

Summary
MYTHE permet la détection sensible des interactions transitoires et stables entre les protéines qui sont exprimées dans l'organisme modèle Saccharomyces cerevisiae. Elle a été appliquée avec succès pour étudier exogènes et de la levure des protéines membranaires intégrales en vue d'identifier leurs partenaires d'interaction de façon à haut débit.
Abstract
L'importance biologique fondamentale et clinique de protéines membranaires intégrales conduit au développement d'un système à base de levure pour l'identification à haut débit des interactions protéine-protéine (PPI) pour les protéines transmembranaires pleine longueur. À cette fin, notre laboratoire a développé le split-ubiquitine levure membrane à base de double-hybride (MYTH) du système. Cette technologie permet la détection sensible des interactions protéiques transitoires et stables à l'aide
Protocol
1. Renseignements généraux
Interactions protéine-protéine (IPP) sont les éléments constitutifs fondamentaux impliqués dans régissant tous les processus cellulaires. Par conséquent, il est essentiel que toutes les interactions sont strictement réglementées afin de maintenir l'homéostasie cellulaire, comme un changement dans cet équilibre biologique joue souvent un rôle dans la maladie et la transformation des cellules cancéreuses. Membrane protéines associées sont parmi la classe la plus importante de protéines biologiquement comme ils peuvent initier des cascades de signalisation complexes, et la médiation à la fois l'importation et l'exportation de diverses molécules, y compris les médicaments, qui a été d'une importance récents dans le domaine des soins de santé comme des problèmes liés à résistance aux médicaments sont devenus plus fréquents. Gagner un aperçu de la complexité de cette classe de protéines nécessite l'identification de leurs partenaires d'interaction. Découverte des partenaires tels s'est révélé difficile, comme souvent, il exige des conditions rigoureuses qui doivent être optimisés pour chaque protéine membranaire [1].
L'importance biologique fondamentale et clinique de protéines membranaires intégrales conduit au développement d'un système à base de levure pour l'identification à haut débit des IPP pour les protéines transmembranaires pleine longueur. À cette fin, nous avons développé la levure split-ubiquitine membrane à base de double-hybride (MYTH) du système [2-4]. Cet outil permet la détection sensible des interactions protéiques transitoires et stables. Elle a été appliquée avec succès pour étudier les protéines exogènes et endogènes exprimées dans le modèle d'organisme de Saccharomyces cerevisiae [3-7]. MYTHE tire parti de l'observation que l'ubiquitine peut être séparée en deux moitiés:. La moitié C-terminale (C UB) et la moitié N-terminale (Nubi) dans des études in vivo ont montré que ces fragments de reconstituer spontanément en raison de leur forte affinité pour un autre. (Figure. 1a) Toutefois, l'introduction d'une isoleucine 13 à mutation ponctuelle de glycine dans la moitié N-terminale de l'ubiquitine (produisant un fragment appelé N ous G) empêche cette ré-association spontanée [8] (1b Figure.).
Nous utilisons ce principe dans le système de mythe (Figure. 1c et 2). En bref, la protéine membranaire intégrale appât est fusionnée à un fragment de C ub qui est lié à un facteur de transcription artificiels constitués de l'Escherichia coli DNA-binding protéine LexA et le domaine d'activation VP16 du virus herpès simplex. Les proies sont générés par la fusion des fragments d'ADN génomique provenant d'ADNc ou à la fraction de NubG. Une interaction entre les protéines appâts et proies dans un hôte levure conduit à la reconstitution de «pseudo-ubiquitine 'une molécule entière, avec la reconnaissance ultérieure par des enzymes cytosoliques désubiquitination (DUB) et la libération protéolytique du facteur de transcription. Le facteur de transcription peut alors pénétrer dans le noyau de la cellule, et d'activer un système de gène rapporteur (impliquant généralement l'expression de la HIS3, lacZ et gènes ADE2) permettant la croissance des souches de levures sur des milieux sélectifs, ce qui est indicatif de l'appât et l'interaction des proies [ 2-4].
L'identification et la caractérisation des interactions protéine membranaire intégrale fournira des informations pour aider à comprendre pleinement leur fonction. Comme nous, plus précisément à comprendre et à disséquer les rôles des protéines qui interagissent avec les protéines membranaires intégrales, nous pouvons mieux comprendre l'interaction dynamique impliqués dans la régulation de ces protéines et de découvrir de nouvelles cibles qui pourraient avoir un potentiel thérapeutique.
2. Sélection d'appâts et de système approprié MYTHE
- Avant de procéder à l'analyse MYTHE, vérifiez que votre protéine a son extrémité N-et / ou C-terminale dans le cytosol de la cellule. Il est essentiel que le C-ub LexA-VP16 tag être fusionné à votre protéine à un tel terminal, car les enzymes nécessaires pour la libération désubiquitination facteur de transcription sont situés dans le cytosol [4].
