

Summary
この作品は、染色体レベルでのRNA / DNAハイブリッドを形成し、酵母細胞内でRNAからゲノムDNAに遺伝情報の伝達を明らかにする方法を示しています。
Abstract
合成短い核酸ポリマー、オリゴヌクレオチド(オリゴ)は、分子生物学の最も機能的かつ広範なツールです。オリゴは、任意の所望のDNAまたはRNA配列を含むように作製することができると、ベースと糖の修飾のさまざまなを含むように調製することができる。また、オリゴは、特定の核酸の変化を模倣するため、DNA損傷と修復のメカニズムの効果を調べるために重要なツールとして使用できるように設計することができます。我々は、50〜80ヌクレオチドの間の長さと熱科学Dharmacon RNAを含むオリゴは、染色体RNA / DNAハイブリッドの in vivo、機能や結果でとDNAに埋め込 まれたリボヌクレオチドの、勉強するのに特に適していることが分かった。 RNA / DNAハイブリッドが容易にDNA複製、修復と転写の間に形成することができる、しかし、非常に小さな細胞のRNA / DNAハイブリッドの安定性についてはほとんど知られており、どの程度これらの雑種は、細胞の遺伝的整合性に影響を及ぼす可能性がする。 RNAを含むオリゴは、従って、染色体DNAへのリボヌクレオチドを導入し、選択した長さと塩基組成のRNA / DNAハイブリッドを生成するのに最適なベクトルを表します。ここでは、真核生物のモデル系の酵母/ サッカロマイセスセレビシエ /のゲノム中へのリボヌクレオチドの取り込みのためのプロトコルを提示する。まだ、私たちの研究室が利用されているサーモ科学Dharmacon RNAを含むオリゴは、細菌からヒトの細胞に、種々の細胞系の染色体レベルでのRNA / DNAハイブリッドを生成する。
Protocol
論理的根拠
80量体のサーモ科学Dharmaconオリゴ、50の使用を悪用し、ここでは、我々は、オリゴが標的染色体DNAにアニーリング以下、酵母細胞のゲノムDNAに遺伝情報を転送できることによって、遺伝子補正アッセイに基づいて、手順を示します。オリゴが標的の成功は予想される表現型を表示する酵母のコロニーの出現によって得点が記録されます。所望の遺伝子改変はオリゴ配列に組み込まつまたは複数のヌクレオチドの中に運ばれている場合、遺伝情報は、ターゲットプロセス中に染色体レベルでは、このように、染色体の標的DNAへのRNAの消化管から直接RNA / DNAハイブリッド形態を流れる。サーモ科学Dharmacon RNA含有オリゴリボヌクレオチド/秒は、DNA修復合成を介して遺伝子ターゲティングのためのテンプレートとして機能することができますまたはDNAに埋め込むことができ、DNA複製中にテンプレートとして機能する。これらの実験では、酵母/ 出芽酵母 /真核生物のモデル系を使用しますが、同様のアプローチは、他の生物や細胞型にも応用することができます。このビデオでは、我々はターゲットの酵母のゲノムの遺伝子座に相同で設計され、標的遺伝子の変異を修正するには真中の1リボヌクレオチドを含むサーモ科学Dharmaconオリゴを使用してください。 RNAを含むオリゴによる形質転換後、我々は、オリゴのRNA管が染色体DNAに組み込まれ、DNA複製時のDNA合成のテンプレートとして機能することができることを示す特定の周波数で標的部位の補正を観察。 RNA気道内に運ば遺伝情報を安定的に次のセルの世代に伝えられる。ここでは、染色体DNA中に転送するRNAの情報を検出するためにサーモ科学Dharmacon RNA含有オリゴとアプローチによって、酵母細胞の形質転換の手順を説明します。
この実験で使用されているRNAを含むオリゴのA.デザイン
