

Summary
ويرد وصف المبادئ التوجيهية للكمبيوتر يستند توصيف الهيكلية والوظيفية للبروتين باستخدام خط أنابيب I - TASSER. بدءا من تسلسل البروتين الاستعلام ، يتم إنشاؤها باستخدام نماذج 3D التحالفات خيوط متعددة ومتكررة المحاكاة التجمع الهيكلي. بعد ذلك يتم رسمها بناء على الاستدلالات وظيفية للبروتينات مباريات مع هيكل معروف ووظائفها.
Abstract
وقد مشفر مشاريع تسلسل الجينوم الملايين من تسلسل البروتين ، والتي تتطلب معرفة هيكلها وظيفة لتحسين فهم دورها البيولوجي. على الرغم من الأساليب التجريبية يمكن أن توفر معلومات مفصلة عن جزء صغير من هذه البروتينات ، لا بد من النمذجة الحاسوبية لمعظم جزيئات البروتينات التي uncharacterized تجريبيا. خادم I - TASSER هو طاولة العمل على الخط لصالح عالية الدقة نمذجة بنية البروتين ووظيفته. تسلسل معين من البروتين ، وإخراج نموذجية من خادم I - TASSER يتضمن بنية التنبؤ الثانوية ، وتوقع الوصول المذيبات من كل رواسب والبروتينات قالب مثلي الكشف عنها بواسطة خيوط والتحالفات الهيكل ، ما يصل الى خمسة كامل طول نماذج هيكلية التعليم العالي ، وهيكل قائم على شروحه للتصنيف الوظيفي الانزيم ، حيث أنتولوجيا الجينات والبروتينات ويجند مواقع الربط. يتم تمييزها عن التوقعات بنتيجة الثقة التييروي مدى دقة التوقعات من دون الاطلاع على البيانات التجريبية. لتسهيل الطلبات الخاصة للمستخدمين النهائيين ، والخادم توفر قنوات لقبول المحددة من قبل المستخدم بين بقايا المسافة والاتصال لتغيير الخرائط بشكل تفاعلي النمذجة I - TASSER ، بل يسمح أيضا للمستخدمين لتحديد أي البروتينات والقالب ، أو لاستبعاد أي قالب البروتينات خلال محاكاة هيكل التجمع. يمكن جمع المعلومات الهيكلية من قبل المستخدمين على أساس الأدلة التجريبية أو رؤى البيولوجية بهدف تحسين نوعية I - TASSER التنبؤات. تم تقييم الملقم كما أفضل البرامج لبنية البروتين والتنبؤات وظيفة في التجارب الأخيرة CASP المجتمع ككل. هناك حاليا> 20000 مسجل من العلماء أكثر من 100 دولة الذين يستخدمون على الخط I - TASSER الخادم.
Protocol
نظرة عامة على الأسلوب
النموذج التالي تسلسل إلى بنية إلى وظيفة ، الإجراء I - TASSER 1-4 لهيكل والنمذجة الدالة ينطوي على أربع خطوات متتالية : (أ) تحديد قالب LOMETS 5 ، (ب) إعادة تجميع الهيكل جزء بواسطة متماثلة ، تبادل مونتي كارلو المحاكاة 6 ، (ج) هيكل المستوى الذري الصقل باستخدام REMO 7 و MD - FG 8 ، و (د) تفسيرات وظيفة الهيكل القائم على استخدام العامل المساعد 9.
تحديد قالب : للحصول على تسلسل الاستعلام المقدمة من قبل المستخدم ، هي الخيوط الأولى التي تتابع من خلال مكتبة هيكل PDB ممثل من قبل LOMETS مثبتة محليا التلوي الترابط الملقم. الترابط هو إجراء المحاذاة تسلسل بنية استخدامها لتحديد البروتينات التي قد يكون لها قالب بنية مشابهة أو تحتوي على هيكلية مماثلة لعزر البروتين الاستعلام. لزيادة تغطية templ مثليأكلت المكتشفة ، LOMETS يجمع متعددة للدولة من بين أحدث خوارزميات تغطي خيوط منهجيات مختلفة. منذ خيوط برامج مختلفة لديها أنظمة مختلفة الدرجات والحساسيات والمحاذاة ، ويتم تقييم نوعية التحالفات والترابط المتولدة من كل برنامج خيوط بواسطة تطبيع Z - درجة ، والتي يتم تعريفها على النحو التالي : 
حيث Z - النتيجة هي النتيجة في وحدات الانحراف المعياري النسبي لمتوسط الإحصائية لجميع التحالفات التي تم إنشاؤها بواسطة البرنامج ، و0 Z هو برنامج محدد Z - درجة قطع مصممة على أساس اختبارات واسعة النطاق مؤشر الترابط من 5 إلى التفريق "جيدة 'و' سيئة 'القوالب. قالب برصيد - Z عالية يعني أن يكون لها قوالب أعلى درجة محاذاة أعلى بكثير من معظم القوالب الأخرى ، والتي عادة ما يعني أن محاذاة يناظر نموذجا جيدا. إذا كان معظم القوالب والترابط الأعلى مرحباغ تطبيع Z - العشرات ، ودقة من نموذج I - TASSER النهائية هي عادة ما تكون مرتفعة. ومع ذلك ، إذا كان البروتين هو كبير ويقتصر على تغطية خيوط التحالفات على منطقة صغيرة من البروتين الاستعلام ، وارتفاع درجة تطبيع Z - لا يعني بالضرورة دقة النمذجة عالية لنموذج كامل طول. يتم جمع خيوط التحالفات اثنين من كبار من كل برنامج والترابط المستخدمة في الخطوة التالية من تجميع الهيكل.
