

Summary
Questo video illustra un metodo per analizzare e cultura neuroni commissurali dalla E13 del midollo spinale dorsale di ratto. Dissociato neuroni commissurali sono utili per studiare i meccanismi cellulari e molecolari della crescita degli assoni e di orientamento.
Abstract
Commissurali neuroni sono stati ampiamente utilizzati per studiare i meccanismi alla base di guida degli assoni durante lo sviluppo embrionale del midollo spinale. I corpi cellulari di questi neuroni si trovano nel midollo spinale dorsale e la loro assoni seguire traiettorie stereotipati durante lo sviluppo embrionale. Assoni commissurali inizialmente progetto ventralmente verso il fondello. Dopo aver attraversato la linea mediana, questi assoni svolta anteriormente e verso il progetto del cervello. Ognuno di questi passi è regolata dall'azione di spunti di guida diversi. Culture altamente arricchito nei neuroni commissurali sono ideali per molti esperimenti affrontare i meccanismi di pathfinding assone, tra cui saggi di tornitura, immunochimica e biochimica. Qui, descriviamo un metodo per analizzare e cultura neuroni commissurali dalla E13 del midollo spinale dorsale di ratto. In primo luogo, il midollo spinale è isolato e strisce dorsali sono sezionati fuori. Il tessuto dorsale è poi dissociata in una sospensione cellulare da tripsinizzazione e distruzione meccanica. I neuroni sono placcati su poli-L-lisina rivestite coprioggetto di vetro o dei tessuti-cultura piatti. Dopo 30 ore
Protocol
1. Dissezione del cavo embrionale dorsale spinale di ratto
Raccomandazioni generali
Tenere L-15 di media su ghiaccio e cambiare spesso il medium nel piatto dissezione per mantenere gli embrioni freschi. Questo aiuta a preservare l'integrità dei tessuti. Tutti i passaggi vengono eseguiti con due paia di pinzette Dumont # 5, a meno indicato. Per evitare la contaminazione, spruzzare tutti gli strumenti e le superfici di lavoro con il 70% di etanolo e mantenere medie bottiglia dissezione chiuso. Per trasferire embrioni tra piatti, usare una pipetta di plastica tagliata o un mestolo forato. E 'fondamentale non danneggiare il midollo spinale (intaccare, stretching) per completare con successo la dissezione.
Preparazione
- Freddo L-15 di media
- 50 mL di L-15 + inattivato al calore del 10% siero di cavallo (HiHS). Tenere su ghiaccio.
Dissezione del midollo spinale
- Euthanize uno E13 gravidanza scena ratto (E0 = primo giorno giorno dopo l'accoppiamento) con una camera di CO 2 secondo le linee guida istituzionali.
- Spray etanolo al 70% sul ventre. Pizzicare e tirare la pelle della regione addominale inferiore con una pinza e taglio con le forbici chirurgiche. Ripetere l'operazione per tagliare il muscolo e strati peritoneale per raggiungere la cavità addominale. Creare una forma a V incisione dal taglio del tessuto lungo i lati del ventre, fino al torace. Sollevare e tirare indietro il tessuto per esporre la cavità addominale.
- L'utero è fissato in tre sedi: al centro dell'addome inferiore, e ad entrambi gli angoli laterali superiori. Sollevare l'utero afferrando il tessuto tra sacche embrionali. Tagliare il tessuto connettivo per rimuovere l'utero e posto in una capsula di Petri riempite con L-15 su ghiaccio.
- I passi successivi sono fatta sotto un microscopio dissezione. Per separare un embrione da extraembrionali tessuti e delle membrane embrionali, afferrare il tessuto tra sacche uterina con un paio di pinze,. Con l'altra coppia, pizzicare la parte più trasparente del sacco per tagliare attraverso le membrane superficiali (il lato oscuro è la placenta). Strizzare l'embrione premendo delicatamente la placenta lato del sacco. Rimuovere tutti gli embrioni e posto in una capsula di Petri riempite con L-15 su ghiaccio.
