

Summary
Cette vidéo montre une méthode de disséquer et de la culture des neurones commissuraux de la moelle épinière de rats E13 dorsale. Dissocié neurones commissuraux sont utiles pour étudier les mécanismes cellulaires et moléculaires de la croissance de l'axone et d'orientation.
Abstract
Neurones commissuraux ont été largement utilisées pour étudier les mécanismes sous-jacents guidage axonal pendant le développement embryonnaire, la moelle épinière. Les corps cellulaires de ces neurones sont situés dans la moelle épinière dorsale et leurs axones suivent des trajectoires stéréotypées pendant le développement embryonnaire. Axones commissuraux initialement projet ventralement vers la plaque de sol. Après avoir traversé la ligne médiane, ces axones tourner vers l'avant et le projet vers le cerveau. Chacune de ces étapes est réglementée par l'action de plusieurs signaux de guidage. Cultures hautement enrichi dans les neurones commissuraux sont idéalement adaptés à de nombreuses expériences aborder les mécanismes de pathfinding axone, y compris les essais de tournage, l'immunochimie et la biochimie. Ici, nous décrivons une méthode de disséquer et de la culture des neurones commissuraux de la moelle épinière de rats E13 dorsale. Tout d'abord, la moelle épinière est isolé et bandes dorsales sont disséquées. Le tissu dorsale est alors dissocié en une suspension de cellules par trypsinisation et les perturbations mécaniques. Les neurones sont étalées sur des lamelles en verre poly-L-lysine ou de culture de tissus plats. Après 30 heures
Protocol
1. Dissection de la moelle épinière de rats dorsale embryonnaire
Les recommandations générales
Gardez milieu L-15 sur la glace et changer fréquemment la moyenne dans le plat de dissection de garder les embryons frais. Cela permet de préserver l'intégrité des tissus. Toutes les étapes sont réalisées avec deux paires de pinces # 5 Dumont sauf indication. Pour éviter la contamination, par pulvérisation tous les outils et les surfaces de travail avec 70% d'éthanol et de garder bouteille moyenne de dissection fermé. Pour transférer des embryons entre les plats, utiliser une pipette en plastique coupé ou une cuillère perforée. Il est essentiel de ne pas endommager la moelle épinière (entailler, étirement) pour mener à bien la dissection.
Préparation
- Froid milieu L-15
- 50 ml de L-15 + inactivé à la chaleur de 10% de sérum de cheval (HiHS). Restez sur la glace.
Dissection de la moelle épinière
- Euthanasier un E13 grossesse scène rat (E0 = premier jour suivant le jour d'accouplement) avec une chambre de CO 2 en fonction de directives institutionnelles.
- Vaporiser l'éthanol à 70% sur l'abdomen. Pincez et tirez la peau de la région abdominale basse avec une pince et coupé avec des ciseaux chirurgicaux. Répéter pour couper à travers le muscle et les couches péritonéale pour atteindre la cavité abdominale. Créer une incision en forme de V en coupant le tissu sur les côtés de l'abdomen, jusqu'à la poitrine. Soulevez et tirez le tissu pour exposer la cavité abdominale.
- L'utérus est fixé à trois endroits: à l'abdomen bas au centre, et à deux coins supérieurs latéraux. Soulever l'utérus en le saisissant par le tissu entre les sacs embryonnaires humaines. Coupez le tissu conjonctif pour enlever l'utérus et le placer dans une boîte de Pétri remplie de L-15 sur la glace.
- Les prochaines étapes sont effectuées sous un microscope de dissection. Pour séparer un embryon à partir des tissus extra-embryonnaires et membranes embryonnaires, attraper le tissu entre les sacs de l'utérus avec une paire de pinces,. Avec l'autre paire, pincer les côtés plus transparente du sac de couper à travers les membranes superficielles (le côté sombre est le placenta). Pressez l'embryon en appuyant doucement sur le côté du placenta du sac. Retirez tous les embryons et les placer dans une boîte de Pétri remplie de L-15 sur la glace.