- Ensuite, décider laquelle des deux variantes principales du mythe est approprié. Pour les protéines de la levure non-indigènes, mythe traditionnel (tMYTH) peut être utilisé, où les appâts sont surexprimés ectopique d'un plasmide. Pour les protéines de la levure indigène, MYTHE intégré (iMYTH) est la méthode de choix. En iMYTH appâts sont marqués de façon endogène avec le C-ub LexA-VP16 tag, les laissant sous le contrôle de leur promoteur natif. Ceci est avantageux, comme le niveau d'expression de type sauvage d'appâts contribue à éliminer les problèmes associés à la surexpression de la protéine, comme une augmentation du nombre de faux positifs [3]. À l'exception de la construction d'appât initial et les milieux sélectifs utilisés, les deux formes du mythe sont réalisées d'une manière essentiellement identique. Pour plus de clarté, nous allons nous concentrer principalement sur l'utilisation de tMYTH dans ce rapport, que cette variante peut, en principe, être utilisés avec des protéines membranaires à partir de pratiquement n'importe quel organisme et estdonc plus largement applicables.
3. Génération d'appâts et de validation
- Média nécessaire et les solutions
- Stérile ddH 2 O préparé par autoclave à 121 ° C, 15 psi pendant 30 minutes.
- 3-amino-1 ,2,4-triazole (3-AT) solution préparée comme une solution stock 1M dans le trou DDH 2 O. Stériliser par passage à travers un filtre de 0,2 um.
- Médias croissance YPAD composé d'extrait de levure 1% p / v, 2% p / v peptone, 2% p / v de glucose et 100 uM adénine préparés dans le trou DDH 2 O. Stériliser à l'autoclave à 121 ° C, 15 psi pendant 30 minutes.
- 10x acide aminé / Mélanger base nucléotidique: Le mélange complet contient 1,0 mM Adénine, Uracile 1,8 mM, 1,0 mM d'arginine, 1,0 mM histidine, 2,3 mM isoleucine, la leucine 7,6 mm, 1,6 mm Lysine, Méthionine 10,1 mm, 3,0 mm phénylalanine, 16,8 mm thréonine, tryptophane 2,0 mM, 1,7 mM tyrosine et valine 12,8 mm, préparé dans le trou DDH 2 O. Pour abandon omettre les acides aminés nécessaires (s) et / ou de bases nucléotidiques (s). Stériliser à l'autoclave à 121 ° C, 15 psi pendant 30 minutes.
- Synthétique Drop-Out (SD) Médias croissance composé de 0,67% p / v base azotée de levure (sans acides aminés, mais avec du sulfate d'ammonium), 2% p / v de glucose, 2% p / v agar et 1x acide aminé / nucléotides Mix, préparés dans le trou DDH 2 O. Préparez les deux liquides et solides (contenant de l'agar 2%) SD-leucine médias. Préparez aussi solide SD-tryptophane-Leucine et SD-tryptophane-Leucine-adénine-Histidine médias. Stériliser à l'autoclave à 121 ° C, 15 psi pendant 30 minutes. Verser les médias solides dans des boîtes de Pétri 100x15 mm.
- Synthétique Drop-Out (SD) Croissance supports contenant 3-AT. Préparez SD-tryptophane-Leucine-adénine-Histidine média tel que décrit, mais contenant du 3-AT à des concentrations de 25, 50, 75 et 100 mm. Ajouter la quantité appropriée de 3-AT de la solution stock de stériles 1M pour les médias après qu'il a été autoclavé et refroidi (mais n'a pas encore solidifié). Verser dans des boîtes de Pétri 100x15 mm.
- Mélanger Acétate PEG / Lithium composé de 40% p / v PEG-3350, 120 mM d'acétate de lithium et de 167 mg / ml d'ADN de sperme de saumon (Sel de sodium de type III), préparé dans le trou DDH 2 O. Afin de garantir la stérilité de ce mélange le préparer à partir de l'eau stérile et des solutions (c'est à dire autoclavé 50% de PEG-3350, autoclavés acétate de lithium 1M et 2 mg / ml de sperme de saumon ADN de type III de sel de sodium préparés stériles ddH 2 O).
- Enzymes et réactifs pour mener PCR.
- Commercial miniprep kit.
- Billes en verre sodocalcique (0,5 mm).
- Compétente cellules d'Escherichia coli adapté à la propagation de plasmide (par exemple, DH5a, XL10 or) et les médias standard adapté à la propagation des bactéries et la sélection du plasmide.
- Souches de levures spécifiques, des plasmides et des amorces telles que décrites dans le protocole.
- Génération d'appâts tMYTH par la réparation de Gap
- L'appât doit être cloné dans un vecteur approprié pour le marquage et l'expression. Une variété de vecteurs tMYTH sont actuellement disponibles pour une utilisation dans la construction d'appât. Vecteurs tels que le pCMBV4, pAMBV4 et pTMBV4 permettre la construction de C-terminale des appâts marqués (BAIT-C UB-LexA-VP16) sous le contrôle de la CYC1 (faible), ADH1 (fort) et TEF1 (très fort) les promoteurs, respectivement . Appâts N-terminale marqués (LexA-VP16-C ub-Bait) peuvent être générés en utilisant des vecteurs tels que pTLB-1 et pBT3-N, sous le contrôle de l'TEF1 et CYC1 promoteurs, respectivement. Le choix dépend de vecteur de l'appât et doivent être déterminées empiriquement. Dans certains cas, l'expression élevée appât est nécessaire afin de détecter les interactions, alors que dans d'autres cas, la surexpression d'appât peut en fait être préjudiciable, conduisant à une augmentation du nombre de faux positifs.
- Restriction digérer le plasmide sélectionné sur le site de restriction approprié (s). Décolleté devrait se produire que dans le voisinage immédiat de la C-ub LexA-VP16 tag (en amont de la balise pour le C-terminal de marquage ou en aval pour les N-terminale de marquage). Par exemple, lorsque vous utilisez le vecteur pAMBV4, Sfil est un choix idéal. Stocker le plasmide digéré à -20 ° C jusqu'à utilisation.