تكرارية محاكاة هيكل التجمع : بعد إجراء الترابط ، يتم تقسيم الاستعلام إلى تسلسل خيوط الانحياز والمناطق الصغيرة المحايدة. يتم استئصال شظايا المستمر في محاذاة خيوط من القوالب واستخدامها مباشرة لتجميع الهيكل ، في حين يتم بناؤها في المناطق الصغيرة المحايدة حلقة عبر الاقتداء أساسه. يتم تنفيذ الإجراء تجميع الهيكل على نظام شعرية تسترشد تبادل محاكاة مونتي كارلو 6 متماثلة. مجال القوة I - TASSER يشمل الهيدروجين بوnding التفاعلات 10 ، القائم على المعرفة حيث الطاقة المستمدة من الهياكل الإحصائية البروتين المعروف في PDB 11 ، تسلسل التنبؤات المستندة إلى اتصال من SVMSEQ 12 ، والقيود المكانية التي تم جمعها من 5 قوالب LOMETS الترابط. تتجمع في أشراكا متعلق بتكوين ولدت في النسخ المتماثلة درجات الحرارة المنخفضة خلال المحاكاة بواسطة SPICKER 13 إلى التعرف على هياكل الدول الحرة انخفاض الطاقة. ويتم الحصول على centroids مجموعة من المجموعات التي بلغ متوسطها الأعلى إحداثيات 3D أشراكا من جميع المسافات والهيكلية المستخدمة لتوليد النموذج النهائي. يتم تكرار المحاكاة وإجراء التجميع مرتين لإزالة اشتباكات الفراغية وزيادة تحسين البنية العالمية.
المستوى الذري نموذج البناء والصقل : يتم تخفيض centroids العنقودية التي تم الحصول عليها بعد تجميع SPICKER نماذج البروتين (كل المخلفات التي يمثلها C α وسلسلة جانبية مركز الكتلة) و حافي محدودة التطبيق البيولوجي. ويتم بناء نموذج كامل ذرية من نماذج انخفاض في خطوتين. في الخطوة الأولى ، يتم استخدام REMO 7 إلى بناء كامل الذرية نماذج من طراز C - ألفا آثار عن طريق تحسين شبكات H - السندات. في الخطوة الثانية ، هي مزيد من الصقل REMO كامل ذرية من النماذج من تصميم MD - FG 14 ، مما يحسن من زوايا التواء العمود الفقري ، وأطوال السندات ، والتوجهات rotamer الجانب السلسلة ، من خلال المحاكاة الحيوية الجزيئية ، على نحو يستهدي شظايا هيكلية البحث عن هياكل PDB بواسطة TM - محاذاتها. وتستخدم النماذج MD - FG المكرر والنماذج النهائية للتنبؤات بنية التعليم العالي من خلال I - TASSER.
وتقدر نوعية النماذج ولدت على أساس درجة الثقة (C - درجة) ، والتي يتم تعريفها على أساس Z - درجة من الترابط والتحالفات LOMETS تقارب I - TASSER المحاكاة ، التي صيغت رياضيا على النحو التالي : 
حيث
سي النتيجة لديه علاقة قوية مع نوعية النماذج I - TASSER. من خلال الجمع بين C - درجة وطول البروتين ، يمكن تقدير دقة النماذج I - TASSER الأولى مع وجود خطأ في المتوسط 0.08 لدرجة TM - 2 و 15 ألف لRMSD. بشكل عام ، والنماذج مع درجة C -> -- من المتوقع أن يكون 1.5 أضعاف الصحيح. هنا ، وRMSD TM - درجة كلاهما تدابير معروفة طوبولوجي التشابه بين النموذج والهيكل الأصلي. TM - درجة valuوفاق النطاق في [1 ، 0] ، حيث يشير إلى ارتفاع درجة هيكل أفضل مباراة 16،17. ولكن لأقل مرتبة النماذج (أي 2 الموديلات الثانية عشرة -5) ، ودرجة الترابط بين - C - TM مع درجة وRMSD أضعف بكثير (~ 0.5) ، والتي لا يمكن استخدامها لتقدير موثوق به للجودة نموذج مطلقة.
النموذج الأول هو النموذج الأفضل دائما في I - TASSER المحاكاة؟ الجواب على هذا السؤال يتوقف على نوع الهدف. لأهداف سهلة ، والنموذج الأول هو النموذج الأفضل عادة والخمسين درجة مئوية ، عادة ما يكون أعلى بكثير من بقية النماذج. ومع ذلك ، من أجل الأهداف الصعبة ، حيث لا يملك خيوط يضرب قالب كبير ، والنموذج الأول ليس بالضرورة هو النموذج الأفضل وI - TASSER بالفعل صعوبة في اختيار أفضل قالب والنماذج. ينصح بالتالي لتحليل جميع نماذج (5) لأهداف صلبة وحدد لهم استنادا إلى المعلومات التجريبية والمعرفة البيولوجية.
PRED الدالةictions : في الخطوة الأخيرة ، وتستخدم النهائي 3D - النماذج المولدة من FG - MD التنبؤ ثلاثة جوانب وظيفة البروتين ، وهما : أ) لجنة الإنزيمات (EC) أرقام 18 و (ب) أنتولوجيا الجينات (GO) 19 شروط و( ج) مواقع ملزمة ليغاندس جزيء صغير. لجميع الجوانب الثلاثة ، ويتم إنشاؤها باستخدام العامل المساعد التفسيرات الوظيفية ، وهو نهج جديد للتنبؤ وظيفة البروتين على أساس التشابه العالمية والمحلية للبروتينات في قالب PDB مع هيكل معروف ووظائفها. أولا ، يتم مطابقة الطبولوجيا من النماذج العالمية وتوقع ضد المكتبات قالب الوظيفية باستخدام برنامج المواءمة الهيكلية TM - محاذاة 20. المقبل ، ويتم اختيار مجموعة من البروتينات الأكثر مشابهة للنماذج الهدف من المكتبة على أساس تشابهها الهيكل العالمي ، ويتم تنفيذ عملية بحث واسعة النطاق المحلي لتحديد هيكل وتسلسل التشابه موقع بالقرب من منطقة نشطة / ملزمة. وتستخدم عشرات التشابه الناتجة العالمية والمحلية إلى رتبةالبروتينات القالب (المتماثلات وظيفية) ونقل هذا الشرح (أرقام المفوضية الأوروبية والوجود حيث جين 19) استنادا إلى مشاهدات سجل أعلى. وبالمثل ، فإن الاستدلال على بقايا يجند موقع ملزمة ووضع يجند ملزمة على أساس المواءمة المحلية للاستعلام مع بقايا يجند موقع الملزمة المعروفة في قوالب وظيفة أعلى الدرجات 9.
