

Summary
MITO permite la detección sensible de las interacciones transitoria y estable entre las proteínas que se expresan en el organismo modelo Saccharomyces cerevisiae. Se ha aplicado con éxito para estudiar las proteínas integrales de membrana exógenos y la levadura con el fin de identificar a sus socios que interactúan de un modo de alto rendimiento.
Abstract
La importancia biológica fundamental y clínica de las proteínas integrales de membrana impulsado el desarrollo de un sistema a base de levadura para la identificación de alto rendimiento de las interacciones proteína-proteína (PPI) para las proteínas transmembrana de larga duración. Con este fin, nuestro laboratorio desarrolló la levadura split-ubiquitina membrana base de dos híbridos (MITO) del sistema. Esta tecnología permite la detección sensible de las interacciones proteína transitoria y estable con
Protocol
1. Antecedentes
Interacciones proteína-proteína (IBP) son los pilares fundamentales que participan en el gobierno de todos los procesos celulares. En consecuencia, es esencial que todas las interacciones son estrictamente regulados con el fin de mantener la homeostasis celular, como un cambio en este equilibrio biológico común juega un papel en la enfermedad y la transformación de las células cancerosas. Membrana de proteínas asociadas se encuentran entre la clase de mayor importancia biológica de las proteínas, ya que puede iniciar complejas cascadas de señalización, y mediar en la importación y exportación de diversas moléculas, como las drogas, que ha sido de importancia reciente en el campo de la salud como problemas relacionados con la resistencia a los medicamentos se han convertido en cada vez más común. La percepción de la complejidad de esta clase de proteínas requiere la identificación de sus socios que interactúan. El descubrimiento de tales socios ha demostrado ser un reto, ya menudo requiere de las duras condiciones que debe ser optimizado para cada proteína de membrana [1].
La importancia biológica fundamental y clínica de las proteínas integrales de membrana impulsado el desarrollo de un sistema a base de levadura para la identificación de alto rendimiento de PPI de proteínas transmembrana de larga duración. Con este fin, hemos desarrollado la levadura split-ubiquitina membrana base de dos híbridos (MITO) del sistema [2-4]. Esta herramienta permite la detección sensible de las interacciones proteína transitoria y estable. Se ha aplicado con éxito para estudiar las proteínas exógenas y endógenas expresado en el organismo Saccharomyces cerevisiae modelo [3-7]. MITO se aprovecha de la observación de que la ubiquitina se pueden separar en dos mitades:. La mitad C-terminal (C UB) y la mitad N-terminal (Nubi) en estudios in vivo han demostrado que estos restos de reconstituir de forma espontánea debido a su alta afinidad por unos a los otros. (Figura. 1 bis) Sin embargo, la introducción de una isoleucina 13 a punto de mutación de glicina en la mitad N-terminal de la ubiquitina (que produce un fragmento conocido como N ub G) impide que este re-asociación espontánea [8] (1b Figure.).
Usamos este principio en el sistema de Mito (Figure. 1c y 2). En pocas palabras, la proteína de membrana integral cebo se funde a un resto C ub que está relacionado con un factor de transcripción artificial formado por la Escherichia coli ADN-proteína de unión LexA, y el dominio de activación de VP16 del virus del herpes simple. Presas son generados por la fusión de fragmentos de ADN genómico o de ADNc derivados de la fracción NubG. Una interacción entre las proteínas cebo y la presa en una gran cantidad de levadura conduce a la reconstitución de la "pseudo-ubiquitina" una molécula de larga duración, con el posterior reconocimiento por enzimas citosólicas deubiquitinating (DUBS) y la liberación proteolítica del factor de transcripción. El factor de transcripción se puede entrar en el núcleo de la célula, y activar un sistema de gen reportero (por lo general implican la expresión de la HIS3, y los genes lacZ ade2) permitiendo el crecimiento de cepas de levadura en medios selectivos, lo cual es indicativo de cebo y la interacción presa [ 2-4].
La identificación y caracterización de las interacciones de proteínas de membrana integral proporcionará información para ayudar a entender completamente su función. A medida que más precisa comprender y analizar el papel de las proteínas que interactúan con las proteínas integrales de membrana, podemos comprender mejor la interacción dinámica que participan en la regulación de estas proteínas y descubrir nuevas dianas que puedan tener un potencial terapéutico.
2. Selección de cebo y el sistema adecuado MITO
- Antes de realizar análisis del mito, verifique que su proteína tiene su N-y / o C-terminal en el citosol de la célula. Es esencial que el C-ub serie LEXA-VP16 etiqueta se fusiona con la proteína en una terminal, ya que las enzimas necesarias para la liberación deubiquitinating factor de transcripción se encuentran en el citosol [4].
- A continuación, decida cuál de las dos principales variantes del mito es adecuado. Para las proteínas de la levadura no nativos, mito tradicional (tMYTH) puede ser utilizado, en el que los cebos se sobreexpresan ectópica de un plásmido. Para las proteínas de levaduras nativas, MITO integrado (iMYTH) es el método de elección. En iMYTH cebos son endógenamente etiquetados con la C-ub serie LEXA-VP16 etiqueta, dejándolos bajo el control de su promotor nativo. Esto es ventajoso, ya que el nivel de expresión de tipo salvaje de cebos ayuda a eliminar los problemas asociados con la sobreexpresión de la proteína, tales como un mayor número de falsos positivos [3]. Con la excepción de la construcción inicial de cebo y los medios selectivos utilizados, tanto en las formas de MITO se llevan a cabo de una manera esencialmente idéntica. Para mayor claridad, se centrará principalmente en el uso de tMYTH en este informe, ya que esta variante puede, en principio, ser utilizada con proteínas de la membrana de prácticamente cualquier organismo y espor tanto, más ampliamente aplicable.