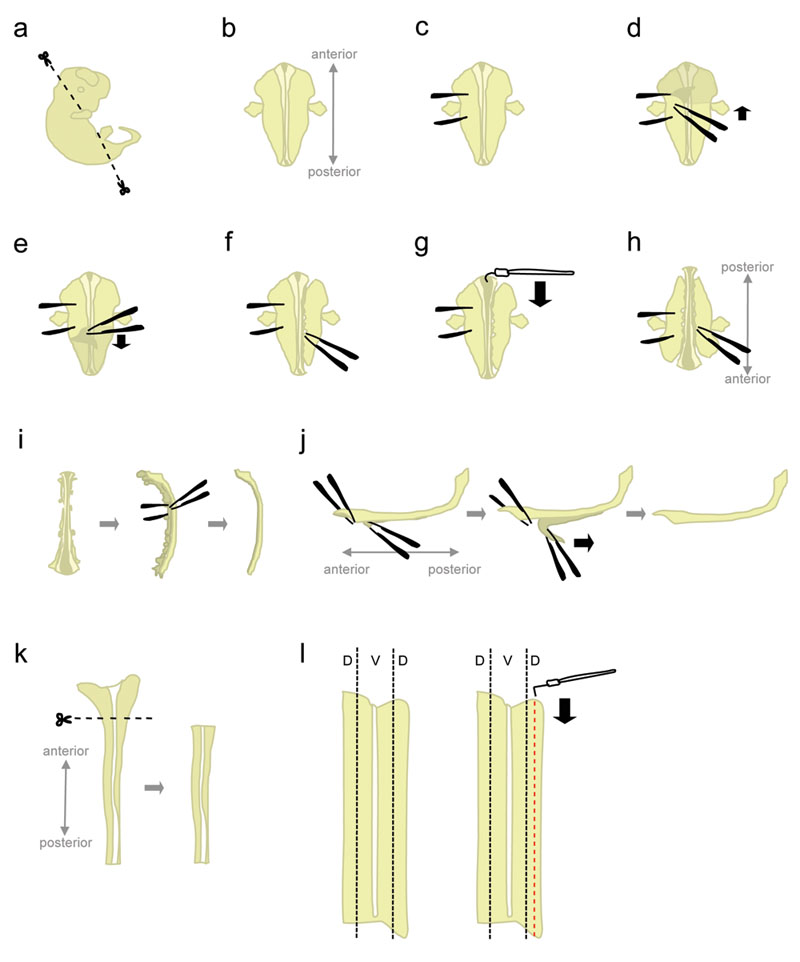
- Placer un embryon dans un plat de 10 cm de Pétri remplie de glace froide L-15. Utiliser microciseaux, couper la tête et parties postérieures à angle montré dans la figure 1a.
Découpage à ces angles permettra de positionner l'embryon lorsqu'il est placé "face ventrale vers le bas". - Position de l'embryon "face ventrale vers le bas" (antérieure pointant loin, postérieure pointant vers l'expérimentateur) (Fig. 1b).
- Maintenez fermement l'embryon d'un côté, en utilisant la pince pour "épingler" l'embryon (figure 1C). Ne pas déplacer la pince, car elle va déchirer le tissu. Avec l'autre paire de pince, retirez la peau qui recouvre le dos de l'embryon à partir de la région entre le "holding" pince (figure 1D-e). Cela permettra d'exposer la moelle épinière qui, à ce stade, est enveloppé par des membranes (les méninges).
Prenez la peau sur les côtés plutôt que d'en haut la moelle épinière pour éviter d'entailler la moelle épinière. - Partiellement détacher le tissu sur le côté droit de la moelle épinière (fig. 1f). Démarrage au niveau de la pince tenant, poke le tissu avec une pince fermée, aussi près que possible de la moelle épinière. Ensuite, ouvrir lentement la pince à déchirer le tissu. Cela devrait se détacher les ganglions de la racine dorsale (DRG) de la moelle épinière, et devraient aussi perturber les organes ventrale. Laissez un peu de tissu attachées aux extrémités antérieures et postérieures. Tissus Laissant attachés ajoute du poids à l'embryon, l'empêchant ainsi d'être tiré à la hausse durant la prochaine étape. De plus, ce tissu est utilisé pour tenir à l'embryon dans les étapes ultérieures.