我々は、酵母の遺伝的欠陥を修正するRNAを含むオリゴを設計しているS. trp5遺伝子座での出芽酵母染色体DNA。プロトコルで使用されている酵母菌株は、2塩基欠失一ナンセンス変異(図1A)を持つ変異体trp5遺伝子が含まれています。このような酵母株はトリプトファン要求性変異体であるとトリプトファンせずにメディア上でコロニーを形成しない。 TRP5遺伝子のDNA配列は、サッカロミセス属ゲノムデータベース(http://www.yeastgenome.org/~~V)で入手可能です。このプロトコルで使用されているサーモ科学Dharmacon RNA含有オリゴは、欠失変異とtrp5対立遺伝子(図1A)のナンセンス変異を修正するために設計された1つリボヌクレオチドを修正するために設計された2塩基のDNAの挿入、は65 - merのです。オリゴのシーケンスは次のように設計されています。
5' - AAAAGGGTTTTGATGAAGCTGTCG - CG - GATCCCACATTCTG - RG - GAAGACTTCAAATCCTTGTATTCT。このオリゴは、200nmのスケールでサーモ科学Dharmacon(ラファイエット、CO)により合成され、そして脱塩され、脱保護し、PAGE精製せずに使用。
酵母のトランスフォーメーションのためのRNAを含むオリゴ(Storici らから変更。、2007年)のB.の準備
- すべての開始前に潜在的なRNaseのコンタミを除去するためのRNase除染液と捜査官が着用しているオリゴチューブ、ピペット、渦、ラック、実験エリアと手袋などの実験に使用される材料を拭きます。すべてのステップでRNaseフリー水、化学試薬、チューブやピペットチップを使用してください。この実験ではすべてのステップでは、RNaseフリーにする必要があります。
- サーモ科学Dharmacon RNA含有オリゴは、会社から250ピコモル/ RNaseフリー水とペレットを溶解するために精力的に渦を持つμlのストック溶液に、受信した懸濁します。 -80℃で保存する
- すぐに変換する前に、氷上でRNAを含むオリゴを解凍し、RNaseフリーのチューブのRNaseフリー水を50ピコモル/μlに希釈する。各変換には、RNAを含むオリゴの1ナノモルが必要です。
- オリゴの二次構造を除去するために2分間100℃のヒートブロック上のRNAを含むオリゴの選択された量の変性。
- すぐに変性した後、チューブを氷上に置きます。変換まで氷上で保管してください。
- 実験のコントロールとして対応するDNAは、専用のオリゴが使用されます。このオリゴは、上記のように、-20℃から融解し、RNAを含むオリゴなどの変換用に準備されています。
RNA含有オリゴを用いて酵母のC.変換
- trp5変異酵母細胞と豊富なYPD液体培地5mlに接種し、30℃で一晩(参照物質)で成長する。
- YPD液体培地50mlに一晩培養液1.5 mlを。
- 4時間、30℃でシェーカー(225 rpmの)で細胞をインキュベート。
- 解決策1と解決策2 IMMを準備ediately RNaseフリーチューブの変換の前に(資料を参照してください)。
- 50ミリリットルのRNaseフリーのチューブと2分間1562グラムに相当する3000回転、、でスピンして細胞培養を転送します。細胞の降水量のペレットは約です。 0.3センチメートル3。
- 上清を除去し、2分間3000rpmでRNaseフリー水とスピンの50mlで細胞を洗浄。
- 可能な限り培地に存在する可能性がある培地とのRNaseを取り除くために5回ごとに手順6を繰り返します。
- 解決策1と2分間3000rpmでスピン5mlに上清と再懸セルを削除します。
- ソリューション1の250μlの上清と再懸セルを削除します。細胞のこの量は7-8に変換するのに十分です。
- RNaseフリーのマイクロチューブに細胞懸濁液のアリコート50μlを、RNAを含むオリゴワーキング溶液(1ナノモル)、またはDNAのみのオリゴワーキング溶液(1ナノモル)、または滅菌水20μlを20μl20μlを追加ネガティブコントロールのための無オリゴある。次に、各形質転換反応のために解決策2を300μlを加える。キャリア自体とオリゴの行為として、変換の過程でサケ精子DNAを追加する必要はありません。