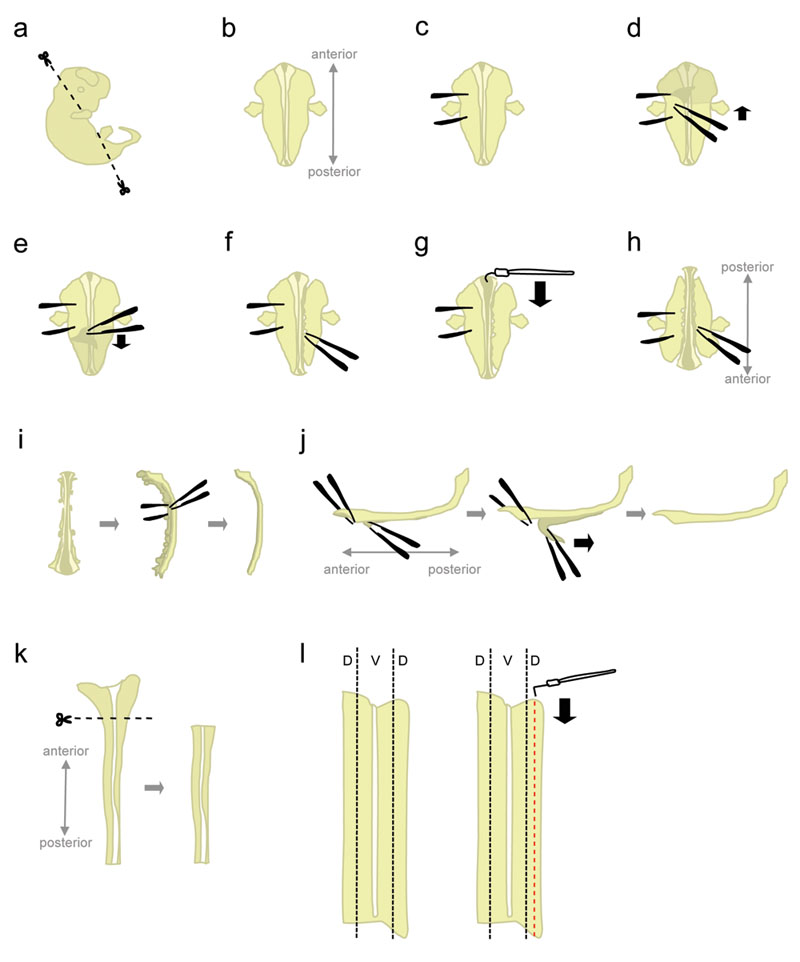
- Mettete un embrione in un piatto da 10 cm Petri riempite con ghiaccio-freddo L-15. Utilizzando microscissors, tagliare via la testa e le parti posteriori ad angolo mostrato in figura 1a.
Taglio a questi angoli aiuterà posizione l'embrione, quando sono immessi "faccia ventrale giù". - Posizionare l'embrione "faccia ventrale giù" (anteriore rivolta in direzione opposta, posteriore rivolta verso lo sperimentatore) (fig. 1b).
- Tengono saldamente l'embrione da un lato, utilizzando le pinze a "fissare" l'embrione (fig. 1c). Non spostare le pinze, in quanto strappare il tessuto. Con l'altra coppia di pinze, togliere la pelle che copre la parte posteriore dell'embrione a partire dalla regione tra la "holding" pinze (fig. 1d-e). Questo esporre il midollo spinale che, a questo punto, è avvolto da membrane (le meningi).
Prendete la pelle dai lati anziché da sopra il midollo spinale per evitare di intaccare il midollo spinale. - Parzialmente staccare il tessuto sul lato destro del midollo spinale (fig. 1f). Partendo a livello della pinza detenzione, colpire il tessuto con una pinza chiusa, il più vicino possibile al midollo spinale. Poi, aprire lentamente le pinze per strappare il tessuto. Questo dovrebbe staccare i gangli della radice dorsale (DRG) dal midollo spinale, e dovrebbe anche disturbare gli organi ventrale. Lasciare un po 'di tessuto attaccati alle estremità anteriore e posteriore. Tessuti Lasciando collegato aggiunge peso per l'embrione, impedendo così di essere tirato verso l'alto durante il passo successivo. Inoltre, che il tessuto utilizzato per contenere al embrione nelle fasi successive.