3. Generación y validación de cebo
- Medios necesarios y Soluciones
- Estéril ddH2O preparado en autoclave a 121 ° C, 15 psi durante 30 minutos.
- 3-amino-1 ,2,4-triazol (3-AT) preparada como una solución 1M en ddH 2 O. Esterilizar por el paso a través de un filtro de 0,2 micras.
- YPAD medios de cultivo que consiste en 1% de extracto de levadura w / v, 2% w / v de peptona, glucosa al 2% w / v adenina y 100 mM preparado en ddH 2 O. Esterilizar en autoclave a 121 ° C, 15 psi durante 30 minutos.
- 10 veces más aminoácidos / Mix de bases de nucleótidos: La mezcla completa contiene 1,0 mM de adenina, uracilo 1,8 mM, 1,0 mM arginina, histidina 1,0 mM, 2,3 mM isoleucina, leucina 7,6 mM, 1,6 mM Lisina, Metionina 10,1 mm, 3,0 mm de fenilalanina, un 16,8 mM treonina, triptófano 2,0 mM, 1,7 mM de tirosina y valina de 12,8 mm, elaborado en ddH2O. Por abandono de omitir el ácido amino necesario (s) y / o de bases de nucleótidos (s). Esterilizar en autoclave a 121 ° C, 15 psi durante 30 minutos.
- Sintética de deserción escolar (SD) los medios de cultivo que consiste en un 0,67% w / v base nitrogenada de levadura (sin aminoácidos, pero con sulfato de amonio), el 2% w / v de glucosa, 2% w / v agar y 1x mezcla de aminoácidos / nucleótidos, preparado en ddH 2 O. Preparar tanto líquidos como sólidos (que contiene 2% de agar) SD-leucina los medios de comunicación. También preparan sólida SD-triptófano, leucina y los medios de comunicación SD-triptófano, leucina-adenina-histidina. Esterilizar en autoclave a 121 ° C, 15 psi durante 30 minutos. Vierta en medios sólidos 100x15 mm placas de Petri.
- Sintética de deserción escolar (SD) de crecimiento medio que contiene 3-AT. Prepare SD-triptófano, leucina-adenina-Histidina los medios de comunicación como se ha descrito, pero que contienen 3-AT a concentraciones de 25, 50, 75 y 100 mm. Añadir la cantidad apropiada de 3-A de la solución madre 1M estériles a los medios de comunicación después de haber sido el autoclave y se enfría (pero aún no ha solidificado). Vierta la mezcla en 100x15 mm placas de Petri.
- PEG / acetato de litio mezcla compuesta por 40% w / v de PEG-3350, 120 mM de acetato de litio y 167 mg / mL de ADN de esperma de salmón (Tipo de sal sódica III), preparado en ddH 2 O. Para garantizar la esterilidad de esta mezcla se preparan a partir de agua destilada y soluciones (es decir, en autoclave 50% de PEG-3350, autoclave acetato de litio 1M y 2 mg / ml de ADN de esperma de salmón tipo III sal de sodio preparado en estériles ddH 2 O).
- Enzimas y reactivos para la realización de PCR.
- Miniprep kit comercial.
- La cal sodada cuentas de vidrio (0,5 mm).
- Escherichia coli competentes células adecuadas para la propagación del plásmido (por ejemplo, DH5a, XL10 oro) y los medios de comunicación estándar adecuado para la propagación de bacterias y la selección del plásmido.
- Determinadas cepas de levadura, los plásmidos y las cartillas como se describe en el protocolo.
- Generación de Cebos tMYTH por reparación brecha
- El cebo debe ser clonado en un vector adecuado para el etiquetado y la expresión. Una variedad de vectores tMYTH están disponibles para su uso en la construcción de cebo. Vectores, como el pCMBV4, pAMBV4 pTMBV4 y permitir la construcción de C-terminal cebos etiquetado (cebo-C ub-la serie LEXA-VP16) bajo el control de la CYC1 (débil), los promotores ADH1 (fuerte) y TEF1 (muy fuerte), respectivamente . Cebos N-terminal etiquetados (la serie LEXA-VP16-C ub-cebo) se pueden generar utilizando vectores como pTLB-1 y N-pBT3, bajo el control de la TEF1 y CYC1 promotores, respectivamente. La elección del vector depende de la trampa y debe ser determinada empíricamente. En algunos casos, la expresión más elevada de cebo es necesario con el fin de detectar las interacciones, mientras que en la sobreexpresión de otros cebos casos realmente puede ser perjudicial, dando lugar a un mayor número de falsos positivos.
- Análisis de restricción del plásmido seleccionado en el sitio de restricción apropiado (s). División debe darse sólo en las inmediaciones de la C-ub serie LEXA-VP16 etiqueta (aguas arriba de la etiqueta de marcado C-terminal o aguas abajo de la N-terminal de marcado). Por ejemplo, cuando se utiliza el vector pAMBV4, SfiI es una opción ideal. Guarde el plásmido digerido a -20 ° C hasta que esté listo para su uso.