- 均質成分を混合する激しくボルテックス。
- 30℃シェーカーで30分間の変換反応をインキュベートする。
- 42℃のヒートショック℃で15分間。
- 4分間2236グラムに相当する5000回転、、で細胞をスピンダウン。
- 滅菌水100μlの上清と再懸セルを削除します。
- この細胞懸濁液のアリコートを取ると10万倍と約を使用して1つのYPDプレート上にプレートの細胞によって滅菌水で希釈する。 30で15滅菌ガラスビーズとインキュベート℃で2日間C。
- プレート約を使用してトリプトファン(SC - TRP)のない合成完全な固体培地のいずれかのペトリ皿上の各形質転換反応液からすべて懸濁している細胞。 30で15滅菌ガラスビーズとインキュベートします4-5日のためのC(図2)。
RNA含有オリゴによる遺伝子補正のD.解析
- RNAを含むオリゴ、DNAのみのオリゴおよびno -オリゴ制御のための遺伝子の補正周波数を計算するために選択培地(図2)だけでなく、YPD培地上に生育したコロニーの数を数えます。得られた数値を比較します。 2塩基の欠失やナンセンス変異をもつtrp5対立遺伝子の自然復帰率は、このように我々はオリゴを細胞に添加されていない選択培地上にコロニーの形成を期待していない、使用する酵母菌株の10月9日よりも小さいです。
- いくつか出ストリークは、無作為にシングルコロニーの分離株を取得するYPD培地に形質転換体のコロニーをピックアップ。コロニーの成長のための二日を待って、その後いくつかの(少なくとも5)の単一コロニーを取り、YPD上と選択培地上でパッチを作る。
- コロニーPCR(図1B)によってRNAを含むオリゴの標的領域(250-1,000 bp)を増幅するプライマー対を設計します。コロニーPCRのための手順は、(Storiciとレズニック、2006 2から変更)、次のとおりです。
- 再懸濁し、細胞(約1立方ミリメートルは)水50μlの個々のパッチから取得しlyticaseの1単位を追加します。 100℃ヒートブロックでインキュベーションに続いて10分、°溶液中の細胞とリリースゲノムDNAを切断する5分間室温でインキュベートする。
- PCR条件:PCR反応は、細胞懸濁液10μlを含む、50ピコモルフォワードプライマーとリバースプライマーの各々は、10mMのdNTPを、Taqポリメラーゼの1単位、10倍のバッファとの5μlを1μl、滅菌水で調整されます50μlの最終容積。 95℃30秒の30サイクル° C、55℃30秒° C、および72℃で1分間° C; 72℃で7分間の最終伸長時間は℃、その後のサンプルのPCRプログラムは95℃で3分間です。 4℃で保存されています1分/ kbの延長時間は、この反応のために仮定されます。
- PCRに続いて、サンプルはPCR産物(図3)の観察のために1%または2%(PCR産物の予想サイズに応じて)アガロースゲル上で実行されています。
- 遺伝情報が転送される場合、RNA含有オリゴは、酵母のゲノムのターゲット領域での新たな制限部位(図1B)を生成することによって、それは適切な制限酵素でPCR産物を消化して情報の正しい伝達を確認することが可能です。制限部位は、RNAを含むオリゴによって生成されていない場合は、手順6に進みます。特定の制限酵素を使用してダイジェストPCR産物。消化反応は15μlに6 PCR産物の液、バッファー、BSA(いくつかの酵素のために必要とされていない可能性があります、使用する酵素のための命令を参照)、制限酵素0.5μlの、および滅菌水を含まれています。サンプルは、使用される酵素の特定の温度で1時間インキュベートする。