- Conception amorces pour l'amplification et le clonage du gène d'intérêt. L'extrémité 5 'de votre Primer doit correspondre à environ 35-40 nucléotides en amont du site de restriction, tandis que l'extrémité 3' doit correspondre à l'18-20 premiers nucléotides du gène cible. L'extrémité 5 'de l'amorce reverse doivent correspondre le complément inverse de environ 35-40 nucléotides en aval du site de restriction, avec l'extrémité 3' correspondant le complément inverse de l'18-20 derniers nucléotides du gène cible (en omettant le codon stop si le C-ub LexA-VP16 tag est placé à la partie C-terminale). Selon que la N-ou C-terminale marquage est effectué, sélectionnez le 35-40 nucléotides de l'amorce avant ou arrière de telle sorte que le gène cible est cloné en phase avec le C-ub LexA-VP16 tag.
- Amplifier le gène d'intérêt par PCR en utilisant des amorces ci-dessus. Paramètres de la PCR dépend de l'enzyme particulière et des amorces spécifiques utilisées.
- Transformer le produit de PCR avec le plasmide digéré dans une souche de laboratoire de la levure appropriée portant une mutation leu2 (par exemple BY4741). Une souche journaliste MYTHE (ex THY.AP4 ou L40) peut être utilisé mais il n'est pas nécessaire, car le but de la levure à ce point est tout simplement de servir comme un environnement dans lequel le gap repair recombinaison homologue peut se produire. Transformation doit être effectuée comme suit:
- Inoculer une seule colonie de votre souche de levure sélectionnée dans 5 ml de milieu stérile et YPAD incuber une nuit à 30 ° C avec une agitation constante (200 rpm).
- Diluer la culture durant la nuit dans 50 ml de médias YPAD frais à une DO600 de ~ 0,15 et incuber à 30 ° C avec agitation (200 rpm). Croître pour environ 3-4 heures jusqu'à ce qu'une DO600 de ~ 0,6 est atteint.
- Centrifuger les cellules à 700xg pendant 5 minutes et retirer le surnageant.
- Resuspendre la cellule dans 25 ml stériles O ddH pastille 2 et centrifuger à 700xg pendant 5 minutes.
- Enlever le surnageant et remettre en suspension les cellules dans 1 ml stériles ddH O. 2 pastilles
- Ajouter 100 ul de cellules, mélangez 300 uL d'acétate PEG / lithium, et le plasmide digéré (50 fmol) et un produit de PCR (250-500 fmol) à un tube de centrifugeuse.
- Incuber à 30 ° C pendant 30 minutes.
- Choc thermique à 42 ° C pendant 1 heure.
- Centrifuger à 3000xg pendant 5 min et retirer le surnageant.
- Resuspendre la cellule dans 200 pi 2 O stérile ddH granulés et la plaque sur la totalité du volume solide, SD-leucine milieux sélectifs. Croître à 30 ° C pendant 2-4 jours.
- Grow up une seule colonie de la souche transformée dans 5 mL SD-leucine milieux liquides à 30 ° C pendant la nuit.
- Centrifuger les cellules à 700xg pendant 5 minutes et retirer le surnageant.
- Isoler l'ADN plasmidique à partir d'appâts les culots cellulaires en utilisant n'importe quel kit commercial miniprep. Suivez le protocole standard avec une modification. Afin d'assurer suffisamment de lyse cellulaire de la levure, ajouter un petit volume de 0,5 mm de verre de soda perles de la chaux pour le culot, après remise en suspension initiale et vortexer vigoureusement pendant 5 minutes. Ensuite, passez avec le protocole commercial normal.
- Transformez ADN de levure isolée dans une compétents E. coli souche appropriée pour la propagation de plasmide (par exemple, DH5a, XL10 or) avec une efficacité de transformation d'au moins 1x10 7 cellules / ug d'ADN. Notez que les plasmides appâts peuvent être sélectionnées pour l'aide à la kanamycine.
- L'ADN plasmidique de la récolte transformée E. coli en utilisant une méthode d'ADN standard d'isolation ou un kit commercial.
- Vérifiez construction proprement dite de l'appât plasmide par séquençage.
- Transformez l'appât vérifié de construire dans une souche journaliste MYTHE appropriés (p. ex THY.AP4, L40). Le protocole de transformation de la levure nous venons de décrire peut être utilisé, en remplaçant l'ADN plasmidique appâts en place du produit plasmide digéré et le PCR.
- Validation Bait - bonne localisation
- Avant l'utilisation, des souches d'appât sont analysés afin de s'assurer qu'ils sont correctement localisés sur la membrane de la levure. Lorsque vous utilisez iMYTH, cette localisation dépendra spécifiquement sur les propriétés de l'appât marqués. Pour tMYTH, plasmides appâts comprennent généralement une séquence signal (par exemple, Mata) diriger la protéine exprimée à la membrane plasmique. La localisation est déterminée en utilisant la microscopie à fluorescence. L'inclusion d'une molécule YFP dans la séquence tag appâts (c. C-ub-YFP LexA-VP16) est l'approche la plus simple et directe, permettant une visualisation directe des cellules vivantes, et est couramment utilisé dans iMYTH. Alternativement, une approche standard d'immunofluorescence utilisant des anticorps contre le LexA ou VP16 composantes de la balise peut être utilisée.