جودة وظيفة (الجماعة الأوروبية ومصطلح GO) هو التنبؤ في تقييم TASSER - I تقوم على درجة التماثل وظيفية (FH - نقاط) والذي هو قياس التشابه بين العالمي والمحلي الاستعلام والقالب ، وتعرف على النحو التالي : 
حيث C - درجة وتقديرا لنوعية النموذج تنبأ على النحو المحدد في المعادلة. (2) ؛ TM - تدابير درجة التشابه الهيكلية العالمية بين النموذج والبروتينات قالب ؛ RMSD علي هو RMSD بين النموذج والبنية القالب في محاذاة المنطقة هيكليا من TM - محاذاة 20؛ COV يمثل تغطية المواءمة الهيكلية (أي نسبة من رواسب الانحياز هيكليا مقسوما على طول الاستعلام) ؛ علي هو معرف هوية التسلسل في محاذاة TM - محاذاتها. درجة الثقة المقدرة للتوقعات المفوضية الأوروبية يتضمن العدد أيضا مصطلح لتقييم المباراة نشط موقع (ACM) بين الاستعلام والقالب داخل المنطقة المحلية المحددة ، وتحسب على النحو التالي : 
حيث N يمثل عدد طن من المخلفات قالب الحالية داخل المنطقة المحلية ، وعلي N هو عدد أزواج محاذاة بقايا الاستعلام القالب ، د ب هي المسافة بين الزوج C α عشر الأولى من مخلفات الانحياز ، د 0 = 3.0 A هو وقطع المسافة ، M الثاني هو عشرات BLOSUM بين الزوج إيث مخلفات الانحياز. بوجه عام ، FH - النتيجة في النطاق [0 ، 5] ، والنتيجة هي ACM بين [0 ، 2] ، حيث تشير إلى أعلى الدرجات الوظيفية تعيينات أكثر ثقة. كما يستخدم لتقييم درجة ACM الهيكل المحلية والتشابه تسلسل بالقرب من مواقع يجند الملزمة ، والتي يشار اليها على انها BS - درجة.
1. تقديم تسلسل البروتين
- زيارة صفحة الويب I - TASSER في http://zhanglab.ccmb.med.umich.edu/I-TASSER أن تبدأ مع هيكل ووظيفة التجربة النمذجة.
- نسخ ولصق تسلسل الأحماض الأمينية في شكل المقدمة أو تحميلها مباشرة من الكمبيوتر بالنقر على زر "تصفح". I - TASSER خادم يقبل حاليا متواليات مع ما يصل إلى 1500 المخلفات. البروتينات أطول من مخلفات 1500 وعادة ما تكون متعددة المجالات البروتينات ، وينصح أن تكون مقسمة إلى مجالات الفردية قبل تقديمها إلى TASSER - I.
- توفير البريد الإلكتروني (إلزامي) واسم لهذا المنصب (اختياري).
- يمكن للمستخدمين تحديد الخارجي اختياريا بين الدقةidue الاتصال / المسافة القيود ، إضافة في قالب إضافية أو استبعاد بعض البروتينات خلال قالب هيكل عملية النمذجة. تعلم المزيد حول استخدام هذه الخيارات في المقطع "مناقشة".
- أن يقدم التسلسل ، انقر على زر "تشغيل I - TASSER". وسيتم توجيه المتصفح إلى صفحة تأكيد عرض معلومات المستخدم المحدد ، وتحديد وظيفة (رقم الوظيفة) عدد وارتباط لصفحة ويب حيث سيتم إيداع النتائج بعد الانتهاء من العمل. يمكن للمستخدمين مرجعية هذا الارتباط أو يدون رقم تعريف العمل للرجوع اليها في المستقبل.
2. توافر نتائج
- التحقق من حالة عملك المقدمة من خلال زيارة صفحة قائمة انتظار I - TASSER في http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php . انقر فوق علامة التبويب بحث واستخدام رقم هوية الوظيفة أو تسلسل الاستعلام للبحث وظيفتك المقدمة.
- بعد هيكل وظيفة موالانتهاء deling ، سيتم إرسال إشعار بريد إلكتروني تحتوي على صورة للهياكل وتوقع وشبكة الربط لك. انقر على هذا الرابط أو افتح الرابط المرجعية في الخطوة 1.5 لعرض وتحميل النتائج.
3. هيكل الثانوي والتنبؤات الوصول المذيبات
- التحقق من تسلسل FASTA الاستعلام تنسيق المعروضة على الجزء العلوي من صفحة نتائج. ويمكن إذا تم تحديد أي ضبط النفس إضافية / قالب خلال تقديم تسلسل ، وصلة لعرض صفحة ويب المحددة من قبل المستخدم المعلومات أيضا أن ينظر إلى (الشكل 1A).