- bを転送した遺伝子改変を観察するために、2%アガロースゲルの同じ行上に消化サンプルと一緒に未消化サンプルを実行するRNA含有オリゴのy RNA路(図3)。
- PCR精製キットを用いてPCR産物を精製し、DNAシーケンシングのためにそれらを準備する。製品を増幅するために使用したものと同じプライマーでシークエンシング用サンプルを提出する。
- 選択された参照配列(図4)と複数の配列のアライメントを可能にするソフトウェアを使用してDNAシーケンシングの結果を分析する。
RNA含有オリゴのE.アルカリ処理(図5)
- 各反応について、1.5 mlチューブにRNA含有オリゴ、またはDNA -オリゴの1ナノモル(250ピコモル/μLストック溶液4μl)を添加。
- 加水分解のために1 M NaOHを4μlのを追加し、あるいは1時間、ネガティブコントロールとして、4μlのH 2 Oを追加し、水浴で65℃でインキュベートするその後、水浴から氷へ移動する。
- 1.2MのHClを、H 2 Oの1 Mトリス-塩酸、4μlの4μlの、あるいはH 2 Oの6μlのとネガティブコントロール用の1 Mトリス- HClを4μlの2μlので中和する。変換まで氷上で保管してください。

図1。欠陥trp5遺伝子のおよびRNAを含むオリゴで補正TRP5対立遺伝子の模式図。)trp5変異遺伝子は、2塩基欠失(黒い三角形)と1ベースのナンセンス変異を(アスタリスク)が含まれています。一本鎖RNAを含む2塩基挿入(青ループ)とVan91 I制限酵素部位を(ブラケットによって示される)を生成する1 - RNAの塩基置換(赤の矩形)とオリゴは、遺伝的欠陥を修正するための酵母細胞に形質転換されtrp5遺伝子の。 TRP5遺伝子はRNA含有オリゴ(訂正拠点が青い矩形で示されている)によって修復された後にB)、Van91私のサイトはTRP5遺伝子に生成されます。 TRP5遺伝子で唯一Van91 I制限部位を含む278 bpの断片は、プライマー(P1とP2)のペアによって増幅されたPCRです。 Van91でP1とP2のPCR産物の消化後に生成された177 bpと101 bpの断片を、私も表示されます。

図2。 RNAを含むオリゴ型の変換結果。)無オリゴで形質転換した酵母細胞は、SC - TRPの培地上の任意のコロニーを形成しない、プレートABを参照してください。 B)細胞の後にSC - TRPに成長しているプレートCGショーの酵母のコロニーは、RNAを含むオリゴの1ナノモルで形質転換されています。 C)のセルの後にSC - TRPに成長しているプレートHLショーの酵母のコロニーは、対応するDNAのみのコントロールオリゴの1ナノモルで形質転換されています。

図3。からの遺伝情報の伝達RNAを含む標的ゲノム領域を増幅するPCR産物の制限消化により酵母染色体DNAにオリゴをの検出。PCRサンプルの2%アガロースゲル電気泳動P1とP2によって増幅し、Van91 I制限酵素で消化。レーン1、8、15、100のサイズは、200、300、400と左に表示されている500塩基のDNAラダー、レーン2、trp5変異株のゲノムDNAから増幅さtrp5遺伝子座のPCR産物、レーン3、PCRレーン4-7、RNAを含むオリゴの標的のTrp +コロニーに由来するゲノムDNAから増幅したPCR産物;レーン9〜14、Van91 I制限消化DNAのみのオリゴがターゲットとのTrp +コロニーに由来するゲノムDNAから増幅産物レーン2から7までのPCR産物の。レーン10〜14でカットされていないPCR産物のバンドの存在はTRP5軌跡(CCACATTCTGG)でVan91 Iで部分消化によって説明することができます。 Van91私(CCANNNN NTGG)のための切断部位は、複数のシーケンスを持つことができることを考慮すると、TRP5で生成されたサイトは、酵素のための最適なターゲットになる場合があります。我々はオリゴによって運ばれたもの以外の追加変更を検出しないすべての上記のPCR産物のDNA配列に続く、実際には(図4を参照)。 Van91でP1とP2プライマーと消化の産物のバンドが増幅された278 bpのPCRバンドは177 bpおよび101 bpの私は、右上の矢印で示されています。