- Cartillas de diseño para la amplificación y clonación del gen de interés. El extremo 5 'del cebador deben coincidir aproximadamente 35-40 nucleótidos aguas arriba del sitio de restricción, mientras que el extremo 3' deben coincidir con los primeros 18 a 20 nucleótidos del gen objetivo. El extremo 5 'del primer inversa debe ser igual al complemento reverso de aproximadamente 35-40 nucleótidos río abajo del sitio de restricción, con el extremo 3' coincida con el complemento reverso de los últimos 18-20 nucleótidos del gen diana (omitiendo el codón de parada si el C-ub serie LEXA-VP16 etiqueta se coloca en el extremo C-terminal). Dependiendo de si la N-o C-terminal de marcado se realiza, seleccione el 35-40 nucleótidos del cebador como avance o retroceso que el gen de interés se clona en el marco de la C-ub serie LEXA-VP16 etiqueta.
- Amplificar el gen de interés mediante PCR utilizando los cebadores anterior. Parámetros de PCR dependerá de la enzima en particular y los primers específicos utilizados.
- Transformar el producto de PCR, junto con el plásmido digerido en una cepa de levadura de laboratorio adecuados que lleva una mutación leu2 (por ejemplo, BY4741). Una cepa reportero MITO (por ejemplo, THY.AP4 o L40) se puede utilizar pero no es necesario, ya que el propósito de la levadura en este momento es simplemente servir como un entorno en el que la brecha de reparación de recombinación homóloga puede ocurrir. Transformación debe realizarse de la siguiente manera:
- Inocular una colonia de la cepa de levadura seleccionada en 5 ml de los medios de comunicación YPAD estéril y se incuba durante la noche a 30 ° C con agitación constante (200 rpm).
- Diluir el cultivo de una noche en 50 ml de los medios de comunicación YPAD fresco a un OD 600 de ~ 0,15 y se incuba a 30 ° C con agitación (200 rpm). Crecer durante aproximadamente 3-4 horas hasta una OD 600 de ~ 0.6 se alcanza.
- Centrifugar las células a 700xg durante 5 minutos y retirar el sobrenadante.
- Resuspender el botón celular en 25 ml estéril O ddH 2 y centrifugar a 700xg durante 5 minutos.
- Eliminar el sobrenadante y resuspender el botón celular en 1 ml estéril ddH 2 O.
- Añadir 100 ml de las células, 300 L PEG / litio mezcla de acetato, y el plásmido digerido (50 fmol) y del producto de PCR (250-500 fmol) a un tubo de microcentrífuga.
- Se incuba a 30 ° C durante 30 minutos.
- De choque térmico a 42 ° C durante 1 hora.
- Centrifugar a 3000xg durante 5 minutos y separar el sobrenadante.
- Resuspender el botón celular en 200 l estéril ddH2O y la placa de todo el volumen en sólidos, SD-leucina medios selectivos. Crecer a 30 ° C durante 2-4 días.
- Crecer una sola colonia de la cepa transformada en 5 ml SD-leucina medio líquido a 30 ° C durante la noche.
- Centrifugar las células a 700xg durante 5 minutos y retirar el sobrenadante.
- Aislar el ADN del plásmido cebo de los gránulos de las células utilizando cualquier kit comercial minipreparación. Siga el protocolo estándar con una modificación. A fin de garantizar suficiente lisis celular de la levadura, añadir una pequeña cantidad de bicarbonato de 0,5 mm perlas de vidrio de cal para la pastilla después de la resuspensión inicial y agitar vigorosamente durante 5 minutos. A continuación, proceder con el protocolo comercial normal.
- Transformar el ADN de levaduras aisladas en un competente E. coli cepa adecuada para la propagación del plásmido (por ejemplo, DH5a, XL10 Oro) con una eficiencia de transformación de al menos 1x10 7 células / g de ADN. Tenga en cuenta que los plásmidos cebo puede ser seleccionado para el uso de la kanamicina.
- Cosecha del ADN del plásmido de la transformada E. coli utilizando un método de aislamiento del ADN estándar o un kit comercial.
- Verificar la construcción adecuada de la carnada de la secuenciación del plásmido.
- Transformar el cebo verificado construir en una cepa MITO reportero apropiada (por ejemplo, THY.AP4, L40). El protocolo de transformación de levadura se acaba de describir se pueden utilizar, sustituyendo el ADN del plásmido cebo en lugar del producto plásmido digerido y PCR.
- Validación de cebo - Localización adecuada
- Antes de su uso, las cepas de cebo son analizados para asegurarse de que están correctamente localizados en la membrana de la levadura. Cuando se utiliza iMYTH, esta localización dependerá específicamente de las propiedades del cebo etiquetados. Para tMYTH, plásmidos cebo generalmente incluyen una secuencia de señales (por ejemplo, Matα) la dirección de la proteína expresada en la membrana plasmática. La localización se determina mediante microscopía de fluorescencia. La inclusión de una molécula de YFP en la secuencia de etiqueta cebo (es decir, C ub-YFP, la serie LEXA-VP16) es el método más simple y directa, lo que permite la visualización directa de las células vivas, y se utiliza comúnmente en iMYTH. Por otra parte, un método estándar de inmunofluorescencia utilizando anticuerpos contra la serie LEXA VP16 o componentes de la etiqueta se puede utilizar.