- A partir de l'extrémité antérieure, utilisez l'aiguille de tungstène en forme de crochet pour couper les méninges et la moelle épinière ouverte le long de la roofplate (fig. 1g).
- Tournez l'embryon de 180 degrés (postérieur pointant loin, pointant antérieure vers l'expérimentateur) (fig. 1 h).
Cela permet à l'expérimentateur d'utiliser la même main pour détacher le tissu de l'autre côté de l'embryon. - Tenir l'embryon en saisissant sur le côté préalablement détaché, et perturber le tissu du côté restant en utilisant la même méthode (voir 1.8.). Quand c'est fait, détacher complètement le tissu des deux côtés de la moelle épinière.
Alternative: détacher complètement du reste du tissu sur le côté gauche, mais laisser un peu de tissu attaché à l'extrémité antérieure et postérieure du côté droit. Cela peut parfois aider à maintenir la moelle épinière en position à l'étape 1.12. - Placez la moelle épinière de son côté et supprimer la plupart des tissus restants mésenchymateuses et les DRG (fig. 1i).
- À cette étape, les méninges et la moelle épinière sont deux "feuilles" de tissu apposé sur l'autre. Pin bas la plus grande, la plus antérieure-portion de la moelle épinière, et de décoller d'un court segment des méninges de la cor épinièreD (Fig. 1j, gauche). Maintenant, maintenez les deux segments séparés en saisissant à travers avec une pince, et de décoller les méninges avec une surface lisse, un mouvement constant (fig. 1j).
Inégale "peeling" conduira à la rupture de la moelle épinière et / ou la couche méninges. Il n'est généralement pas possible de récupérer la portion de la moelle épinière encore attaché à méninges. - Avec une pipette en plastique, le transfert de la moelle épinière isolée à une boîte de Petri contenant de la L-15 HiHS + 10% et de laisser sur la glace. Typiquement, la moelle épinière de tous les embryons sont recueillis avant la dissection des portions dorsale.
Dorsale de la moelle épinière de dissection
- Placez une moelle épinière dans une boîte de Pétri contenant de la L-15 HiHS +10%, à plat dans un "livre ouvert" de configuration. Coupez le plus large, la partie antérieure (qui comprend une partie du cerveau postérieur) (fig. 1K).
- Le tissu dorsale est située dans les parties latérales plus de la moelle épinière (figure 1l, gauche). Bien épinglant le cordon avec une aiguille de tungstène droite, utilisez l'aiguille de tungstène en forme de L pour couper une bande qui est 1/5ème de la largeur de la moitié de la moelle épinière. (Fig. 1l, droite). Placez les bandes dorsales dans un tube plastique 15 ml contenant de la L-15 + HiHS 10% sur la glace.
Dorsale sections du tube neural d'embryons E13 ~ 12 ~ sera le rendement de 3,5 à 4.000.000 cellules pour le placage après dissociation. Plus les cellules peuvent être obtenues en effectuant une dissection grossière de la moelle épinière dorsale (découpe des bandes plus larges dorsale), mais la pureté des neurones commissuraux sera réduite.
2. Commissurales neurone de la culture
Les recommandations générales
Toutes les étapes doivent être effectuées dans des conditions stériles sous une hotte de culture de tissus, sauf indication contraire. Utilisez un milieu frais et les suppléments fraîchement décongelé et réactifs. La dissociation et la trituration des mesures sont effectuées en Ca 2 + / Mg 2 + sans HBSS pour minimiser Ca 2 + / Mg 2 +-dépendante adhérence.
Préparation
- lamelles couché (utiliser du verre Desag allemand) ou des plaques de culture tissulaire (voir procédé de revêtement ci-dessous).
- chaudes des médias Placage Neurobasal (voir ci-dessous), en boîtes de culture tissulaire et de CO 2 dans l'équilibre incubateur de culture tissulaire pendant au moins 1,5 h avant l'ensemencement.
- Trypsine 2,5% dans le bain-marie à 37 ° C
- 2 bouteilles de HBSS Ca 2 + / Mg 2 +-libre, un à 4 ° C, l'un à 37 ° C.