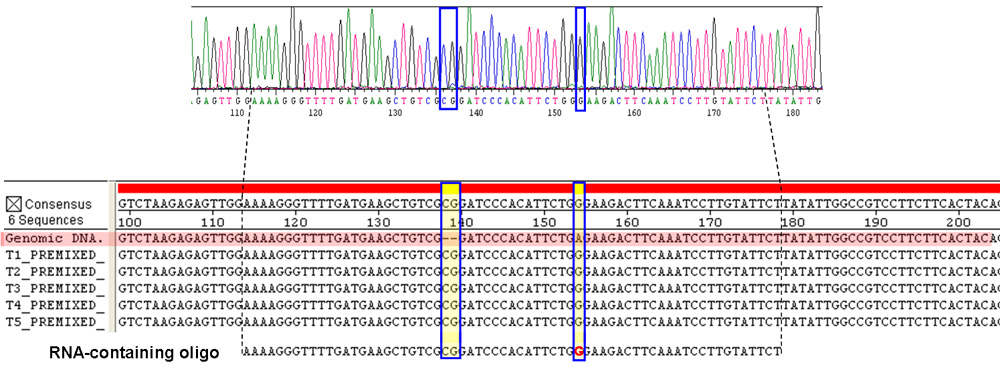
図4。 RNAを含むオリゴによる遺伝子の補正を示すDNA塩基配列決定の結果。)RNAを含むオリゴの標的ゲノム領域のDNAの電気泳動図。 DNA配列(箱入り青)のGは、RNA含有オリゴでRGに由来する。また、箱入りのCG拠点の挿入です。他のすべてのPCR産物からシークエンシングの結果は、よくバックグラウンドを超える蛍光シグナルを、また、これと同等明らかです。 B)のTrp + transfoのTRP5領域の配列は、DNAのみのオリゴ(T1)およびRNAを含むオリゴ(T2 - T5)の上にコンセンサス配列に一致するとの標的rmantsは(ゲノムDNA、赤オリゴが標的とする前にtrp5変異細胞のそれと比較されています網掛け)。底の修理RNAを含むオリゴは2つの塩基のDNAの挿入と赤でマークされた一塩基(G)のRNA置換されています。だけでなく、RNAを含むオリゴその黄色の色調のショーと青の枠で囲ま領域はDNAのみのオリゴは、正確にすべての試験した試料の欠失変異やナンセンス変異を修正。破線は、RNAを含むオリゴ配列の位置をマーク。してくださいここをクリックして図4の拡大バージョンを参照すること。

図5。アルカリ処理は、RNAを含むオリゴによる遺伝子の補正を防ぎます。RNA含有オリゴ(R)で形質転換頻度は赤いバーでとDNAのみのオリゴ(D)で、青色のバーに表示されていることが示されている。エラーバーは各オリゴのための3つの独立した変換のための平均の標準誤差を表しています。 DNAのみのオリゴはなしとNaOH処理と同様の形質転換頻度が表示されます。異なって、RNA含有オリゴによる形質転換頻度は、NaOHで0次の治療に低下します。したがって、RNA含有オリゴと準備がDNAのみのオリゴで汚染されていない、したがって、観察された形質転換頻度は、RNAを含むオリゴに固有のものです。
Discussion
RNAは細胞内でゲノムDNAに直接遺伝情報を転送することができるという事実は、合成RNAを含むオリゴ(Dharmacon合成オリゴ)1の使用を悪用発見された。それは、細胞がDNA合成の鋳型としてRNAを含む分子またはRNAのみのシーケンスを使用することができる原理の証明だ。だけでなく、遺伝情報が逆転写されたDNAのコピーの中間の必要なしにゲノムDNAにRNAから直接転送することができますデモにつながったRNAを含むオリゴの使用だけでなく、RNAが相同テンプレートとしてで使用できることを証明するDNA損傷1,3の修復。我々はここで示した例のようにオリゴは、RNAのみで作らまたはDNA配列に埋め込まれているだけで、単一のリボヌクレオチドを含めることができます。 RNA含有オリゴは、ゲノムの不良品trp5対立遺伝子にナンセンス変異を修正するために設計された1つの埋め込 みリボヌクレオチドを持っています。リボヌクレオチドからゲノムDNAの遺伝情報の伝達は、標的細胞の表現型の変化(細胞がトリプトファンを欠く培地上に生育している)によって明らかにされる。 RNA含有オリゴで得られた遺伝子の補正の周波数を測定し、コントロールDNAのみのオリゴのそれと比較されます。我々は様々な酵母の遺伝子を変異させ、異なるセルの背景、この遺伝子の補正アッセイを行うことにより、我々は、特にRNAを含むオリゴが標的に影響する要因/ sで検出することができます。従って、我々は細胞がRNA / DNAハイブリッドの安定性を制御する方法のメカニズムを明らかにすることができます。単にRNAを含むオリゴの異なる変種を設計することで我々は、DNA修飾でテンプレートとして機能する特定のRNAの消化管に対する尤度を決定することができますし、我々はDNAに組み込まれた特定のRNA配列の安定性を決定することができます。また、我々は、RNA / DNAハイブリッドと相互作用する因子の生体内基質に好まれるかを識別することができます。