- A partire dalla fine anteriore, utilizzare il gancio a forma di ago di tungsteno per tagliare le meningi e aprire il midollo spinale lungo la roofplate (fig. 1g).
- Ruotare l'embrione di 180 gradi (posteriore in direzione opposta, puntando verso l'anteriore sperimentatore) (fig. 1 ora).
Questo permette lo sperimentatore di utilizzare la stessa mano per staccare il tessuto dal lato dell'embrione. - Tenere l'embrione afferrando sul lato precedentemente distaccato, e distruggere il tessuto dalla parte rimanente con lo stesso metodo (vedi 1.8.). Al termine, staccare completamente il tessuto su entrambi i lati del midollo spinale.
Alternativa: staccare completamente il tessuto rimanente sul lato sinistro, ma lasciare un po 'di tessuto allegato alla fine anteriore e posteriore sul lato destro. Questo a volte può aiutare a mantenere la colonna vertebrale in posizione al punto 1.12. - Posizionare il midollo spinale su un lato e rimuovere la maggior parte del rimanente tessuto mesenchimale e DRG (fig. 1 decies).
- A questo punto, le meningi e il midollo spinale sono due "fogli" di tessuto apposto su ogni altro. Definire con precisione il più grande, più a porzione anteriore del midollo spinale, e staccare un breve segmento di meningi dal cor spinaled (fig. 1j, a sinistra). Ora, tenere i due segmenti separati da afferrare tutto con una pinza, e staccare le meningi con un movimento regolare e costante (fig. 1j).
Irregolare "peeling" porterà alla rottura del midollo spinale e / o il livello di meningi. Di solito non è possibile recuperare la parte del midollo spinale ancora attaccato al meningi. - Utilizzando una pipetta di plastica, trasferire il midollo spinale isolato di una piastra di Petri contenente L-15 HiHS + 10% e lasciare sul ghiaccio. Tipicamente, il midollo spinale di tutti gli embrioni vengono raccolti prima sezionare le porzioni dorsale.
Dissezione dorsale del midollo spinale
- Mettete un midollo spinale in una capsula di Petri contenente L-15 HiHS +10%, disteso in una configurazione "libro aperto". Tagliare via più ampio, porzione anteriore (che comprende parte del romboencefalo) (1k fig.).
- Il tessuto dorsale si trova nella maggior parte laterale del midollo spinale (fig 1l, a sinistra). Mentre appuntare il cordone ombelicale con un ago di tungsteno dritta, utilizzare la forma di L ago di tungsteno per tagliare una striscia che è 1/5th la larghezza della metà del midollo spinale. (Fig. 1 litro, a destra). Mettere le strisce dorsale in un tubo di plastica da 15 ml contenente L-15 + HiHS 10% su ghiaccio.
Dorsale sezioni del tubo neurale del ~ 12 E13 embrioni produrrà ~ 3,5-4000000 cellule per la placcatura, dopo dissociazione. Più celle possono essere ottenute eseguendo una dissezione grossolana del midollo spinale dorsale (il taglio più larghe dorsale), ma la purezza neurone commissurali sarà ridotto.
2. Commissurali neurone cultura
Raccomandazioni generali
Tutti i passaggi devono essere eseguite in condizioni di sterilità in una cappa di coltura tissutale salvo diversa indicazione. Usare un mezzo fresco e supplementi appena scongelati e reagenti. La dissociazione e passi triturazione vengono eseguiti a Ca 2 + / Mg 2 + senza HBSS per minimizzare Ca 2 + / Mg 2 +-dipendente adesione.
Preparazione
- coprioggetto patinata (l'uso di vetro tedesco Desag) o piastre di coltura tissutale (vedere la procedura di rivestimento sotto).
- terreni in piastra calda Neurobasal (vedi sotto), in piatti coltura tissutale e CO 2 equilibrato nel tessuto culturale incubatore per almeno 1,5 ore prima della placcatura.
- 2,5% tripsina nel 37 ° C a bagno-maria
- 2 bottiglie di HBSS Ca 2 + / Mg 2 +-free, una a 4 ° C, uno a 37 ° C.
