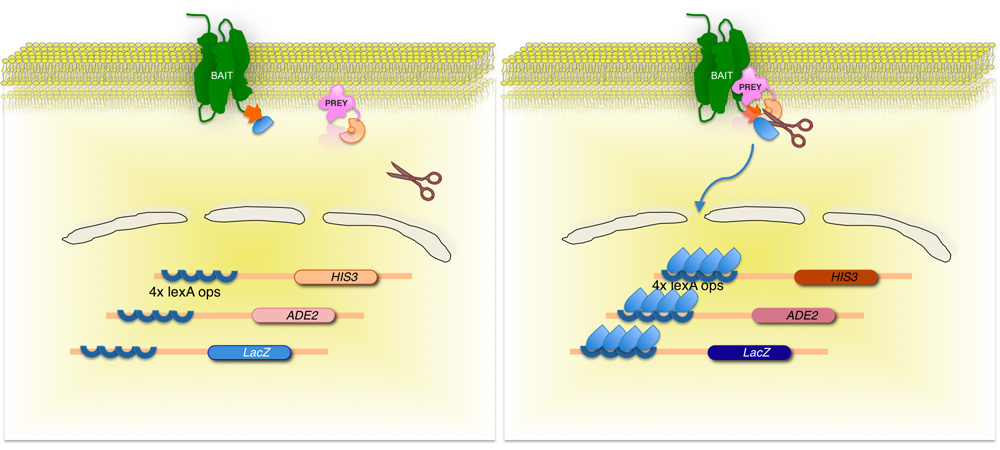
図2。スプリットユビキチン系膜の酵母ツーハイブリッド(神話)システム。
膜タンパク質の利子(餌)は、ユビキチン酵母のC末端側半分(C UB)に転写因子に結合した融合している。 cDNAまたはゲノムDNAライブラリーを使用して、ライブラリ(獲物)でエンコードされた各タンパク質は、ユビキチン部分の対応するN末端 (N ユビキチン G)に融合させる。 2つのタンパク質が相互作用していない場合、転写因子は、膜のインタフェース(左パネル)のままです。しかし、タンパク質が相互作用する場合、2つのユビキチン部分は、ユビキチン特異的プロテアーゼによる切断の結果、参加する。開裂は、レポーター遺伝子の発現(右パネル)で、その結果、転写因子を解放

図3。神話のパイプライン。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
神話は、完全長の膜タンパク質と細胞質または膜結合パートナー間の相互作用の同定を可能にする最初のハイスループットシステムです。それは生物[3-7]の範囲から膜タンパク質を研究するために使用されています。タンパク質の-利益を確保するために精査する必要があるかもしれない具体的な詳細は、しかし、存在する神話と一緒に勉強するために適している。
多くの膜結合タンパク質は、続いて成熟タンパク質を生成するために切断されるシグナル配列を介して細胞膜に送られます。このシーケンスは、生物固有であり、シグナル配列が認識されないので、それが酵母内で発現する場合は、このネイティブのシグナル配列はタンパク質の利子の誤局在を引き起こす可能性があります。この問題を回避するために、我々は、接合因子α(MATα)から派生した、酵母のシグナル配列に融合させるこれらの特定のタンパク質を設計した。このペプチド配列(MFα- SSいわゆる)酵母の形質膜へのタンパク質の再局在化と重要なことは、酵母のシグナルペプチダーゼによって切断される。このペプチド配列は、プラスミドpTMBV -MFαとpAMBV -MFαに含まれています。
強調する必要があるもう一つの重要なパラメータは、餌の発現レベルです。餌の発現を駆動するプロモーターは、このパラメータを調節する。それはNubG / NubIテストと餌が(すなわち、それが無差別に多くの非特異的な獲物のタンパク質と相互作用する)、"自己活性化"である過剰発現のアーティファクトを、排除するために必要な3 - ATの量を使用して餌の発現レベルを最適化するために必要となる場合があります。酵母のタンパク質を調べると、iMYTHが適用されているほとんどの生理学的に関連する餌の濃度を生成します。この場合、目的遺伝子は、ゲノムDNA内にカブ- TFタグが付けられています。また、外因性の蛋白質は低い餌の式になりますCYC1プロモーターを運ぶpBT3 - STEとpCMBVプラスミドから発現することができます。プラスミドpTMBVとpTLB1港TEF1プロモーターpAMBVはADH1プロモーターを持っている間、両方のbaitタンパク質のそのドライブの強い表現。 baitタンパク質のレベルはさらに最適化が必要な場合は、それがTEF1プロモーターを運ぶことpTLB - 1プラスミドを使用する必要があるかもしれません、しかし、のLexA DNA結合ドメインは、最終的には減少し、外因性レポーター遺伝子のプロモーターの方に親和性を和らげるためにR156Gで変異している自己活性化の確率[5]。
神話の成功に重要な役割を果たしているもう一つの要因は、スクリーニングのプロセスで使用される最適なライブラリです。これは、内因性の餌の発現プロファイルに依存します。たとえば、餌は特定の組織で発現し、したがって、この特定の組織から構成されているライブラリを使用することが重要です可能性があります。これは生理学的に関連する相互作用が検出されるようになります。
神話のシステムは、研究が困難であったタンパク質のクラスに関する情報を豊富に提供するシンプルかつ迅速なツールです。これらの同定された相互作用は、膜タンパク質の完全な生物学的機能の解明に助けることができる。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
イゴールStagljarはDualSystemsバイオテック、スイスの共同創立者です。
Acknowledgments
我々は、この原稿の重要な読書のために夜明けのエドモンズに感謝します。 Stagljarラボは、イノベーションのためのカナダの財団(CFI)、健康研究(CIHR)、心臓や脳卒中財団、カナダの癌協会、およびノバルティスのためのカナダの研究所からの資金によってサポートされています。
Materials
| Name | Company | Catalog Number | Comments |
| Polyethenlene Glycol (PEG3350) | BioShop Canada | PEG335 | |
| Lithium Acetate Bihydrate | BioShop Canada | LIA001 | |
| X-Gal (5-Bromo-4-Chloro-3-Indolyl-b-D-galactopyranoside) | BioShop Canada | XGA001 | |
| N`,N-dimethyl formamide | BioShop Canada | DMF 451 | |
| 3-amino-1,2,4-triazole (3-AT) | BioShop Canada | ATT124 | |
| Sodium phosphate dibasic | BioShop Canada | SPD307 | |
| Sodium phosphate monobasic | Fisher Scientific | BP329-500 | |
| Salmon Sperm DNA | VWR international | CA80601-120 | |
| D-Glucose | BioShop Canada | GLU501 | |
| LB Broth LENOX | BioShop Canada | LBL405 | |