- Validation Bait - N ous G / I Contrôle de test
- Une fois la localisation correcte de l'appât a été établie, il est nécessaire de s'assurer que les appâts ne pas activer le système rapporteur seul ou en présence de non-interaction proies (c.-à vérifier que les appâts ne sont pas auto-activation). Ceci est accompli en utilisant la N ous g / essai I, où l'appât est transformé par l'interaction (positif) et sans interaction proies de contrôle (négatif), et la croissance est évaluée sur des milieux sélectifs. Un appât doit croître sur des milieux sélectifs in la présence du contrôle positif, et ne poussent pas dans la présence du contrôle négatif, afin d'être adapté pour une utilisation dans le mythe.
- Commencez en transformant la souche appât pour être testés dans 100-200 ng de proie plasmide contrôle. Le protocole décrit précédemment la transformation de la levure peut être utilisée, en substituant SD-leucine des médias à la place de YPAD et en utilisant SD-tryptophane-Leucine des médias pour l'étape de placage finale. Les constructions suivantes proies de contrôle sont couramment utilisés:
- POST1-N ous I (contrôle positif)
- POST1-N ous G (contrôle négatif)
- pFUR4-N ous I (contrôle positif)
- pFUR4-N ous G (contrôle négatif)
- OST1 est un composant du complexe oligosaccharyl et est localisée à la membrane du réticulum endoplasmique [9] tandis FUR4 est une perméase uracile et est localisée à la membrane plasmique [10]. Bien que ces protéines sont généralement notre premier choix pour une utilisation non-interaction proies, leur aptitude à varier au cas par cas. Dans le cas improbable où votre appât est prévu pour véritablement interagir avec deux de ces contrôles, les proies alternatives devront être sélectionnés. Rappelons que N ous I est la forme sauvage de l'extrémité N-terminale de l'ubiquitine, et interagit spontanément avec C ub indépendant d'une interaction entre les protéines à laquelle l'UB C et N ous sont fusionnés. Ainsi N ous proies I constituent des contrôles positifs, alors que le N ous G proies (portant le Isoleucine 13 à la mutation Glycine qui empêche l'association spontanée de N et C ub ub) servent de témoins négatifs.
- Resuspendre colonies uniques de chaque appât transformé en 100 uL stériles O. ddH 2
- Série diluer les cellules remises en suspension dans stériles ddH 2 O pour produire des dilutions de 1 / 10, 1 / 100 et 1 / 1000.
- Spot 5 volumes ul de cellules non dilué et dilué sur SD-tryptophane-Leucine et SD-tryptophane-Leucine-adénine-Histidine médias avec et sans 3-AT à une gamme de concentrations. 3-AT agit comme un inhibiteur compétitif du gène HIS3 journaliste, et sert à augmenter la rigueur du processus de sélection. Il peut être utile dans certains cas, pour inhiber la croissance non-spécifique de faiblement à modérément auto-activation des appâts.
- Laisser les gouttes de sécher et ensuite incuber les plaques à 30 ° C pendant 2-4 jours.
- Tous les transformants devrait croître sur les plaques SD-tryptophane-Leucine, ce qui indique qu'ils ont été transformées avec succès avec des proies plasmide. Les appâts qui ne l'auto-activation va croître sur le DD-tryptophane-Leucine-adénine-Histidine médias seulement lorsqu'ils sont transformés avec N ous j'ai proies des constructions, et non pas avec N ous proies G. Notez ce que la concentration de 3-AT est requise dans les médias (le cas échéant) que cela devra être utilisé lors du dépistage.
4. Dépistage
- Média nécessaire et les solutions
- Stérile ddH 2 O préparé par autoclave à 121 ° C, 15 psi pendant 30 minutes.
- 0,9% de NaCl préparés dans le trou DDH 2 O et stérilisé à l'autoclave à 121 ° C, 15 psi pendant 30 minutes.
- Solution de phosphate de sodium composée de 493 mm phosphate de sodium dibasique et 250 mM phosphate de sodium monobasique dans le trou DDH 2 O. Stériliser à l'autoclave à 121 ° C, 15 psi pendant 30 minutes.
- X-Gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) solution préparée comme une de 100 mg / ml solution mère dans le N, N-diméthylformamide.
- Médias croissance 2xYPAD contenant 2% de l'extrait de levure p / v, 4% p / v peptone, 4% p / v de glucose et 100 uM adénine, préparé dans le trou DDH 2 O. Stériliser à l'autoclave à 121 ° C, 15 psi pendant 30 minutes.
- Dropout synthétique (SD) de croissance, préparés comme décrit précédemment. Préparer liquides SD-leucine et solide SD-tryptophane-Leucine. Verser le milieu solide dans les deux plats de Pétri 100x15 mm. Préparer solide SD-tryptophane-Leucine-adénine-Histidine médias dans des plaques de 150 mm de diamètre, 16 planches pour chaque écran, contenant 3-AT, si nécessaire, à la concentration déterminée à partir du G N UB / test de contrôle, je.
- Dropout synthétique (SD) + 5-bromo-4-chloro-3-β-Indoyl-D-galactopyranoside (X-Gal). Préparer SD-tryptophane-Leucine-adénine-Histidine média contenant de l'agar comme décrit précédemment. Après autoclavage, laisser refroidir, ajouter 3-AT (si nécessaire), suivi par volume 1/10ème of solution stérile de phosphate de sodium. Ensuite, ajoutez X-Gal solution à une concentration finale de 80 pg / mL. Bien mélanger et verser dans des plaques de 150 mm ronde.