- due incendi lucidato vetro pipette Pasteur uno con un diametro grande la metà di consueto, e uno con un diametro leggermente inferiore alla metà. Usa sterilizzati pipette Pasteur. A fuoco lucidare le pipette, utilizzare un becco Bunsen (la parte superiore della fiamma luce blu) per fondere la punta a diminuire leggermente il diametro. Perché questo passo è eseguito al di fuori della cappa coltura tissutale, spruzzare le pipette con etanolo al 70% prima di mettere sotto il cofano coltura di tessuti. Per un recupero più efficiente dei neuroni, cappotto le pipette Pasteur con supporti contenenti siero prima dell'inizio della dissociazione o poco prima del passo triturazione (pipette riempire con i media e mantenere per 30 secondi). Questo consentirà di evitare le cellule di attaccarsi alla parte interna della pipetta Pasteur durante la triturazione.
- acqua sterile per il lavaggio MilliQ PLL rivestite piatti o coprioggetto
- MgSO 12,5% 4 soluzione in HBSS
Poli-L-lisina rivestimento
Se si utilizza coprioggetto di vetro, acido-lavaggio per 24 ore e sterilizzare prima della placcatura (vedi Kaech e Banker, 2006). Usare la lingua tedesca coprioggetto di vetro Desag.
Per rivestire poli-L-lisina su coprioggetto di vetro o di plastica piatti coltura dei tessuti:
- sotto una cappa colture di tessuti, ricoprire le superfici con una piccola cupola di 100 ug / ml soluzione PLL per 1,75-2 ore.
- lavare due volte con acqua MilliQ, almeno 5 minuti per lavaggio (può essere effettuata durante la fase cellula dissociazione di seguito).
- conservare in acqua fino al momento dell'uso. Non lasciate che il PLL rivestita superficie asciutta.
Per ridurre gli sprechi PLL per il rivestimento coprioggetto, collocare il coprioggetto in sterili piatti batterica Petri. Cappotto e lavare i coprioggetti mettendo una cupola di liquido sul coprioggetto. I piatti batterica Petri sono idrofobiche, in modo che il liquido deve rimanere sul coprioggetto di vetro. Trasferire i coprioggetti ai piatti coltura dei tessuti poco prima placcatura le cellule.
Per i neuroni placcato su piatti di plastica coltura tissutale, l'adesione è normalmente più alto, così allungamento dei neuriti potrebbe essere diminuita.
Dissociazione e placcatura
- Verificare che le strisce dorsale si sono stabiliti fino al fondo della provetta. Eliminare la maggior parte HiHS L-15 +10% con una pipetta Pasteur. Lavare rapidamente le strisce dorsale del tubo neurale, una volta con l'aggiunta di 3 ml di freddo (4 ° C) HBSS.
- Lasciate che le strisce dorsale del tubo neurale accontentarsi di 2 minuti, quindi rimuovere la HBSS con una pipetta Pasteur.
- Aggiungi caldo (37 ° C) HBSS ad un volume di 4,7 ml. Tgallina aggiungere 0,3 ml di tripsina 2,5% per ottenere una concentrazione finale di 0,15% tripsina.
- Incubare a 37 ° C in bagnomaria per 7 min. Mescolare delicatamente una volta a metà strada attraverso l'incubazione.
- Aggiungere 30 microlitri DNAsi (25 000 U / mL) per una concentrazione finale di 150 U / mL. Aggiungere 60 ml di MgSO 4 e mescolare brevemente, per una concentrazione finale di 0,15%.
Per il tessuto da dissezioni grossolani, incubare per un extra di 1 minuto a 37 ° C in bagnomaria.
In questa fase, le sezioni del tubo neurale dorsale dovrebbe essere frammentato. Se le sezioni dorsale del tubo neurale non hanno cominciato a frammento, in genere significa che lo stock di tripsina 2,5% è vecchio, e archivi fotografici dovrebbero essere scongelati, o il lavaggio con HBSS freddo non rimuovono efficacemente il HiHS dal campione. - Centrifugare i frammenti di tessuto a 200 g per 4 min.