- deux extincteurs polie en verre Pipettes Pasteur une avec un diamètre de la moitié de la taille habituelle, et une avec un diamètre légèrement inférieur à la moitié. Utilisez stérilisés pipettes Pasteur. Pour le feu-polonais des pipettes, utilisez un brûleur Bunsen (en haut de la flamme bleu clair) pour faire fondre la pointe à diminuer légèrement le diamètre. Parce que cette étape est réalisée en dehors de la hotte de culture tissulaire, les pipettes de pulvérisation avec de l'éthanol à 70% avant de placer sous le capot de culture de tissus. Pour la récupération plus efficace des neurones, enduire les pipettes Pasteur avec des milieux contenant du sérum avant le début de la dissociation ou juste avant l'étape de trituration (remplir les pipettes avec les médias et de garder pendant 30 secondes). Cela permettra d'éviter les cellules de coller à l'intérieur de la pipette Pasteur pendant la trituration.
- l'eau stérile pour le lavage milliQ PLL-vaisselle ou des lamelles de revêtement
- 12,5% MgSO 4 solution dans HBSS
Poly-L-lysine revêtement
Si vous utilisez des lamelles de verre, de l'acide de lavage pendant 24 heures et stériliser avant le placage (voir Kaech et Banker, 2006). Utilisez allemande lamelles de verre Desag.
Pour manteau de poly-L-lysine sur des lamelles en verre ou en plastique des boîtes de culture de tissu:
- sous une hotte de culture de tissus, recouvrir des surfaces avec un petit dôme de 100 ug / ml solution pour PLL de 1,75 à 2 heures.
- laver deux fois avec de l'eau milliQ, au moins 5 minutes par lavage (peut être effectuée pendant l'étape de dissociation cellulaire ci-dessous).
- stocker dans l'eau jusqu'à son utilisation. Ne laissez pas la surface PLL revêtement sec.
Afin de réduire le gaspillage PLL pour le revêtement de lamelles, placez les lamelles de boîtes de Petri stériles bactérienne. Manteau et laver les lamelles en plaçant un dôme de liquide sur les lamelles. Les plats de Pétri bactériennes sont hydrophobes, de sorte que le liquide doit rester sur les lamelles de verre. Transférer les lamelles de boîtes de culture tissulaire, juste avant plaquage des cellules.
Pour les neurones plaquée sur le plastique des boîtes de culture tissulaire, l'adhésion est normalement plus élevé, donc un allongement des neurites pourrait être diminuée.
La dissociation et le placage
- Vérifiez que les bandes dorsales se sont installés vers le fond du tube. Enlever la plupart des HiHS L-15 10% avec une pipette Pasteur. Laver rapidement les bandes dorsales du tube neural fois en ajoutant 3 ml de froid (4 ° C) HBSS.
- Laissez les bandes dorsales du tube neural reposer pendant 2 minutes, puis retirez le HBSS avec une pipette Pasteur.
- Ajouter au chaud (37 ° C) HBSS à un volume de 4,7 ml. Torsque ajouter 0,3 ml de trypsine à 2,5% pour donner une concentration finale de 0,15% de trypsine.
- Incuber à 37 ° C au bain-marie pendant 7 min. Mélanger délicatement fois à mi-chemin de l'incubation.
- Ajouter 30 ul DNAse (25 000 U / ml) pour une concentration finale de 150 U / ml. Ajouter 60 ul de MgSO 4 et mélanger brièvement, pour une concentration finale de 0,15%.
Pour les tissus à partir de dissections grossière, incuber pour un supplément de 1 minute à 37 ° C au bain-marie.
A ce stade, les sections du tube neural dorsal doit être fragmenté. Si les sections dorsale du tube neural n'ont pas commencé à se fragmenter, cela signifie généralement que le stock de la trypsine à 2,5% est vieux, et de nouvelles actions doivent être décongelés, ou les laver avec HBSS froid n'a pas d'éliminer efficacement les HiHS de l'échantillon. - Centrifuger les fragments de tissus à 200 g pendant 4 min.
- Enlever le surnageant avec une pipette Pasteur, laissant ~ 50-100 ul de liquide au fond du tube.