- PEG / Lithium solution d'acétate II contenant 40% de PEG-3350, 100 mM d'acétate de lithium, EDTA 1 mM et 10 mM Tris pH 7,5. Préparer cette solution à l'aide stériles ddH 2 O et de solutions (par exemple autoclave de 50% de PEG-3350, 1 M d'acétate de lithium, 100 mM Tris pH 7,5 et EDTA 500 mM pH 8,0).
- L'acétate de lithium / Tris EDTA Solution contenant l'acétate de lithium 110 mM, 11 mM Tris pH 7,5 et 1,1 mM d'EDTA. Préparer cette solution à l'aide stériles ddH 2 O et de solutions (par exemple l'acétate de lithium 1M autoclave, 100 mM Tris pH 7,5 et EDTA 500 mM pH 8,0).
- 10x Tris EDTA solution composée de 100 mM de Tris pH 7,5 et EDTA 10 mM préparée dans le trou DDH 2 O. Stériliser à l'autoclave à 121 ° C, 15 psi pendant 30 minutes.
- Solution d'ADN simple brin porteur (ADNsb) contenant 2 mg / ml de sperme de saumon ADN de type III de sel de sodium, préparés stériles O. ddH 2
- Commercial miniprep kit.
- Billes en verre sodocalcique (0,5 mm).
- Compétente cellules d'Escherichia coli adapté à la propagation de plasmide (par exemple, DH5a, XL10 or) et les médias standard adapté à la propagation des bactéries et la sélection du plasmide.
- Souches de levures spécifiques et les plasmides comme décrit dans le protocole
- Transformation à grande échelle
- Inoculer une seule colonie de la souche journaliste MYTHE contenant votre appât dans 5 mL de SD-leucine médias et incuber une nuit à 30 ° C avec agitation (200 rpm).
- Diluer la culture durant la nuit dans 200 ml SD-leucine médias à une DO600 = 0,15 et incuber à 30 ° C avec agitation (200 rpm). Croître jusqu'à la DO600 = 0,6 - 0,7 (environ 4-5 heures).
- Peu avant la DO600 cible est atteinte, le dégel d'une partie aliquote de la solution ADNsb. Faire bouillir à 100 ° C pendant 5 minutes puis refroidir sur glace. Répéter une fois.
- Lorsque la cible a été atteinte DO600, récolte les cellules par centrifugation à 700xg pendant 5 minutes (diviser la culture de 200 mL entre les tubes ml 4x50 centrifugeuse à bouchon à vis).
- Laver chaque culot avec 30 ml stériles ddH 2 O et brièvement au vortex l'échantillon. Centrifuger à 700xg pendant 5 minutes.
- Jeter le surnageant et remettre chaque culot dans 1 ml d'acétate de lithium / Tris EDTA. Transférer dans un tube de micro stérile de 1,5 ml et centrifuger à 700xg pendant 5 minutes.
- Jeter le surnageant et remettre chaque culot dans 600 ul d'acétate de lithium / Tris EDTA.
- Ajoutez ce qui suit 4x15 mL tubes à centrifuger à bouchon à vis:
- 2,5 ml de PEG / acétate de lithium Solution II
- 600 uL cellules resuspendues
- Solution 100 uL ADNss
- 7 pg d'ADN de la banque proies
- Bibliothèques contenant des proies marqués à l'extrémité N-ou C-terminale de N ous G, et préparés à partir d'une variété de sources d'ADNc ou génomique, sont disponibles dans le commerce ( www.dualsystems.com ). La bibliothèque spécifique utilisé doit être déterminée au cas par cas, dépend de l'appât sélectionnés et les objectifs expérimentaux.
- Vortex les tubes pendant 1 minute pour bien mélanger puis incuber dans un bain-marie à 30 ° C pendant 45 minutes. Mélanger brièvement toutes les 15 minutes.
- Ajouter 160 uL sulfoxyde de diméthyle (DMSO) à chaque tube et mélanger immédiatement en inversant les tubes.
- Incuber dans un bain-marie à 42 ° C pendant 20 minutes.
- Recueillir les transformants par centrifugation à 700xg pendant 5 minutes.
- Jeter le surnageant. Récupérer les transformants par resuspension de chaque culot dans 3 ml 2xYPAD. Piscine tous les échantillons ensemble dans un seul tube à centrifugation de 50 mL à bouchon à vis.
- Incuber à 30 ° C pendant 90 minutes pour la récupération des cellules.
- Centrifuger à 700xg pendant 5 minutes et jeter le surnageant.
- Resuspendre les culots cellulaires dans 4,9 ml de NaCl 0,9% stérile.
- En utilisant 100 ul de cellules resuspendues préparer 10 dilutions en série dans stérile de NaCl 0,9% allant de 10x à 1000x.
- Plate 100 pi de la dilution 100x et 1000x sur SD-tryptophane-Leucine médias et incuber à 30 ° C pendant 2-3 jours. Ces plaques servent de contrôle et sont utilisés pour calculer l'efficacité de la transformation.