- Eliminare il surnatante con una pipetta Pasteur, lasciando ~ 50-100 ml di liquido sul fondo della provetta.
- Flick il tubo dolcemente per allentare il pellet, poi lavare le cellule con l'aggiunta di 5 ml di HBSS caldo. Lasciare riposare a temperatura ambiente per 2 min.
- Centrifugare a 200 g per 5 min.
- Eliminare il surnatante con una pipetta Pasteur, lasciando ~ 50-100 ml di liquido sul fondo della provetta.
- Flick il tubo dolcemente per allentare il pellet e risospendere le cellule in parte. Poi aggiungere 2 ml di HBSS caldo.
- Utilizzare il piccolo (metà diametro) fuoco lucido vetro pipetta Pasteur di dissociare le cellule lentamente pipettando su e giù 4-6 volte. Evita di fare bolle, e pipetta il liquido contro la parete del tubo. Non troppo triturare.
- Utilizzare il più piccolo incendio lucidato vetro pipetta Pasteur di dissociarsi ulteriormente le cellule lentamente pipettando su e giù 3-4 volte. Evita di fare bolle, e pipetta il liquido contro la parete del tubo. Non troppo triturare.
Per il tessuto da dissezioni grossolana, aggiungere un ulteriore 1 ml di HBSS caldo al tubo al termine della dissociazione.
Quando dissociare le cellule, non è necessario dissociare tutti i grumi di cellule e aggregati. Fermare pipettando su e giù o passare a una pipetta Pasteur con un diametro inferiore se si vede non diminuire ulteriormente le dimensioni degli aggregati di cellule con pipettaggio ulteriormente. - Lasciate che i frammenti residui di tessuto stabilirsi nella provetta per 1 min. Non è necessario trasferire le cellule in una nuova provetta.
- Prelevare 20 ml di sospensione cellulare e aggiungere 5 ml di trypan blu. Contare le celle su un emocitometro.
I neuroni devono essere ≥ 95% valida per esclusione trypan blu. - Piastra i neuroni in Media Placcatura Neurobasal (vedi ricette di seguito).
- Densità di placcatura suggerito per l'ottenimento di neuroni isolati (a bassa densità culture al fine di evitare i neuroni aggregazione o sovrapposti tra loro):
- 120 000 - 180 000 cellule / pozzetto di una piastra ben 6
- 60 000 - 75 000 cellule / pozzetto di una piastra 12 pozzetti
- 16-18 ore più tardi, cambiare il supporto a Media crescita Neurobasal (vedi ricette di seguito)
Non usare una pompa a vuoto per aspirare il contenuto del piatto quando si cambia la cultura dei media; delicatamente con una pipetta. Questo evita sloggiare i neuroni.
Rappresentante dei risultati:
Quattro ore dopo la placcatura, i neuroni devono avere aderito al poli-L-lisina (PLL)-superficie rivestita. Sotto illuminazione a contrasto di fase, corpi cellulari aderito sono in genere relativamente piatto e di forma ovale (fig. 2a). Cellule che non hanno ben aderito appaiono come sfere che si muovono leggermente quando il piatto è molto delicatamente colpita sul lato. Molti fattori potenzialmente in grado di impedire l'adesione delle cellule (vedi discussione).
Dopo 30 ore in vitro, la maggior parte dei neuroni hanno esteso un assone con un cono di crescita visibile (fig 2c, d). Se la scarsa crescita assonale si osserva, verificare che i mezzi di crescita Neurobasal è stato fatto con terreno fresco e supplementi. I neuroni restano sani per almeno 6 giorni in queste condizioni. Questa procedura ha dimostrato di resa affidabile preparazioni altamente arricchito nei neuroni commissurali, con ~ 90% dei neuroni che esprimono DCC (Yam et al. 2009). La larghezza della striscia dorsale del midollo spinale che viene utilizzato per la preparazione della sospensione cellulare influenzerà la purezza della cultura, con una maggiore purezza quando sottili strisce sono utilizzati. Un esempio di applicazione è mostrato in figura 3 (immunofluorescenza). Vedi l'articolo di Yam et al. (2009) per ulteriori esempi.