- Flick délicatement le tube pour desserrer le culot, puis lavez les cellules en ajoutant 5 ml de HBSS chaud. Laisser reposer à température ambiante pendant 2 min.
- Centrifuger à 200 g pendant 5 min.
- Enlever le surnageant avec une pipette Pasteur, laissant ~ 50-100 ul de liquide au fond du tube.
- Flick délicatement le tube pour desserrer le culot et partiellement remettre en suspension les cellules. Puis ajouter 2 ml de HBSS chaud.
- Utilisez la petite (demi-diamètre) polie au feu pipette Pasteur en verre de dissocier les cellules par pipetage lentement jusqu'à-et-vient 4-6 fois. Évitez de faire des bulles, et la pipette le liquide contre la paroi du tube. Ne pas trop triturer.
- Utilisez la plus petite polie au feu pipette Pasteur en verre de continuer à dissocier les cellules par pipetage lentement jusqu'à-et-vient 3-4 fois. Évitez de faire des bulles, et la pipette le liquide contre la paroi du tube. Ne pas trop triturer.
Pour les tissus à partir de dissections grossière, ajouter un supplément de 1 ml de HBSS chaud pour le tube à la fin de la dissociation.
Lorsque dissocier les cellules, il n'est pas nécessaire de dissocier les amas cellulaires et de granulats. Arrêtez de pipetage va-et-ou le changement d'une pipette Pasteur avec un diamètre plus petit si vous voyez pas de diminution supplémentaire de la taille des agrégats cellulaires avec pipetage plus loin. - Laissez tous les fragments de tissu restant s'installer dans le tube pendant 1 min. Il n'est pas nécessaire de transférer les cellules dans un nouveau tube.
- Prenez 20 pl de suspension cellulaire et ajouter 5 l de bleu trypan. Compter les cellules sur un hémocytomètre.
Les neurones doivent être ≥ 95% viables par exclusion du bleu trypan. - Assiette des neurones dans les médias Placage Neurobasal (voir recettes ci-dessous).
- Densités plaquage suggérée pour l'obtention de neurones isolés (faible densité des cultures pour éviter l'agglutination des neurones ou qui se chevauchent les uns les autres):
- 120 000 - 180 000 cellules / puits d'une plaque de 6 puits
- 60 000 - 75 000 cellules / puits d'une plaque de 12 puits
- 16-18 heures plus tard, le changement des médias pour les médias de croissance Neurobasal (voir recettes ci-dessous)
Ne pas utiliser une pompe à vide pour aspirer les médias de la boîte de culture lors d'un changement des médias; délicatement l'utilisation d'une pipette. Cela évite déloger les neurones.
Les résultats représentatifs:
Quatre heures après l'étalement, les neurones doivent avoir adhéré à la poly-L-lysine (PLL) à revêtement de surface. Sous un éclairage en contraste de phase, les corps cellulaires sont généralement respectés relativement plat et de forme ovale (fig. 2a). Les cellules qui n'ont pas bien adhéré apparaissent comme des sphères qui se déplacent légèrement lorsque le plat est très doucement tapé sur le côté. De nombreux facteurs peuvent potentiellement entraver l'adhésion des cellules (voir la discussion).
Après 30 heures in vitro, la plupart des neurones ont prolongé d'un axone avec un cône de croissance visible (figure 2c, d). Si le pauvre croissance axonale est observée, vérifiez que le milieu de croissance Neurobasal a été faite avec un milieu frais et suppléments. Les neurones restent en bonne santé pendant au moins 6 jours dans ces conditions. Cette procédure s'est avérée fiable le rendement des préparations hautement enrichi dans les neurones commissuraux, avec ~ 90% des neurones exprimant DCC (Yam et al. 2009). La largeur de la bande de la moelle épinière dorsale qui est utilisé pour la préparation de la suspension cellulaire aura une incidence sur la pureté de la culture, avec une plus grande pureté où minces bandes sont utilisées. Un exemple d'application est montré dans la figure 3 (immunofluorescence). Voir l'article de Yam et al. (2009) pour plus d'exemples.

Figure 1. Schéma de la colonne vertébrale étapes de dissection du cordon. D = dorsal, V = ventrale. Cliquez ici pour voir une figure plus grande.