主に短期利用、in vitro試験のいくつかのRNAを含むオリゴは、同様のRNase Hの生体機能に 、RNアーゼHの酵素4として、DNAとのハイブリッドのRNAを認識することができる要因の機能を特徴づけるために実施されているが、 RNA / DNAハイブリッド安定性はほとんど未知のままに影響を与えることができる他のタンパク質の同一性として。かなりの長さ(50〜80量体)と、最適な品質のRNAを含むオリゴを利用する可能性が(そのようなサーモ科学DharmaconオリゴRNAを含有など)は直接の in vivo での分子プロセスの広い範囲を調べる方法を開いています目的の細胞。このビデオに示すように、RNAを含むオリゴを使用した変換は、いくつかの追加手順がRNaseにによる劣化を防ぐために、DNA分子を用いて変換に比べて本質的に必要になります。このように、RNAを含むオリゴを使用して変換が酵母のシステムに限定されるものではなく、DNAオリゴによる変換が堪能である任意の生物や細胞のタイプに適用することができます。
結論では、RNAを含むオリゴで細胞を標的とする細胞にin vivoで DNAに埋め込 まれているRNA / DNAハイブリッドとリボヌクレオチドを生成するための機会を提供しています。 in vivoでの安定性、機能とこれらの結果は、RNAを生成/ DNAハイブリッドは、潜在的にDNA修復の未知のメカニズムを明らかにして標的遺伝子のために新しい戦略を明らかに、分析し、特徴づけることができる。
Disclosures
この記事の映像制作は、このマニュアルで使用する試薬や器具を生産するサーモフィッシャーサイエンティフィック、後援した。
Acknowledgments
この作品は、グルジアがん連合助成金- R9028によってサポートされていました。
Materials
A. Transformation reagents and media (modified from Storici and Resnick, 2006 |
|||
|
|||
B. Colony PCR materials. |
|||
|
|||
C. PCR purification. |
|||
|
|||
D. Gel Electrophoresis. |
|||
|
|||
E. Restriction digestion. |
|||
|
|||
F. Alkali treatment for the RNA-containing oligo. |
|||
|
References
- Storici, F., Bebenek, K., Kunkel, T. A., Gordenin, D. A., Resnick, M. A. RNA-templated DNA repair. Nature. 447, 338-3341 (2007).
- Storici, F., Resnick, M. The delitto perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol. 409, 329-345 (2006).
- Storici, F. RNA-mediated DNA modifications and RNA-templated DNA repair. Curr Opin Mol Ther. 10, 224-230 (2008).
- Cerritelli, S. M., Crouch, R. J. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 276, 1494-1505 (2009).

{kind=link}