Figura 1. Schemi di passi dissezione del midollo spinale. D = dorsali, V = ventrale. Clicca qui per vedere una figura più grande.

Figura 2. Risultato Rappresentante del commissurali isolatoneuroni placcato su un PLL rivestita coprioggetto di vetro. a, b) 4 ore dopo la placcatura, i neuroni hanno aderito alla superficie. Bar = 20 micron. c, d) 30 ore dopo la placcatura, la maggior parte dei neuroni hanno esteso un assone con un cono di crescita visibile. Bar = 20 micron.

Figura 3. Un neurone commissurali immunostained di gamma-tubulina (verde), con F-actina etichettati da falloidina (F-actina, rosso) e il nucleo di DAPI (blu).
Ricette e commenti
Terreni in piastra Neurobasal
- Neurobasal
- 10% di siero FBS (HiFBS)
- 2 mM L-glutammina (da 200 mM soluzione stock)
Opzionale: antibiotici penicillina / streptomicina (usa la metà della concentrazione normale)
La crescita media Neurobasal
- Neurobasal
- B27 (1 / 50 di diluizione a magazzino)
- 2 mM L-glutammina (da 200 mM soluzione stock)
- Opzionale: antibiotici penicillina / streptomicina (usa la metà della concentrazione normale)
Una volta che i media è fatta, può essere conservato a 4 ° C per due settimane. Per equilibrare la temperatura e il pH del supporto prima placcatura le cellule, i media luogo in piastre di coltura tissutale Petri e posto in una coltura di tessuti incubatore per almeno 1,5 ore.
Neurobasal
Dopo una bottiglia di medio Neurobasal è stato aperto, può essere conservato per un mese a 4 ° C al buio. Smaltire Neurobasal che è stato aperto per più di un mese altrimenti la sopravvivenza delle cellule sarà più bassa.
Inattivati al calore siero fetale bovino (HiFBS) o siero di cavallo (HiHS)
Per riscaldare-inattivare calore FBS o HS, a 56 ° C a bagnomaria per 30 minuti. Agitare il flacone ogni 10 minuti circa o giù di lì. (Per la precisione l'uso di una bottiglia di dimensioni simili riempito d'acqua. Inserire un termometro nella bottiglia d'acqua per vedere quando 56 ° C è stata raggiunta. Begin tempi a questo punto.) Calore-inattivato FBS può essere necessario centrifugare per cancellare precipitati, e può essere aliquotati e ri-congelato a -20 ° C.
L-Glutammina
Sempre disgelo una nuova aliquota di L-glutamina per ciascun esperimento.
B27
Aliquote di B27 può essere mantenuta a 20 ° C per una conservazione a lungo termine, oppure a 4 ° C per un massimo di un mese.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Abbiamo descritto un metodo per analizzare e cultura neuroni commissurali dal midollo spinale di ratto embrionale. Questa procedura è stata utilizzata di routine nel nostro laboratorio per la preparazione affidabile neuroni per studiare i meccanismi cellulari e molecolari di orientamento degli assoni. Per la biologia cellulare e gli esperimenti immunochimica, dissezione di una cucciolata produce i neuroni sufficienti. Quando le cellule sono più necessari, come in molti esperimenti di biochimica, la dissezione di due cucciolate possono essere richiesti. Per una persona formata, dissezione e la dissociazione di ~ 20 embrioni possono essere eseguite in meno di 4 ore. Tempi più lunghi si tradurrà in una dissezione più difficile del midollo spinale a causa di cambiamenti nella integrità dei tessuti, e può anche compromettere il recupero effettivo di neuroni vitali.
Quando si esegue la procedura per la prima volta, le cellule piastra su varie superfici rivestite PLL (lavata con acido coprioggetto di vetro, piatti di coltura di tessuti) per il confronto. Normalmente, le cellule devono allegare robustamente a PLL rivestite piatti di plastica tessuto culturale o multi-pozzetti. Questo può essere usato come un controllo generale per la vitalità cellulare e la manutenzione se non ci sono problemi con l'adesione cellulare e la crescita sul coprioggetto di vetro. Il principale fattore responsabile della scarsa adesione delle cellule al vetro è la qualità della pulizia dei vetri e coprioggetto (acido-lavaggio e sterilizzazione). Questo influenzerà l'adesione cellulare, in parte, riducendo il PLL-efficienza del rivestimento. Altri fattori comuni sono cattivi PLL-coating (rivestimento di tempo troppo breve o troppo vecchia soluzione PLL), la contaminazione batterica o fungina, o l'uso di mezzi o integratori che sono vecchi o scaduti.