- Validación de cebo - N ub G / I prueba de control
- Una vez que la localización adecuada de los cebos se ha establecido, es necesario asegurarse de que el cebo no activar el sistema de reportero solo o en presencia de presas que no interactúan (es decir, verificar que el cebo no es auto-activación). Esto se logra utilizando la N ub G / Test I, donde se transforma el cebo con la interacción (positivo) y no interactúan entre sí (negativo) el control de presas, y se evalúa el crecimiento en medios selectivos. Un cebo debe crecer en medios selectivos in la presencia del control positivo, y no crecer en presencia del control negativo, con el fin de ser apto para su uso en el mito.
- Empezar por transformar la cepa de cebo de la prueba con 100 a 200 ng de presa plásmido de control. El protocolo de transformación de levadura se ha descrito anteriormente se puede utilizar, en sustitución de SD-leucina medios de comunicación en lugar de YPAD y el uso de SD-triptófano, leucina los medios de comunicación para la etapa de revestimiento final. Las construcciones de control después de la presa son de uso general:
- post1 N-ub I (control positivo)
- post1 N-ub G (control negativo)
- pFUR4 N-ub I (control positivo)
- pFUR4 N-ub G (control negativo)
- OST1 es un componente del complejo oligosaccharyl y se localiza en la membrana del retículo endoplasmático [9], mientras que FUR4 es una permeasa de uracilo y se localiza en la membrana plasmática [10]. A pesar de estas proteínas son generalmente la primera opción para su uso como que no interactúan entre presas, su idoneidad variarán en cada caso. En el caso improbable de que su cebo se prevé que interactúan realmente con estos dos controles, presas alternativa tendrá que ser seleccionado. Recordemos que N que ub es la forma de tipo salvaje de la N-terminal de la ubiquitina, y de forma espontánea interactúa con C ub independiente de la interacción entre las proteínas a las que la UB y la UB C N se fusionan. Por lo tanto N ub presas que constituyen controles positivos, mientras que la N G ub presas (que lleva el Isoleucina 13 a la mutación de glicina que impide la asociación espontánea de N UB y la UB C) sirven como controles negativos.
- Volver a suspender las colonias aisladas de cada cebo transformado en 100 L estéril ddH 2 O.
- Serie diluir las células se resuspendieron en agua estéril ddH2O para producir diluciones de 1 / 10, 1 / 100 y 1 / 1000.
- Spot 5 volúmenes l de células sin diluir y diluido en SD-triptófano, leucina y los medios de comunicación SD-triptófano, leucina-adenina-histidina con y sin 3-AT en un rango de concentraciones. 3-AT actúa como un inhibidor competitivo del gen informador HIS3, y sirve para aumentar el rigor del proceso de selección. Puede ser útil en algunos casos, para inhibir el crecimiento no específico de débil a moderado de auto-activación de los cebos.
- Permita que los puntos que se seque y luego se incuban las placas a 30 ° C durante 2-4 días.
- Todos los transformantes que crecen en las placas SD-triptófano, leucina, lo que indica que se han transformado con éxito con la presa plásmido. Los cebos que no se auto-activa crecerá en SD-triptófano, leucina-adenina-Histidina los medios de comunicación sólo cuando se transforma con el N ub que construye la presa, y no con N ub G presas. Tenga en cuenta lo que la concentración de 3-AT se requiere en los medios de comunicación (si existen), ya que tendrá que ser utilizado durante las pruebas.
4. Proyección
- Medios necesarios y Soluciones
- Estéril ddH2O preparado en autoclave a 121 ° C, 15 psi durante 30 minutos.
- 0,9% NaCl preparado en ddH2O y esterilizar en autoclave a 121 ° C, 15 psi durante 30 minutos.
- Solución de fosfato de sodio que consta de 493 mM de fosfato dibásico de sodio y 250 mM de fosfato de sodio monobásico en ddH 2 O. Esterilizar en autoclave a 121 ° C, 15 psi durante 30 minutos.
- X-Gal (5-bromo-4-cloro-3-indolil-β-D-galactopiranósido) preparada como 100 mg / ml de solución de valores de N, N-dimetilformamida.
- 2xYPAD medios de cultivo que contiene 2% w / v de extracto de levadura, 4% w / v de peptona, el 4% w / v de glucosa y M 100 adenina, preparado en ddH 2 O. Esterilizar en autoclave a 121 ° C, 15 psi durante 30 minutos.
- Sintética de deserción (SD) los medios de cultivo, preparado como se describió anteriormente. Prepare líquido SD-leucina y sólido SD-triptófano-leucina. Vierta los medios sólidos en tanto 100x15 mm platos Petri. Prepare sólido SD-triptófano, leucina-adenina-Histidina los medios de comunicación en 150 mm placas redondas, 16 placas para cada pantalla, que contiene 3-AT, si es necesario, a la concentración determinada por la N G ub / I prueba de control.
- Abandono sintéticos (SD) Media + 5-Bromo-4-cloro-3-β-Indoyl-D-galactopiranósido (X-Gal). Prepare SD-triptófano, leucina-adenina-Histidina medio que contiene agar como se describió anteriormente. Después del autoclave, deje que se enfríe, añadir 3-AT (si es necesario), seguido por el volumen o 1/10of estéril solución de fosfato de sodio. A continuación, agregue X-Gal solución a una concentración final de 80 mg / ml. Mezclar bien y verter en placas 150 mm ronda.