- Tout aussi diviser le reste 4,8 ml de cellules remises en suspension et la plaque sur de grandes (150 mm) SD-tryptophane-Leucine-adénine-Histidine plaques,contenant la quantité nécessaire de 3-AT tel que déterminé au sein du G N UB / je teste, et incuber à 30 ° C pendant 3-4 jours.
- Resuspendre colonies uniques (chaque représentant cellules contenant un potentiel d'interagir appât-proie paire) dans 100 ul de NaCl 0,9% et la plaque 5 aliquotes sur SD-tryptophane-Leucine-adénine-histidine + X-Gal médias (et notamment la 3-AT, si nécessaire). Laisser pousser pendant 2-4 jours. Cette étape sert une deuxième ronde de dépistage sélectif, et contribue à l'élimination des faux positifs obtenus dans la ronde initiale. Seules les colonies qui affichent une croissance robuste et une couleur bleue sont sélectionnés pour une analyse ultérieure.
- Isolement et séquençage ADN Prey
- Isoler l'ADN plasmidique des colonies de levures bleue en utilisant un protocole de miniprep avec les modifications décrites précédemment. Soyez sûr de grandir les cellules en SD-Tryptophane médias seulement, de choisir pour la rétention des proies, mais pas les appâts, les plasmides. Pour les écrans qui produisent un très grand nombre de hits, disponible dans le commerce à haut débit kit miniprep peut être avantageux à ce point.
- Transformez ADN de levure isolées plasmide dans une compétents E. coli souche appropriée pour la propagation de plasmide (par exemple, DH5a, XL10 or) avec une efficacité de transformation d'au moins 1x10 7 cellules / ug d'ADN. Notez que les plasmides proies peuvent être sélectionnés pour l'utilisation de l'ampicilline.
- Isoler l'ADN plasmidique de la transformée E. coli en utilisant une méthode d'ADN standard d'isolation ou un kit commercial. Une fois de plus, un kit haut-débit miniprep peut être utile si le numéro de l'échantillon est grand. L'amplification de l'ADN dans E. coli augmente considérablement le rendement plasmide et veille à ce qu'une quantité suffisante d'ADN est présente à la fois pour le séquençage et l'analyse plus loin.
- Séquence des plasmides isolés en utilisant une amorce complémentaire de la séquence dans le G. N ous
- Compiler et analyser toutes les données de séquençage à assembler votre liste préliminaire des interacteurs. Cela peut se faire manuellement ou de manière automatisée en utilisant un logiciel approprié.
- Test de dépendance Bait
- Après l'assemblage de la liste préliminaire des interacteurs il est important de re-vérifier les interactions et d'éliminer les proies promiscuité qui interagissent / activer le système rapporteur de manière indépendante de l'identité d'appât. Ceci est accompli en utilisant le test de dépendance appât. Dans ce test, tous les interacteurs identifiés sont reconverties dans la souche originale appâts, ainsi que d'une souche hébergeant un appât de contrôle artificiel constitué d'un seul domaine transmembranaire fusionnée à la C-ub LexA-VP16 tag. La transformation est réalisée selon le protocole standard décrit précédemment, en utilisant SD-leucine des médias à la place de YPAD et SD-tryptophane-Leucine milieux solides pour l'étape de placage finale.
- Resuspendre colonies isolées à partir des transformations ci-dessus dans 100 ul de stériles ddH 2 O et SPOT 5 volumes uL sur SD-tryptophane-Leucine-adénine-histidine + X-Gal médias (et notamment la 3-AT si nécessaire). Les plaques sont ensuite incubées pendant 2-4 jours à 30 ° C. Idéalement transformants multiples devraient être sélectionnés pour chaque proie, et tant l'appât original et appâts artificiels doivent être déposés sur la même plaque.
- Levure portant l'appât artificiel et la proie que l'activation cause du système rapporteur (croissance et de couleur bleue) sont considérés comme promiscuité et que la proie spécifique est retirée de la liste des interacteurs.
- Proies qui causent la croissance et la coloration bleue dans la levure avec l'appât d'intérêts, mais pas les appâts artificiels, confirme cette interaction spécifique. Si, toutefois, la levure hébergeant la proie et vos appâts d'intérêts ne se développent pas, cette proie est retiré de la liste des interacteurs.
- Les proies restants constituent la liste complète des interacteurs identifiés dans l'écran MYTHE.
5. Poursuite des études
Une fois le dépistage mythe a été achevée, d'autres analyses doivent être effectuées afin de valider et de déterminer la signification biologique des interactions détectées. Les études spécifiques à accomplir varient au cas par cas, et doit être déterminée par le chercheur individuel. Quelques exemples courants de travaux de suivi comprennent la co-immunoprécipitation expériences et des études de suppression dans l'organisme d'origine. De plus, l'analyse informatique des données obtenues peuvent être utiles pour détecter des tendances, et en aidant à déterminer la pertinence et le rôle potentiel de différentes interactions peuvent jouer. Ainsi, la technologie mythe sert comme un puissant première étape »vers l'identification et la compréhension des interactions critiques fonctionnelle des protéines membranaires. Couplé avec un suivi détaillé des études et d'autres récemment développés et émergents TEchnologies, il promet d'être un outil précieux pour élucider les mystères de la cellule.