Abbiamo usato le culture dei neuroni commissurali preparati secondo questa procedura in diversi tipi di esperimenti, tra cui immunochimica, biochimica, e saggi di svolta (Yam et al. 2009). Sorprendentemente, i neuroni commissurali placcato sul PLL rivestita di vetro mantengono la capacità di rispondere agli stimoli di guida, cioè, che cambierà la loro direzione di crescita in risposta a un fattore chemotropic applicata, come Sonic Hedgehog, precedentemente dimostrato di essere uno spunto di guida degli assoni ( Charron et al, 2003; Okada et al, 2006; Yam et al 2009; Fabre et al, 2010).. Pertanto, questo è un potente sistema per studiare l'effetto degli stimoli guida degli assoni in vitro e permette per esperimenti che non sono possibili in vivo.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Questo lavoro è stato sostenuto dalle concessioni dal Canadian Institutes of Health Research (CIHR), il Peter Lougheed Medical Research Foundation, il programma di McGill in Neuroingegneria, Fonds de la Recherche en Santé du Québec (FRSQ), e la Fondazione canadese per l'innovazione (CFI ). D. Sébastien Langlois è stato sostenuto dal Premio La formazione di un Master presso il Fonds de la Recherche en Santé du Québec (FRSQ) e da un Frederick Banting e Charles Premio per la Migliore Canada Laurea Borse di Studio del Maestro dal Canadian Institutes of Health Research (CIHR). Siamo grati a Jessica MT Pham per l'assistenza con figure.
Materials
| Name | Company | Catalog Number | Comments |
| Neurobasal medium, liquid | Invitrogen | 21103-049 | See Recipes and Comments |
| B27 supplement 50X | Invitrogen | 17504 | See Recipes and Comments |
| Poly-L-lysine 0.01% solution | Sigma-Aldrich | P4707 | |
| L-15 medium, powder | Invitrogen | 41300-070 | |
| Trypsin 2.5% (10X) | Invitrogen | 15090-046 | |
| DNAse I, 25000 U/mL | Worthington Biochemical | ||
| MgSO4 | Sigma-Aldrich | M2643 | |
| HBSS, Ca2+/Mg2+-free | Invitrogen | 14170-112 | |
| L-glutamine 200mM, liquid | Invitrogen | 25030-081 | See Recipes and Comments |
Dissection of embryonic rat dorsal spinal cord (see also Table I)
Commissural neuron culture (see also Table I)
|
|||
References
- Charron, F., Stein, E., Jeong, J., McMahon, A. P., Tessier-Lavigne, M. The morphogen sonic hedgehog is an axonal chemoattractant that collaborates with netrin-1 in midline axon guidance. Cell. , 113-1111 (2003).
- Fabre, P., Shimogori, T., Charron, F. Segregation of ipsilateral retinal ganglion cell axons at the optic chiasm requires the Shh Receptor Boc. Journal of Neuroscience. 30, 266-275 (2010).
- Helms, A. W., Johnson, J. E. Progenitors of dorsal commissural interneurons are defined by MATH1 expression. Development. 125, 919-928 (1998).
- Kaech, S., Banker, G. Culturing hippocampal neurons. Nature Protocols. 1, 2406-2415 (2006).
- Okada, A., Charron, F., Morin, S., Shin, D. S., Wong, K., Fabre, P. J., Tessier-Lavigne, M., McConnell, S. K. Boc is a receptor for sonic hedgehog in the guidance of commissural axons. Nature. 444, 369-373 (2006).
- Yam, P. T., Langlois, S. D., Morin, S., Charron, F. Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron. 62, 349-362 (2009).

{kind=link}