| Yeast Nitrogen Base | BioShop Canada | YNB406 | |
| Yeast Extract | BioShop Canada | YEX401 | |
| Peptone | BD Biosciences | 211677 | |
| Bio-Tryptone | BioShop Canada | TRP402 | |
| Adenine Sulphate | BioShop Canada | ADS201 | |
| L-Uracil | BioShop Canada | URA241 | |
| L-Threonine | BioShop Canada | THR002 | |
| L-Histidine | BioShop Canada | HIS200 | |
| L-Methionine | BioShop Canada | MET222 | |
| L-Valine | BioShop Canada | VAL201 | |
| L-Phenylalanine | BioShop Canada | PHA302 | |
| L-Isoleucine | BioShop Canada | ISO910 | |
| L-Tyrosine | BioShop Canada | TYR333 | |
| L-Leucine | BioShop Canada | LEU222 | |
| L-Arginine | BioShop Canada | ARG006 | |
| L-Tryptophane | Fisher Scientific | BP395-100 | |
| L-Lysine | BioShop Canada | LYS101 | |
| L-Alanine | Fisher Scientific | BP369-100 | |
| Agar | BioShop Canada | AGR001 | |
| Soda Lime Galss Beads | Biospec Products | 11079105 | |
| Sodium Chloride | BioShop Canada | SLD002 |
References
- Stagljar, I., Fields, S. Analysis of membrane protein interactions using yeast-based technologies. Trends Biochem Sci. 27 (11), 559-563 (2002).
- Iyer, K. Utilizing the split-ubiquitin membrane yeast two-hybrid system to identify protein-protein interactions of integral membrane proteins. Sci STKE. 275, pl3-pl3 (2005).
- Paumi, C. M. Mapping protein-protein interactions for the yeast ABC transporter Ycf1p by integrated split-ubiquitin membrane yeast two-hybrid analysis. Mol Cell. 26 (1), 15-25 (2007).
- Stagljar, I. A genetic system based on split-ubiquitin for the analysis of interactions between membrane proteins in vivo. Proc Natl Acad Sci U S A. 95 (9), 5187-5192 (1998).
- Gisler, S. M. Monitoring protein-protein interactions between the mammalian integral membrane transporters and PDZ-interacting partners using a modified split-ubiquitin membrane yeast two-hybrid system. Mol Cell Proteomics. 7 (7), 1362-1377 (2008).
- Scheper, W. Coordination of N-glycosylation and protein translocation across the endoplasmic reticulum membrane by Sss1 protein. J Biol Chem. 278 (39), 37998-38003 (2003).
- Thaminy, S. Identification of novel ErbB3-interacting factors using the split-ubiquitin membrane yeast two-hybrid system. Genome Res. 13 (7), 1744-1753 (2003).
- Johnsson, N., Varshavsky, A. Split ubiquitin as a sensor of protein interactions in vivo. Proc Natl Acad Sci U S A. 91 (22), 10340-10344 (1994).
- Kelleher, D. J., Gilmore, R. The Saccharomyces cerevisiae oligosaccharyltransferase is a protein complex composed of Wbp1p, Swp1p, and four additional polypeptides. J Biol Chem. 269 (17), 12908-12917 (1994).
- Chevallier, M. R. Cloning and transcriptional control of a eucaryotic permease gene. Mol Cell Biol. 2 (8), 977-984 (1982).