Figure 1. Principe de la scission d'ubiquitine. A. L'ubiquitine peut être séparée en deux moitiés: la moitié C-terminale (C UB) et la moitié N-terminale (N ous I). Ces fractions spontanément reconstituer en raison de leur forte affinité pour un autre. b. Une mutation ub point de I à la position 13 à partir d'une isoleucine à la glycine (N ous G) empêche cette ré-association spontanée. C. Dans le système de mythe, le UB C est fusionnée à l'appât d'intérêts (B) et la proie est fusionnée à la G N ub (A). Interaction protéine AB reconstitue pseudo-ubiquitine.
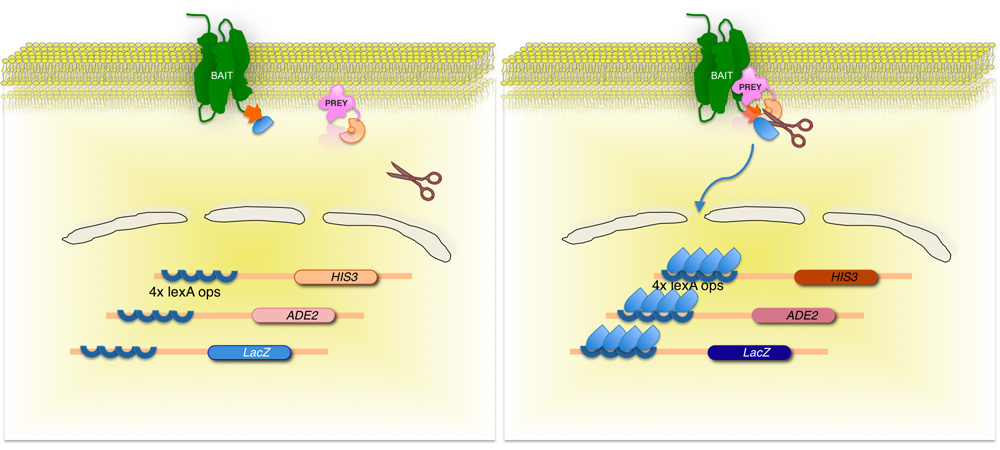
Figure 2. Split-ubiquitine membrane à base de levure de double-hybride (MYTH) du système.
La protéine de la membrane d'intérêts (appât) est fusionnée à la moitié C-terminale de la levure ubiquitine (Ub C), conjugué à un facteur de transcription. En utilisant une banque d'ADNc ou d'ADNg, chaque protéine codée par la bibliothèque (proies) est fusionnée à l'extrémité N-terminale correspondante de la fraction de l'ubiquitine (Ub N G). Si les deux protéines n'interagissent pas, reste le facteur de transcription à l'interface membrane (à gauche). Toutefois, si les protéines interagissent, les deux moitiés ubiquitine rejoindre, résultant en un clivage par l'ubiquitine protéases spécifiques. Décolleté libère le facteur de transcription, résultant de l'expression de gènes rapporteurs (à droite)

Figure 3. MYTHE pipelines.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Le mythe est le système de premier haut débit qui permet l'identification des interactions entre protéines membranaires pleine longueur et cytosoliques ou membranaires partenaires. Il a été utilisé pour étudier des protéines membranaires à partir d'un éventail d'organismes [3-7]. Il ya, cependant, les détails spécifiques qui peuvent avoir besoin d'être examinées attentivement pour s'assurer la protéine d'intérêt est susceptible d'étudier avec le mythe.
Beaucoup de protéines membranaires sont dirigés vers la membrane plasmique par une séquence signal qui est ensuite clivé pour produire la protéine mature. Cette séquence est l'organisme spécifique et il est possible que cette séquence signal natif va provoquer des anomalies de localisation de la protéine d'intérêt lorsqu'il est exprimé dans la levure, car la séquence signal reste méconnue. Pour contourner ce problème, nous avons conçu ces protéines spécifiques à être fusionnée à la séquence signal de la levure, dérivé du facteur alpha accouplement (MATa). Cette séquence peptidique (dits MFα-ss) re-localise la protéine de la membrane plasmique de la levure et surtout, est clivée par des peptidases signal de levure. Cette séquence peptidique est trouvé dans des plasmides pTMBV-MFα et pAMBV-MFα.
Un autre paramètre important qui doit être souligné est l'expression des niveaux d'appât. Le promoteur qui dirige l'expression de l'appât réglemente ce paramètre. Il peut être nécessaire pour optimiser les niveaux d'expression d'appât en utilisant le test NubG / Nubi et la quantité de 3-AT nécessaires pour éliminer les artefacts de la surexpression, où l'appât est «auto-activation» (c'est à dire qu'il interagit avec de nombreuses protéines promiscuité proies non spécifique) . L'examen des protéines de la levure produit des concentrations appâts les plus physiologiquement pertinente lorsque iMYTH est appliquée. Dans ce cas, le gène d'intérêt est étiqueté avec le Cub-TF dans le ADNg. Alternativement, les protéines exogènes peuvent être exprimées à partir pBT3-STE et de plasmides qui portent le pCMBV promoteur CYC1 résultant de l'expression d'appât faible. Les plasmides et pTMBV pTLB1 port du promoteur TEF1 tandis pAMBV a le promoteur ADH1, tant que l'expression forte poussée de la protéine appât. Si les niveaux de protéine appât nécessitent encore l'optimisation, il peut être nécessaire d'utiliser le plasmide pTLB-1 qui porte le promoteur TEF1, cependant, le domaine liant l'ADN LexA est muté au R156G à diminuer l'affinité envers les promoteurs exogènes gène rapporteur, finalement diminuer le probabilité d'auto-activation [5].