Figure 2. Résultat représentatif des commissurale isoléesneurones plaqué sur une lamelle de verre enduit de PLL. a, b) 4 heures après l'étalement, les neurones ont adhéré à la surface. Barre = 20 um. c, d) 30 heures après l'étalement, la plupart des neurones ont prolongé d'un axone avec un cône de croissance visible. Barre = 20 um.

Figure 3. Un neurone commissurale immunocolorées pour la gamma-tubuline (vert), avec F-actine étiquetés par phalloïdine (F-actine, rouge) et le noyau par le DAPI (bleu).
Recettes et commentaires
Médias Placage Neurobasal
- Neurobasal
- 10% de FBS inactivé à la chaleur (HiFBS)
- 2 mM de L-glutamine (à partir de 200 mM solution stock)
En option: les antibiotiques pénicilline / streptomycine (utiliser la moitié de la concentration normale)
Médias croissance Neurobasal
- Neurobasal
- B27 (1 / 50 de dilution du stock)
- 2 mM de L-glutamine (à partir de 200 mM solution stock)
- En option: les antibiotiques pénicilline / streptomycine (utiliser la moitié de la concentration normale)
Une fois que les médias sont faites, il peut être conservé à 4 ° C pendant deux semaines. Pour équilibrer la température et le pH des médias avant l'étalement des cellules, des médias placer dans des boîtes de Pétri de culture de tissu et placer dans un incubateur de culture tissulaire pendant au moins 1,5 heures.
Neurobasal
Après une bouteille de milieu Neurobasal a été ouvert, il peut être conservé pendant un mois à 4 ° C dans l'obscurité. Eliminer Neurobasal qui a été ouvert pour plus d'un mois sinon la survie cellulaire sera plus faible.
Inactivés par la chaleur du sérum de veau fœtal (HiFBS) ou de sérum de cheval (HiHS)
Pour inactiver la chaleur thermique FBS ou HS, à 56 ° C dans un bain d'eau pendant 30 minutes. Agiter le flacon environ toutes les 10 minutes. (Pour plus de précision l'utilisation d'une bouteille de taille semblable rempli d'eau. Placez un thermomètre dans la bouteille d'eau pour voir quand 56 ° C est atteinte. Commencer à chronométrer à ce point.) Chauffer FBS inactivé peut être nécessaire de centrifuger pour effacer précipités, et peuvent être aliquotés et re-congelés à -20 ° C.
L-Glutamine
Faites toujours décongeler une partie aliquote de la L-glutamine pour chaque expérience.
B27
Des aliquotes de B27 peuvent être conservés à 20 ° C pour le stockage à long terme, ou à 4 ° C jusqu'à un mois.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Nous avons décrit une méthode de disséquer et de la culture des neurones commissuraux de la moelle épinière de rats embryonnaires. Cette procédure a été couramment utilisée dans notre laboratoire pour préparer les neurones de manière fiable pour étudier les mécanismes cellulaires et moléculaires du guidage axonal. Pour la biologie cellulaire et des expériences d'immunochimie, la dissection d'une litière suffisante produit des neurones. Lorsque plusieurs cellules sont nécessaires, comme dans de nombreuses expériences de biochimie, la dissection des deux portées peut être nécessaire. Pour une personne formée, la dissection et la dissociation de ~ 20 embryons peuvent être réalisées en moins de 4 heures. Plus longues périodes se traduira par une dissection plus difficiles de la moelle épinière en raison de changements dans l'intégrité des tissus, et peut également compromettre la récupération efficace des neurones viables.
Lorsque vous effectuez la procédure pour la première fois, les cellules plaque sur diverses surfaces enduites PLL (lamelles de verre lavée à l'acide, des boîtes de culture tissulaire) pour la comparaison. Normalement, les cellules devraient attacher solidement à la PLL-vaisselle en plastique recouvert de culture de tissus ou de plaques multi-puits. Ceci peut être utilisé comme un contrôle général de la viabilité cellulaire et de l'entretien s'il ya des problèmes avec l'adhérence cellulaire et la croissance sur des lamelles de verre. Le principal facteur responsable de la mauvaise adhérence des cellules au verre est la qualité du nettoyage de verre et lamelle (acide de lavage et de stérilisation). Cela va affecter l'adhérence cellulaire en partie par la réduction de l'efficacité PLL-revêtement. Autres facteurs communs comprennent mauvaise PLL revêtement (revêtement de temps trop court ou trop vieux solution de PLL), la contamination bactérienne ou fongique, ou l'utilisation des médias ou des suppléments qui sont vieux ou périmés.