- دراسة بنية التنبؤ الثانوية عرضها على النحو التالي : الحلزون ألفا (H) ، بيتا حبلا (S) أو لفائف (C) ودرجة الثقة التنبؤ (0 = منخفض ، 9 = عالية) لكل بقايا. ابحث عن المنطقة مع فترات طويلة من هيكل الثانوية العادية (H او S) التنبؤات ، لتقدير المنطقة الأساسية في البروتين. ويمكن أيضا فئة من البروتين الهيكلي يتم تحليلها على أساس توزيع العناصر الهياكل الثانوية. بنلذا ، والمناطق لفائف طويلة من العناصر في البروتين تشير عادة المناطق غير منظم / المختلين.
- عرض وتوقع الوصول المذيبات (الشكل 1C) إلى مناطق دفن ومذيب التأكد كشفها في الاستعلام. توقعت مجموعة قيم المذيبات الوصول من 0 (بقايا مدفونة) إلى 9 (بقايا التعرض). ويمكن استخدام المنطقة التي تحتوي على مخلفات مدفونة في معظمها لتحديد المنطقة الأساسية في البروتين ، في حين أن المناطق التي تتعرض بقايا المذيبات والماء ماء والمحتملة / مواقع وظيفية.
4. توقعات هيكل التعليم العالي
- انتقل لأسفل لعرض هياكل التعليم العالي من البروتين وتوقع الاستعلام ، عرض في Jmol الصغير التفاعلية (الشكل 2). غادر انقر على الصغير لتغيير مظهر عرض للهيكل ، والتكبير في منطقة محددة ، وتحديد أنواع المخلفات المحددة في نموذج توقع أو حساب المسافات بين البقايا.
- تحليل نماذج عن وجود مناطق غير منظم طويلة. هذه صegions تتوافق عادة إلى المناطق المختلين في البروتين ، أو تشير إلى عدم وجود توافق القالب. هذه المناطق عموما منخفضة دقة النمذجة وإزالة هذه المناطق خلال نماذج من N & C - صول المنطقة وتحسين دقة النمذجة.
- تحميل ملفات PDB هيكل تنسيق للنموذج من خلال النقر على "تحميل الموديل" الروابط. يمكنك فتح هذه الملفات في أي برنامج التصور الجزيئي (مثلا Pymol ، Rasmol الخ) لمزيد من التحليل للخصائص الهيكلية.
- تحليل درجة الثقة (C - درجة) من النمذجة هيكل لتقدير الجودة من الهياكل المتوقعة. C - درجة (Eq. 2) وعادة ما تكون القيم في النطاق [-5 ، 2] ، وفيه أعلى درجة يعكس نموذجا من نوعية أفضل. يظهر المقدرة TM - درجة وRMSD من النموذج الأول باسم "دقة المقدر للنموذج 1". للبروتينات طويلة ، فمن المستحسن لتقييم جودة نموذج يستند إلى TM - النتيجة ، كما TM - النتيجة هي أكثر حساسية للتغيرات طوبولوجي من RMSD. < لى> انقر على "مزيد من المعلومات حول C - درجة" وصلة لتحليل C - درجة ، حجم الكتلة والكثافة كتلة من جميع النماذج. تقدر ترد TM - درجة وRMSD فقط لنموذج I - TASSER الأولى ، لأنه لم يتم C - درجة أقل من النماذج في المرتبة مرتبطة ارتباطا قويا مع TM - درجة أو RMSD. يمكن أن تكون ذات جودة أقل مرتبة من النماذج المقررة جزئيا على أساس كثافة الكتلة وحجم الكتلة النسبية إلى النموذج الأول ، وفيه نماذج من أكبر كتلة وكثافة أعلى في المتوسط أقرب إلى الهيكل الأصلي.
- انخفاض درجة C - التنبؤات تشير عادة التنبؤ بدقة منخفضة. في معظم الحالات من هذا القبيل ، ويفتقر إلى البروتين الاستعلام قالب جيدة في المكتبة ، ويبلغ حجم ما وراء مجموعة من النماذج منذ البداية (أي> بقايا 120). في هذه الحالات ، يمكن للمستخدمين البحث عن القيود المكانية إضافية واستخدامها لتحسين نمذجة I - TASSER (انظر القسم مناقشة). ويشجع أيضا على تقديم متواليات لخادم كوارك لدينا (كوارك / "> http://zhanglab.ccmb.med.umich.edu/QUARK/) للحصول على النمذجة نقية منذ البداية إذا كان حجم البروتين هو أقل من 200 المخلفات.