- PEG / acetato de litio solución II contiene 40% de PEG-3350, 100 mM de acetato de litio, 1 mM EDTA y 10 mM Tris pH 7,5. Prepare esta solución con estériles ddH2O y soluciones (por ejemplo, autoclave 50% de PEG-3350, 1 M de acetato de litio, 100 mM Tris pH 7,5 y 500 mM EDTA pH 8,0).
- Acetato de litio / Tris solución que contiene EDTA 110 mM de acetato de litio, 11 mM Tris pH 7,5 y 1,1 mM EDTA. Prepare esta solución con estériles ddH2O y soluciones (por ejemplo, autoclave acetato de litio 1M, 100 mM Tris pH 7,5 y 500 mM EDTA pH 8,0).
- 10x Tris EDTA solución que consta de 100 mM Tris pH 7,5 y 10 mM EDTA preparada en ddH 2 O. Esterilizar en autoclave a 121 ° C, 15 psi durante 30 minutos.
- ADN de cadena simple portadora (ssDNA) solución que contiene 2 mg / ml de esperma de salmón de ADN de tipo III sal de sodio, preparado en estériles ddH 2 O.
- Miniprep kit comercial.
- La cal sodada cuentas de vidrio (0,5 mm).
- Escherichia coli competentes células adecuadas para la propagación del plásmido (por ejemplo, DH5a, XL10 oro) y los medios de comunicación estándar adecuado para la propagación de bacterias y la selección del plásmido.
- Determinadas cepas de levadura y plásmidos como se describe en el protocolo
- La transformación a gran escala
- Inocular una colonia de la cepa reportero mito que contiene el cebo en 5 ml de SD-leucina los medios de comunicación e incubar una noche a 30 ° C con agitación (200 rpm).
- Diluir el cultivo de una noche en 200 ml SD-leucina medios de comunicación a un OD 600 = 0,15 y se incuba a 30 ° C con agitación (200 rpm). Creciendo hasta que la OD600 = 0,6 a 0,7 (aproximadamente 4-5 horas).
- Poco antes de la OD600 objetivo se alcanza, descongelar una alícuota de la solución de ADN de cadena simple. Hervir a 100 º C durante 5 minutos y luego enfriar en hielo. Repetir una vez.
- Cuando el objetivo OD600 se ha llegado a la cosecha de las células por centrifugación en 700xg durante 5 minutos (división de la cultura entre los tubos de 200 ml 4x50 ml de centrífuga con tapón de rosca).
- Lave cada pellet con 30 mL estéril ddH2O y agitar brevemente la muestra. Centrifugar a 700xg durante 5 minutos.
- Descartar el sobrenadante y resuspender cada pellet en 1 ml de acetato de litio / solución Tris EDTA. Transferir a un tubo estéril de 1,5 ml de microcentrífuga y centrifugar a 700xg durante 5 minutos.
- Descartar el sobrenadante y resuspender cada pellet en 600 l de acetato de litio / solución Tris EDTA.
- Agregue lo siguiente a 4x15 tubos de centrifugación ml con tapa de rosca:
- 2,5 ml de PEG / acetato de litio Solución II
- 600 células resuspendidas l
- 100 L solución ssDNA
- 7 microgramos de ADN presa de la biblioteca
- Bibliotecas que contienen presas etiquetados, ya sea en la N-o C-terminal con N ub G, y se preparó a partir de una variedad de fuentes de cDNA o genómica, están disponibles comercialmente ( www.dualsystems.com ). La biblioteca específica utilizada debe ser determinada sobre una base caso por caso, dependiendo de la carnada seleccionados y los objetivos experimentales.
- Vortex los tubos durante 1 minuto para que se mezcle bien y luego se incuban en un baño de agua a 30 ° C durante 45 minutos. Mezclar brevemente cada 15 minutos.
- Añadir 160 l sulfóxido de dimetilo (DMSO) a cada tubo y mezclar inmediatamente invirtiendo los tubos.
- Incubar en un baño de agua a 42 ° C durante 20 minutos.
- Recoger transformantes por centrifugación a 700xg durante 5 minutos.
- Eliminar el sobrenadante. Recuperar transformantes por la resuspensión de sedimento en cada 2xYPAD 3 ml. Piscina todas las muestras juntas en un solo tubo de centrífuga de 50 ml con tapón de rosca.
- Se incuba a 30 ° C durante 90 minutos para la recuperación celular.
- Centrifugar a 700xg durante 5 minutos y descartar el sobrenadante.
- Resuspender los pellets de células en 4,9 ml estéril al 0,9% de NaCl.
- El uso de 100 l de células resuspendidas preparan 10 diluciones dobles en NaCl estéril al 0,9% que van desde 10x a 1000x.
- Placa de 100 L de las diluciones de 100x y 1000x en SD-triptófano, leucina los medios de comunicación y se incuba a 30 ° C durante 2-3 días. Estas placas sirven como control y se utilizan para calcular la eficiencia de la transformación.