Nous avons utilisé des cultures de neurones commissuraux préparés selon cette procédure en plusieurs type d'expériences, y compris l'immunochimie, biochimie, et des essais de tournage (Yam et al. 2009). Remarquablement, les neurones commissuraux plaqué sur PLL verre enduit de conserver la capacité de répondre aux signaux de guidage, c'est à dire, ils vont changer leur sens de la croissance en réponse à un facteur chemotropic appliquées, telles que Sonic Hedgehog, précédemment révélée être un signal de guidage axonal ( Charron et al, 2003; Okada et al, 2006; Yam et al 2009; Fabre et al, 2010).. Par conséquent, c'est un système puissant pour étudier l'effet des signaux de guidage axonal in vitro et permet à des expériences qui ne sont pas possibles in vivo.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Ce travail a été soutenu par des subventions des Instituts canadiens de recherche en santé du Canada (IRSC), le chercheur Peter Lougheed Medical Research Foundation, le Programme de McGill en neuroingénierie, le Fonds de Recherche en Santé du Québec (FRSQ) et la Fondation canadienne pour l'innovation (FCI ). Sébastien D. Langlois a été soutenue par de bourse de formation à la maîtrise de la Fonds de la recherche en santé du Québec (FRSQ) et par une Frederick Banting et Charles Prix du Meilleur études supérieures du Canada Bourses de maîtrise des Instituts de recherche en santé du Canada (IRSC). Nous sommes reconnaissants à Jessica Pham MT à l'aide de chiffres.
Materials
| Name | Company | Catalog Number | Comments |
| Neurobasal medium, liquid | Invitrogen | 21103-049 | See Recipes and Comments |
| B27 supplement 50X | Invitrogen | 17504 | See Recipes and Comments |
| Poly-L-lysine 0.01% solution | Sigma-Aldrich | P4707 | |
| L-15 medium, powder | Invitrogen | 41300-070 | |
| Trypsin 2.5% (10X) | Invitrogen | 15090-046 | |
| DNAse I, 25000 U/mL | Worthington Biochemical | ||
| MgSO4 | Sigma-Aldrich | M2643 | |
| HBSS, Ca2+/Mg2+-free | Invitrogen | 14170-112 | |
| L-glutamine 200mM, liquid | Invitrogen | 25030-081 | See Recipes and Comments |
Dissection of embryonic rat dorsal spinal cord (see also Table I)
Commissural neuron culture (see also Table I)
|
|||
References
- Charron, F., Stein, E., Jeong, J., McMahon, A. P., Tessier-Lavigne, M. The morphogen sonic hedgehog is an axonal chemoattractant that collaborates with netrin-1 in midline axon guidance. Cell. , 113-1111 (2003).
- Fabre, P., Shimogori, T., Charron, F. Segregation of ipsilateral retinal ganglion cell axons at the optic chiasm requires the Shh Receptor Boc. Journal of Neuroscience. 30, 266-275 (2010).
- Helms, A. W., Johnson, J. E. Progenitors of dorsal commissural interneurons are defined by MATH1 expression. Development. 125, 919-928 (1998).
- Kaech, S., Banker, G. Culturing hippocampal neurons. Nature Protocols. 1, 2406-2415 (2006).
- Okada, A., Charron, F., Morin, S., Shin, D. S., Wong, K., Fabre, P. J., Tessier-Lavigne, M., McConnell, S. K. Boc is a receptor for sonic hedgehog in the guidance of commissural axons. Nature. 444, 369-373 (2006).
- Yam, P. T., Langlois, S. D., Morin, S., Charron, F. Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron. 62, 349-362 (2009).

{kind=link}