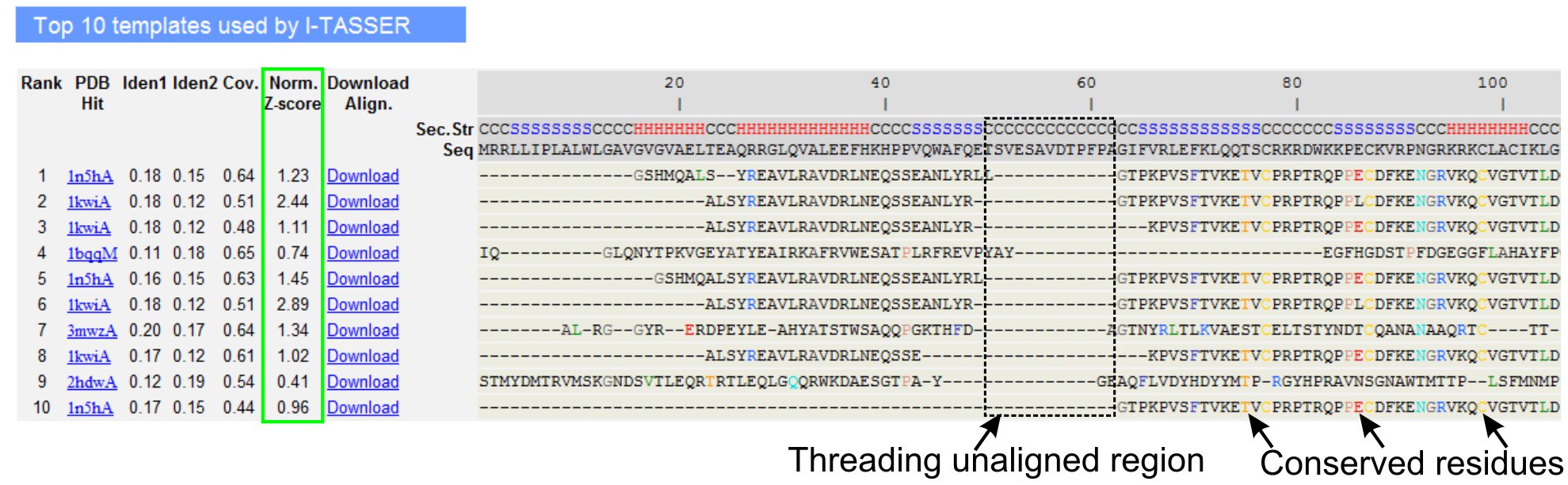
5. LOMETS محاذاة الهدف قالب
- بالتمرير لتحليل العشرة قوالب خيوط من البروتين الاستعلام ، كما حددتها برامج الترابط LOMETS (الشكل 3). عرض تطبيع Z - درجة (Eq. 1) ، كما هو موضح في "نورم. Z - درجة "العمود ، لتحليل نوعية التحالفات الترابط. الاصطفافات برصيد - Z تطبيع> 1 يعكس ثقة المحاذاة وعلى الأرجح لديهم أضعاف نفس البروتين الاستعلام.
- تحليل تسلسل الهوية في المنطقة خيوط الانحياز ('ايدن. 1' العمود) وعلى كامل سلسلة (عمود "ايدن. 2') لتقييم تناظر بين الاستعلام والبروتينات القالب. ارتفاع الهوية تسلسل مؤشرا على الصلة التطورية بين الاستعلام والبروتينات القالب.
- عرض بقايا خيوط ملونة تظهر في محاذاة لتحديد سلبيات بصرياerved مخلفات / الزخارف في الاستعلام والبروتينات القالب. هوية أعلى تسلسل خيوط الانحياز في المنطقة ، بالمقارنة مع كامل سلسلة محاذاة يشير أيضا إلى وجود هيكلية عزر الحفظ / المجالات في الاستعلام.
- تقييم تغطية محاذاة الترابط عن طريق عرض "COV". العمود ويتفقد المحاذاة. إذا كانت التغطية أعلى من التحالفات منخفضة ومحصورة في منطقة صغيرة فقط من البروتين الاستعلام أو غائبة عن قطعة طويلة من تسلسل الاستعلام ، ثم البروتين يحتوي الاستعلام عادة أكثر من مجال واحد وينصح لتقسيم تسلسل ونموذج المجالات بشكل فردي (الشكل 3).
- تحميل ملفات PDB تنسيق المحاذاة تسلسل الهيكل من خلال النقر على "تحميل محاذاة" الروابط. ويمكن فتح هذه الملفات في أي برنامج المواءمة التصور الجزيئي المدرجة في قسم المواد ، ويمكن أن تستخدم أيضا لإضافة قيود إضافية خلال النمذجة هيكل (الخطوة 1.4).
6.الهيكلية النظير في PDB
- عرض الجدول التالي (الشكل 4) من صفحة نتيجة لتحديد العشرة الأوائل النظير الهيكلي للأول نموذج التنبؤ به ، كما حددها برنامج المواءمة الهيكلية TM - محاذاة 20. TM - A درجة> 0.5 يشير إلى أن الكشف عن ونموذج تمثيلي لها طبولوجيا مماثلة ، ويمكن استخدامها لتحديد الهيكلية فئة / البروتين الأسرة من البروتين الاستعلام 16 ، في حين أن أولئك مع TM - درجة <0.3 يعني تشابه بنية عشوائية.
- تحليل تسلسل الهوية وRMSD في المنطقة تتماشى هيكليا هو مبين في "IDEN A' و 'RMSD أ" الأعمدة لتقييم المحافظة على الزخارف في النموذج المكاني والتناظرية الهيكلية. تفقد البصر أزواج بقايا الملونة والانحياز في محاذاة لتحديد هذه البقايا المحفوظة هيكليا والزخارف.
- انقر على رمز PDB هو موضح في عمود "هيت PDB' لزيارة RCSB الموقع ومعرفة المزيد عن تصنيفها الهيكلية (سكوب، كاث وPFAM) ومعلومات وظيفية (EC العدد ، حيث يرتبط GO ويجند ملزمة).
7. التنبؤ به وظيفة العامل المساعد
- انتقل لأسفل في صفحة نتيجة لتحليل التفسيرات الوظيفية للبروتين الاستعلام. وترد وظائف البروتين في سياق الجداول الثلاثة ، عرض : لجنة الإنزيمات (EC) الأرقام ، أنتولوجيا الجينات (GO) الشروط ، ومواقع الربط يجند.
- عرض 'TM - درجة' ، 'RMSD A' ، 'IDEN A' و 'COV". الأعمدة في كل جدول لتحليل المعلمات التشابه الهيكل العالمي والمحافظة على الأنماط المكانية بين المتماثلات وظيفية نموذجية والتي تم تحديدها (قوالب).
8. انزيم التنبؤ اللجنة عدد
- "توقع أرقام المفوضية الأوروبية" عرض أفضل خمسة المتماثلات انزيم المحتملة للبروتين الاستعلام هو مبين في الجدول (الشكل 5). يظهر مستوى الثقة التنبؤ به عدد EC هذه القوالب في العمود 'EC - نقاط. استنادا benchmaيمكن تحليل rking 23 ، التشابه وظيفية (أول 3 أرقام من رقم EC) بين الاستعلام والبروتين قالب تفسر بشكل موثوق به المفوضية الأوروبية ويسجل> 1.1.