- Igualmente se dividen el restante 4,8 ml de células resuspendidas y la placa a gran tamaño (150 mm) SD-triptófano, leucina-adenina-Histidina placas,que contiene la cantidad necesaria de 3-AT como se determina en el G N ub / prueba I, y se incuba a 30 ° C durante 3-4 días.
- Volver a suspender las colonias individuales (cada uno contiene las células que representan un potencial de interacción cebo-presa par) en 100 l de NaCl y la placa de 0,9% en alícuotas de 5 l SD-triptófano, leucina-adenina-Histidina + X-Gal medios de comunicación (e incluyendo 3-AT si es necesario). Dejar crecer durante 2-4 días. Este paso sirve como una segunda ronda de revisión selectiva, y ayuda en la eliminación de falsos positivos obtenidos en la ronda inicial. Únicas colonias que presentan un crecimiento robusto y de color azul son seleccionados para su posterior análisis.
- Presa de aislamiento y secuenciación de ADN
- Aislar el ADN del plásmido de las colonias de levadura azul mediante un protocolo minipreparación con las modificaciones descritas anteriormente. Asegúrese de crecer las células en el SD-triptófano único medio de comunicación, para seleccionar para la retención de la presa, pero no de cebo, los plásmidos. Para las pantallas que producen un gran número de hits, uno disponible en el mercado de alto rendimiento kit minipreparación puede ser ventajoso en este momento.
- Transformar el ADN plásmido de levadura aislada en un competente E. coli cepa adecuada para la propagación del plásmido (por ejemplo, DH5a, XL10 Oro) con una eficiencia de transformación de al menos 1x10 7 células / g de ADN. Tenga en cuenta que los plásmidos presa puede ser seleccionado para el uso de ampicilina.
- Aislar el ADN del plásmido de la transformada E. coli utilizando un método de aislamiento del ADN estándar o un kit comercial. Una vez más, un equipo de alto rendimiento minipreparación puede ser útil si el número de la muestra es grande. La amplificación del ADN en E. coli aumenta en gran medida el rendimiento de plásmidos y asegura que una cantidad suficiente de ADN está presente tanto para la secuenciación y análisis.
- Secuencia de los plásmidos aislados utilizando un cebador complementario a la secuencia dentro de la G. N ub
- Recopilar y analizar todos los datos de secuenciación para armar su lista preliminar de los interactores. Esto puede hacerse manualmente o de forma automatizada mediante un software adecuado.
- Cebo pruebas de dependencia
- Después del montaje de la lista preliminar de los interactores es importante para volver a verificar las interacciones y eliminar presas promiscua que interactúan / activar el sistema de reportero de una manera independiente de la identidad de cebo. Esto se logra mediante el test de dependencia de cebo. En esta prueba, todos los interactores identificados se transforman de nuevo en la cepa original de cebo, así como una cepa albergar un cebo artificial de control que consta de un único dominio transmembrana fundido a la C-ub serie LEXA-VP16 etiqueta. La transformación se lleva a cabo según el protocolo estándar descrito anteriormente, utilizando los medios de comunicación SD-leucina en lugar de YPAD y SD-triptófano, leucina medios sólidos para la etapa de revestimiento final.
- Volver a suspender las colonias aisladas a partir de las transformaciones anteriores en 100 l de agua estéril ddH2O y el punto 5 volúmenes l en SD-triptófano, leucina-adenina-Histidina + X-Gal medios de comunicación (e incluyendo 3-AT, si es necesario). Las placas se incuban durante 2-4 días a 30 ° C. Lo ideal sería que varios transformantes se debe seleccionar para cada presa, y tanto el cebo original y cebos artificiales deben ser vistos en el mismo plato.
- La levadura que lleva el cebo artificial y la presa que la activación causa del sistema de periodista (es decir, el crecimiento y el color azul) se consideran promiscuas y que se aprovechan específico es eliminado de la lista de los interactores.
- Presas que causan el crecimiento y coloración azul en la levadura con el cebo de intereses, pero no el cebo artificial, confirma esta interacción específica. Sin embargo, si la levadura que alberga la presa y su cebo de intereses no crecen, esta presa es eliminada de la lista de los interactores.
- Las presas restantes constituyen la lista completa de los interactores identificado en la pantalla del mito.
5. Los estudios más
Una vez cribado mito ha sido completado, los nuevos análisis se debe realizar con el fin de validar y determinar la importancia biológica de las interacciones detectadas. Los estudios específicos para llevar a cabo variará en cada caso por caso, y debe ser determinado por el investigador individual. Algunos ejemplos comunes de la labor de seguimiento incluyen co-immunoprecipitation experimentos y estudios de eliminación en el organismo de origen. Además, el análisis computacional de los datos obtenidos pueden ser útiles para la detección de patrones, y ayudando a identificar la relevancia y el papel potencial de las diferentes interacciones pueden jugar. Así, la tecnología mito sirve como un poderoso primer paso "hacia la identificación y la comprensión de las interacciones críticas funcional de las proteínas de membrana. Junto con detalle los estudios de seguimiento y otros recientemente desarrollados y emergentes technologies, que promete ser una valiosa herramienta para desentrañar los misterios de la célula.