Un autre facteur qui joue un rôle important pour la réussite le mythe est la bibliothèque de choix utilisé pour le processus de dépistage. Cela dépendra des profils d'expression endogène de l'appât. Par exemple, l'appât peut être exprimé dans des tissus spécifiques, et il est donc important d'utiliser une bibliothèque qui se construit à partir de ce tissu spécifique. Cela permettra d'assurer physiologiquement pertinente des interactions sont détectées.
Le système le mythe est un outil simple et rapide qui fournit une abondance d'informations sur une classe de protéines qui ont été difficiles à étudier. Ces interactions identifiées peuvent aider à l'élucidation de la fonction biologique complet des protéines membranaires.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Igor Stagljar est une co-fondateur de Dualsystems Biotech, en Suisse.
Acknowledgments
Nous tenons à remercier Edmonds Dawn pour une lecture critique de ce manuscrit. Le laboratoire est soutenu par Stagljar fonds de la Fondation canadienne pour l'innovation (FCI), l'Institut canadien de recherche en santé du Canada (IRSC), la Fondation des maladies du cœur, la Société canadienne du cancer, et Novartis.
Materials
| Name | Company | Catalog Number | Comments |
| Polyethenlene Glycol (PEG3350) | BioShop Canada | PEG335 | |
| Lithium Acetate Bihydrate | BioShop Canada | LIA001 | |
| X-Gal (5-Bromo-4-Chloro-3-Indolyl-b-D-galactopyranoside) | BioShop Canada | XGA001 | |
| N`,N-dimethyl formamide | BioShop Canada | DMF 451 | |
| 3-amino-1,2,4-triazole (3-AT) | BioShop Canada | ATT124 | |
| Sodium phosphate dibasic | BioShop Canada | SPD307 | |
| Sodium phosphate monobasic | Fisher Scientific | BP329-500 | |
| Salmon Sperm DNA | VWR international | CA80601-120 | |
| D-Glucose | BioShop Canada | GLU501 | |
| LB Broth LENOX | BioShop Canada | LBL405 | |
| Yeast Nitrogen Base | BioShop Canada | YNB406 | |
| Yeast Extract | BioShop Canada | YEX401 | |
| Peptone | BD Biosciences | 211677 | |
| Bio-Tryptone | BioShop Canada | TRP402 | |
| Adenine Sulphate | BioShop Canada | ADS201 | |
| L-Uracil | BioShop Canada | URA241 | |
| L-Threonine | BioShop Canada | THR002 | |
| L-Histidine | BioShop Canada | HIS200 | |
| L-Methionine | BioShop Canada | MET222 | |
| L-Valine | BioShop Canada | VAL201 | |
| L-Phenylalanine | BioShop Canada | PHA302 | |
| L-Isoleucine | BioShop Canada | ISO910 | |
| L-Tyrosine | BioShop Canada | TYR333 | |
| L-Leucine | BioShop Canada | LEU222 | |
| L-Arginine | BioShop Canada | ARG006 | |
| L-Tryptophane | Fisher Scientific | BP395-100 | |
| L-Lysine | BioShop Canada | LYS101 | |
| L-Alanine | Fisher Scientific | BP369-100 | |
| Agar | BioShop Canada | AGR001 | |
| Soda Lime Galss Beads | Biospec Products | 11079105 | |
| Sodium Chloride | BioShop Canada | SLD002 |
References
- Stagljar, I., Fields, S. Analysis of membrane protein interactions using yeast-based technologies. Trends Biochem Sci. 27 (11), 559-563 (2002).
- Iyer, K. Utilizing the split-ubiquitin membrane yeast two-hybrid system to identify protein-protein interactions of integral membrane proteins. Sci STKE. 275, pl3-pl3 (2005).
- Paumi, C. M. Mapping protein-protein interactions for the yeast ABC transporter Ycf1p by integrated split-ubiquitin membrane yeast two-hybrid analysis. Mol Cell. 26 (1), 15-25 (2007).
- Stagljar, I. A genetic system based on split-ubiquitin for the analysis of interactions between membrane proteins in vivo. Proc Natl Acad Sci U S A. 95 (9), 5187-5192 (1998).
- Gisler, S. M. Monitoring protein-protein interactions between the mammalian integral membrane transporters and PDZ-interacting partners using a modified split-ubiquitin membrane yeast two-hybrid system. Mol Cell Proteomics. 7 (7), 1362-1377 (2008).
- Scheper, W. Coordination of N-glycosylation and protein translocation across the endoplasmic reticulum membrane by Sss1 protein. J Biol Chem. 278 (39), 37998-38003 (2003).
- Thaminy, S. Identification of novel ErbB3-interacting factors using the split-ubiquitin membrane yeast two-hybrid system. Genome Res. 13 (7), 1744-1753 (2003).
- Johnsson, N., Varshavsky, A. Split ubiquitin as a sensor of protein interactions in vivo. Proc Natl Acad Sci U S A. 91 (22), 10340-10344 (1994).
- Kelleher, D. J., Gilmore, R. The Saccharomyces cerevisiae oligosaccharyltransferase is a protein complex composed of Wbp1p, Swp1p, and four additional polypeptides. J Biol Chem. 269 (17), 12908-12917 (1994).
- Chevallier, M. R. Cloning and transcriptional control of a eucaryotic permease gene. Mol Cell Biol. 2 (8), 977-984 (1982).