- ابحث عن الآراء وظيفة (أرقام EC) بين القوالب التي حظيرة مماثلة (أي درجة TM -> 0.5) ، والبروتين الاستعلام. إذا قوالب متعددة ونفس العدد وEC EC - درجة> 1.1 ، ومستوى الثقة التنبؤ عالية جدا. ومع ذلك ، إذا كانت الجماعة الأوروبية نقاط عالية ولكن هناك عدم توافق في الآراء بين الزيارات التي تم تحديدها ، ثم التنبؤ يصبح أقل موثوقية وينصح للمستخدمين الاطلاع على التوقعات GO الأجل.
- انقر على الرابط المقدم على الأرقام الأوروبية لزيارة قاعدة بيانات أنزيم ExPASy وتحليل وظيفة ، بما في ذلك حفز رد فعل ، ومتطلبات التعاون عامل والمسار الأيضي ، قالب من البروتين بالتفصيل.
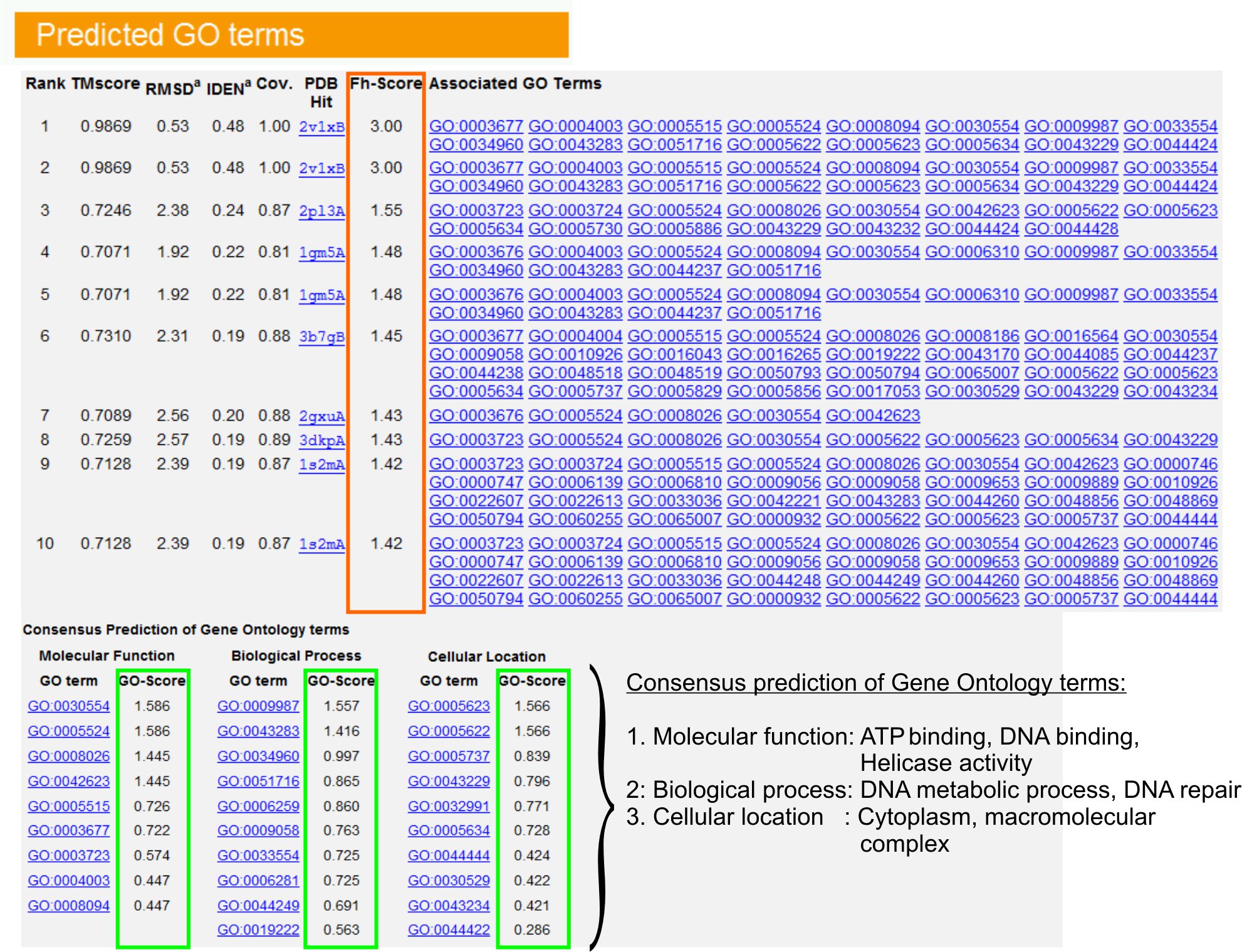
9. الجين التنبؤات الوجود مصطلح (GO)
- عرض "GO حيث توقع" الجدول (الشكللدى عودتهم 6) لتحديد العشرة الأوائل المتماثلات من البروتين الاستعلام في المكتبة PDB ، المشروح مع أنتولوجيا الجينات (GO) المصطلحات. عادة ما يرتبط البروتين مع كل شروط GO متعددة ، واصفا ظائفها الجزيئي (وسط) ، والعمليات البيولوجية (بي بي) والمكون الخلوية (CC). فوق كل فترة لزيارة الموقع وتحليل أميغو تعريفه والنسب.
- تحليل العمود FH - درجة (درجة وظيفية تماثل) للوصول إلى التشابه الوظيفي بين الاستعلام والبروتينات القالب وتقدير مستوى الثقة في نقل الشرح وظيفية من هذه البروتينات. في دراسة مقارنة لدينا 23 ، يمكن أن يكون 50 ٪ من حيث GO الأصلية التي تم تحديدها بشكل صحيح من القالب الأول تحديدها باستخدام قطع FH - 0.8 درجة ، مع دقة 56 ٪ من الإجمالي.