Figura 1. Principio de la separación de la ubiquitina. A. La ubiquitina puede ser separado en dos mitades: la mitad C-terminal (C UB) y la mitad N-terminal (N ub I). Estos restos de reconstituir espontáneamente debido a su alta afinidad por los unos a los otros. b. Una mutación que ub punto en la posición 13 de un isoleucina a la glicina (G N ub) impide que este re-asociación espontánea. c. En el sistema de mito, la UB C se funde con el cebo de intereses (B) y la presa se fusiona con la N G ub (A). Interacción AB proteína reconstituye pseudo-ubiquitina.
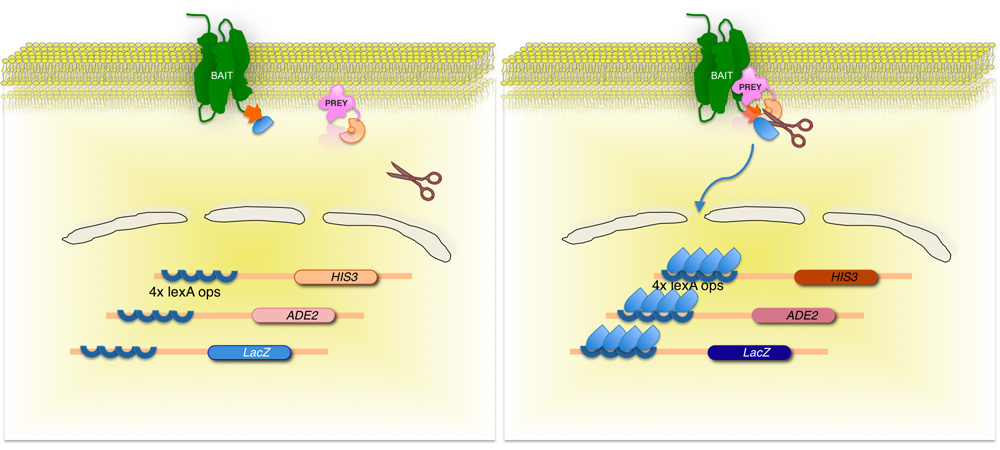
Figura 2. Split-ubiquitina levadura membrana base de dos híbridos (MITO) del sistema.
La membrana de proteína de interés (cebo) se fusiona con la mitad C-terminal de la levadura ubiquitina (Ub C), conjugado con un factor de transcripción. Utilizando una biblioteca de cDNA o ADNg, cada proteína codificada por la biblioteca (la presa) se fusiona con el correspondiente N-terminal de la fracción de ubiquitina (Ub N G). Si las dos proteínas no interactúan, el factor de transcripción se mantiene en la interfaz de la membrana (panel izquierdo). Sin embargo, si las proteínas interactúan, las dos mitades de la ubiquitina unirse, dando lugar a la escisión por la ubiquitina proteasas específicas. División libera el factor de transcripción, lo que resulta en la expresión de genes reporteros (panel derecho)

Figura 3. MITO tubería.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Mito es el primer sistema de alto rendimiento que permite la identificación de las interacciones entre las proteínas de membrana de larga duración y citosólica o socios de la membrana. Se ha utilizado para estudiar la proteína de la membrana de una serie de organismos [3-7]. Hay, sin embargo, los detalles específicos que deben ser examinados para asegurar que la proteína de interés puede ser objeto de estudio con el mito.
Muchas proteínas de membrana se dirigen a la membrana plasmática a través de una secuencia de señales que posteriormente se escinde para producir la proteína madura. Esta secuencia es organismo específico y es posible que esta secuencia señal nativa causará errores de localización de la proteína de interés cuando se expresa en la levadura porque la secuencia de la señal sigue sin ser reconocido. Para evitar este problema, diseñamos estas proteínas específicas que se fusiona con la secuencia señal de la levadura, que se deriven del factor alfa de apareamiento (MATα). Esta secuencia de péptidos (los llamados MFα-ss) re-localiza la proteína de la membrana plasmática de la levadura y lo más importante, se rompe por peptidasas señal de la levadura. Esta secuencia del péptido se encuentra en plásmidos pTMBV-MFα y pAMBV MFα.
Otro parámetro importante que hay que destacar es el nivel de expresión de cebo. El promotor que dirige la expresión de la carnada regula este parámetro. Puede que sea necesario para optimizar los niveles de expresión de cebo mediante la prueba de NubG / Nubi y la cantidad de 3-AT necesarias para eliminar los artefactos sobreexpresión, donde el cebo es "auto-activación" (es decir, promiscuamente interactúa con muchas proteínas de la presa no específicos) . El examen de las proteínas de levadura produce la concentración de cebo más fisiológicamente relevantes cuando iMYTH se aplica. En este caso, el gen de interés está marcado con el Cub-TF en el ADNg. Por otra parte, las proteínas exógenas se puede expresar de pBT3-STE y los plásmidos pCMBV que llevan el promotor CYC1 resultante en la expresión de cebo baja. El pTMBV plásmidos y pTLB1 puerto el promotor TEF1 mientras pAMBV tiene el promotor ADH1, tanto que la expresión de un fuerte impulso de la proteína cebo. Si los niveles de cebo proteínas requieren una mayor optimización, puede ser necesario utilizar el plásmido pTLB-1 que lleva el promotor TEF1, sin embargo, el dominio de ADN de unión LexA se encuentra mutado en R156G para disminuir la afinidad hacia los promotores de genes exógenos reportero, en última instancia, la disminución de la probabilidad de auto-activación [5].