- عرض "التنبؤ توافق من حيث GO" جدول لتحليل موافقة من وظيفة بين القوالب. وتستخدم هذه الوظائف المشتركة للتنبؤ حيث GO (MF ، BP وCC) للاستعلامالبروتين وتقييم مستوى الثقة (GO - درجة) من التنبؤات GO الأجل. استنادا إلى اختبار قياس 23 ، ويتم الحصول على أفضل معدلات سلبية كاذبة وزائفة إيجابية للتنبؤات مع GO - = 0.5 درجة قطع ، مع تناقص التغطية للتنبؤ في مستويات أعمق الأنطولوجيا.
10. البروتين يجند التنبؤات موقع ملزم
- انتقل لأسفل إلى أسفل الصفحة لعرض أكبر عشرة تنبؤات يجند موقع ملزم للبروتين الاستعلام. يتم تصنيف مواقع الربط وتوقع استنادا إلى عدد من التشكل يجند تنبأ بأن حصة جيب ملزمة المشتركة. يتم عرض بالفعل تحديد موقع أفضل ملزمة في Jmol الصغير. انقر على أزرار الراديو لتحليل التوقعات الأخرى وتصور مخلفات التفاعل يجند.
- تحليل العمود BS - رصيده الى تقييم المحلية التشابه بين النموذج وقالب موقع ملزمة. استنادا إلى معيار 9 ، BS - درجة> 1.1 يشير تسلسل عالية وبنية سيمilarity بالقرب من موقع الربط المتوقع في نموذج ملزم وموقع معروف في القالب.
- تحميل ملف PDB هيكل تنسيق للمجمع من خلال النقر على رابط "حمل". يمكن للمستخدمين فتح هذه الملفات في أي برنامج التصوير الجزيئي وعرض تفاعلي للموقع وتوقع ملزمة ويجند البروتين التفاعلات على أجهزتهم المحلية.
11. ممثل النتائج

الشكل 1 مقتطفات من صفحة نتائج تظهر I - TASSER (أ) مهيأ FASTA تسلسل الاستعلام ؛ (B) وتوقع هيكل الثانوية وعشرات الثقة المرتبطة بها ، و (C) وتوقع الوصول مذيب للمخلفات. ويسلط الضوء على تحليل المنطقة الأساسية والمحتملة في ترطيب موقع الاستعلام في سماوي والمستطيلات الحمراء ، على التوالي.

الشكل 2.

الشكل 3. مثال على صفحة نتائج I - TASSER تظهر خيوط قوالب العشرة التي تم تحديدها من قبل البرامج والتحالفات 5 LOMETS الترابط. يتم تقييم نوعية التحالفات القائمة على الترابط تطبيع Z - درجة (أبرزت باللون الأخضر) ، حيث قيمة> 1 يعكس ثقة المحاذاة. ويسلط الضوء على مخلفات الانحياز في القالب الذي يتم مطابقة للمخلفات الاستعلام المناظرة في اللون للإشارة إلى وجود بقايا الحفظ / عزر ، في حين أن عدم وجود التوافق في معظم قوالب أعلى يشير وجود مجالات متعددة في البروتين الاستعلام والبقايا الصغيرة المحايدة تتطابق مع المناطق رابط المجال. انقر هنا للاطلاع على النسخة الكاملة الحجم من الرقم 3.

الشكل 4. مثال على صفحة نتائج تظهر العشرة النظير الهيكلية التي تم تحديدها والتحالفات الهيكلي ، والتي حددها TM - 20 محاذاة برنامج المواءمة الهيكلية. ويستند هذا الترتيب من النظير هو مبين في TM - على درجة (سلط عليها الضوء في الزرقاء) من المواءمة الهيكلية. TM - A درجة> 0.5 يشير إلى أن اثنين من الهياكل ومقارنة طبولوجيا مماثلة ، في حين أن درجة TM - <0.3 يعني التشابه بين هيكلين عشوائي. ويسلط الضوء على أزواج بقايا محاذاة هيكليا في اللون على أساس الملكية الأحماض الأمينية ، في حين يشار إلى المناطق الصغيرة المحايدة من قبل "--".ove.com/files/ftp_upload/3259/3259fig4large.jpg "> اضغط هنا للاطلاع على النسخة الكاملة الحجم من الرقم 4.

الشكل 5. مثال على صفحة نتائج I - TASSER تظهر المتماثلات انزيم البروتين التي يتم تحديدها من الاستعلام في المكتبة PDB. ويتم تحليل مستوى الثقة التنبؤ على أساس عدد EC EC - درجة (وأبرزت باللون الأخضر) ، حيث يسجل EC -> 1.1 يشير التشابه وظيفية (نفس أول 3 أرقام من رقم EC) بين الاستعلام والبروتين القالب.

الشكل 6. مثال على صفحة نتائج I - GO TASSER تظهر توقعات الأجل للبروتين الاستعلام. يتم تصنيف المتماثلات وظيفية للبروتين الاستعلام في قالب مكتبة جين الوجود على أساس درجة ، فتحي حسن بهم (في المستطيل البرتقالي). وتستمد الملامح الفنية المشتركة من هذه الزيارات إلى أعلى الدرجات خينير أكلت النهائي التنبؤات المدى GO للبروتين الاستعلام. وتشير التقديرات تستند نوعية الشروط وتوقع GO - GO على درجة (كما هو موضح باللون الأخضر) ، حيث يسجل GO -> 0.5 يشير إلى التنبؤ موثوق بها. اضغط هنا للاطلاع على النسخة الكاملة الحجم من الرقم 6.