Otro factor que juega un papel importante para el éxito El mito es el de la biblioteca de la opción utilizada para el proceso de selección. Esto dependerá de los perfiles de expresión endógeno cebo. Por ejemplo, el cebo puede ser expresado en tejidos específicos, por lo que es importante usar una biblioteca que se construye a partir de este tejido específico. Esto asegurará que fisiológicamente se detectan interacciones relevantes.
El sistema de MITO es una herramienta sencilla y rápida que proporciona una gran cantidad de información acerca de una clase de proteínas que han sido difíciles de estudiar. Estas interacciones identificadas pueden ayudar en el esclarecimiento de la función biológica completa de proteínas de membrana.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Igor Stagljar es co-fundador de DualSystems Biotech, Suiza.
Acknowledgments
Nos gustaría dar las gracias a Edmonds amanecer para una lectura crítica de este manuscrito. El laboratorio Stagljar es apoyado por fondos de la Fundación Canadiense para la Innovación (CFI), el Instituto Canadiense de Investigación en Salud (CIHR), la Fundación Heart and Stroke, de la Sociedad Canadiense del Cáncer y Novartis.
Materials
| Name | Company | Catalog Number | Comments |
| Polyethenlene Glycol (PEG3350) | BioShop Canada | PEG335 | |
| Lithium Acetate Bihydrate | BioShop Canada | LIA001 | |
| X-Gal (5-Bromo-4-Chloro-3-Indolyl-b-D-galactopyranoside) | BioShop Canada | XGA001 | |
| N`,N-dimethyl formamide | BioShop Canada | DMF 451 | |
| 3-amino-1,2,4-triazole (3-AT) | BioShop Canada | ATT124 | |
| Sodium phosphate dibasic | BioShop Canada | SPD307 | |
| Sodium phosphate monobasic | Fisher Scientific | BP329-500 | |
| Salmon Sperm DNA | VWR international | CA80601-120 | |
| D-Glucose | BioShop Canada | GLU501 | |
| LB Broth LENOX | BioShop Canada | LBL405 | |
| Yeast Nitrogen Base | BioShop Canada | YNB406 | |
| Yeast Extract | BioShop Canada | YEX401 | |
| Peptone | BD Biosciences | 211677 | |
| Bio-Tryptone | BioShop Canada | TRP402 | |
| Adenine Sulphate | BioShop Canada | ADS201 | |
| L-Uracil | BioShop Canada | URA241 | |
| L-Threonine | BioShop Canada | THR002 | |
| L-Histidine | BioShop Canada | HIS200 | |
| L-Methionine | BioShop Canada | MET222 | |
| L-Valine | BioShop Canada | VAL201 | |
| L-Phenylalanine | BioShop Canada | PHA302 | |
| L-Isoleucine | BioShop Canada | ISO910 | |
| L-Tyrosine | BioShop Canada | TYR333 | |
| L-Leucine | BioShop Canada | LEU222 | |
| L-Arginine | BioShop Canada | ARG006 | |
| L-Tryptophane | Fisher Scientific | BP395-100 | |
| L-Lysine | BioShop Canada | LYS101 | |
| L-Alanine | Fisher Scientific | BP369-100 | |
| Agar | BioShop Canada | AGR001 | |
| Soda Lime Galss Beads | Biospec Products | 11079105 | |
| Sodium Chloride | BioShop Canada | SLD002 |
References
- Stagljar, I., Fields, S. Analysis of membrane protein interactions using yeast-based technologies. Trends Biochem Sci. 27 (11), 559-563 (2002).
- Iyer, K. Utilizing the split-ubiquitin membrane yeast two-hybrid system to identify protein-protein interactions of integral membrane proteins. Sci STKE. 275, pl3-pl3 (2005).
- Paumi, C. M. Mapping protein-protein interactions for the yeast ABC transporter Ycf1p by integrated split-ubiquitin membrane yeast two-hybrid analysis. Mol Cell. 26 (1), 15-25 (2007).
- Stagljar, I. A genetic system based on split-ubiquitin for the analysis of interactions between membrane proteins in vivo. Proc Natl Acad Sci U S A. 95 (9), 5187-5192 (1998).
- Gisler, S. M. Monitoring protein-protein interactions between the mammalian integral membrane transporters and PDZ-interacting partners using a modified split-ubiquitin membrane yeast two-hybrid system. Mol Cell Proteomics. 7 (7), 1362-1377 (2008).
- Scheper, W. Coordination of N-glycosylation and protein translocation across the endoplasmic reticulum membrane by Sss1 protein. J Biol Chem. 278 (39), 37998-38003 (2003).
- Thaminy, S. Identification of novel ErbB3-interacting factors using the split-ubiquitin membrane yeast two-hybrid system. Genome Res. 13 (7), 1744-1753 (2003).
- Johnsson, N., Varshavsky, A. Split ubiquitin as a sensor of protein interactions in vivo. Proc Natl Acad Sci U S A. 91 (22), 10340-10344 (1994).
- Kelleher, D. J., Gilmore, R. The Saccharomyces cerevisiae oligosaccharyltransferase is a protein complex composed of Wbp1p, Swp1p, and four additional polypeptides. J Biol Chem. 269 (17), 12908-12917 (1994).
- Chevallier, M. R. Cloning and transcriptional control of a eucaryotic permease gene. Mol Cell Biol. 2 (8), 977-984 (1982).