الرقم 7. مثال على صفحة نتائج I - TASSER تظهر أعلى يجند البروتين ten التنبؤات موقع ملزم باستخدام العامل المساعد 9 الخوارزمية. ويستند ترتيب المواقع ملزمة توقع على عدد من التشكل يجند تنبأ بأن حصة شائعة في جيب ملزمة الاستعلام. BS - درجة (الضوء الاحمر) هو مقياس لتسلسل المحلية والتشابه بين هيكل وتوقع موقع القالب ملزمة ، وغير مفيدة لتحليل الحفاظ على جيوب موقع ملزمة.
les/ftp_upload/3259/3259fig8.jpg "/>
الرقم 8. مثال على الملفات الخارجية ضبط النفس استخدامها لتحديد بقايا فضلات الاتصال / قيود المسافة.

الرقم 9. مثال لضبط النفس الملفات المستخدمة لتحديد بروتين القالب إلى خادم I - TASSER. يمكن للمستخدم تحديد محاذاة الاستعلام إما في شكل قالب FASTA (A) أو (B) شكل 3D.

الرقم 10. ملف سبيل المثال تستخدم لاستبعاد قالب الهيكل الداخلي خلال النمذجة I - TASSER. العمود الأول يحتوي على معرف PDB من البروتينات قالب سيتم استبعادها. ويستخدم العمود الثاني لتحديد هوية قطع تسلسل التي سيتم استخدامها لقوالب أخرى مماثلة في مكتبة القالب.
Discussion
بروتوكول الواردة أعلاه هو توجيهي عام لهيكل والنمذجة الدالة باستخدام ملقم I - TASSER. على الرغم من أن هذا الإجراء الآلي يعمل بشكل جيد جدا بالنسبة لمعظم البروتينات ، والتدخلات البشرية غالبا ما يساعد بشكل كبير على تحسين دقة النمذجة ، وخاصة بالنسبة للبروتينات التي تفتقر إلى القوالب وثيقة في مكتبة PDB. يمكن للمستخدمين التدخل خلال النمذجة I - TASSER في الطرق التالية : (أ) تقسيم المجال متعددة البروتينات ، (ب) توفير القيود الخارجية لتحسين هيكل التجمع ، و (ج) إزالة القوالب خلال النمذجة.
تقسيم متعددة المجالات البروتين :
العديد من سلاسل طويلة تحتوي على البروتين في كثير من الأحيان مجالات متعددة وفقا للمناطق المربوطة رابط مرنة ، مما يجعل من الصعب توضيح هيكلها على حد سواء باستخدام التقنيات التجريبية والحاسوبية. ومع ذلك ، والمجالات هي كيانات مستقلة قابلة للطي ويمكن أن تؤدي وظيفة الجزيئية متميزة ، بل هو مرغوب فيه لتقسيم طويلة متعددة المجال البروتينات ونموذج كل مجال على حدة. مجالات النمذجة وليس فرديا فقط سرعة وتيرة عملية التنبؤ ، لكنه يزيد أيضا نوعية الاستعلام قالب المحاذاة ، مما أدى إلى هيكل أكثر موثوقية والتنبؤات وظيفة.
ويمكن توقع حدود المجال في تسلسل البروتين باستخدام برامج متاحة مجانا على الانترنت الخارجية مثل CDD NCBI 24 ، 25 أو PFAM InterProScan 26. أيضا ، إذا LOMETS التحالفات الترابط متوفرة للاستعلام البروتين ، يمكن أن تكون موجودة من خلال تحديد نطاق حدود البصر فترات طويلة من مخلفات الصغيرة المحايدة في قوالب والترابط العلوي (انظر الخطوة 5.4). تقابل هذه المناطق الصغيرة المحايدة في الغالب إلى مناطق رابط المجال. إذا كان المجال قوالب متعددة متوفرة بالفعل في مكتبة قالب PDB مع جميع المجالات الاستعلام الانحياز ، ومن ثم يمكن للبروتين غرار الاستعلام كما الكامل طول.
توفير القيود الخارجية
الطبقة = "jove_content"> وتسترشد أساسا محاكاة هيكل التجمع في TASSER - I القيود المكانية التي تم جمعها من خيوط LOMETS القوالب. الاستعلام عن البروتينات التي ضربت خيوط جيدة (درجة Norm. Z -> 1) في مكتبة القالب ، والقيود المكانية مستمدة في معظمها من الدقة العالية وI - TASSER سوف تولد نماذج عالية الدقة الهيكلي لهذه البروتينات. النقيض من ذلك ، الاستعلام عن البروتينات التي بلغت ضعف أو عدم الترابط (Z - Norm. درجة <1) ، والقيود المكانية التي تم جمعها غالبا ما تحتوي على أخطاء بسبب عدم التيقن من القالب والمحاذاة. لهذه الأهداف من البروتين ، يمكن للمستخدم تحديد المعلومات المكانية أن تكون مفيدة جدا لتحسين نوعية نموذج التنبؤ بها. يمكن للمستخدمين توفير القيود الخارجية لخادم I - TASSER بطريقتين :
ألف حدد الاتصال / المسافة القيود
تتميز تجريبيا بين بقايا اتصالات / المسافات ، على سبيل المثال من الرنين المغناطيسي أوعبر الربط بين التجارب ، يمكن تحديد عن طريق تحميل ملف ضبط النفس. ويظهر ملف سبيل المثال في الشكل 8 ، حيث العمود 1 يحدد نوع من ضبط النفس ، أي "DIST" أو "الاتصال". لضبط النفس بعد (DIST) ، والأعمدة 2 و 4 مواقع تحتوي على مخلفات (ط ، ي) ، والأعمدة 3 و 5 تحتوي على ذرة وأنواع المخلفات في العمود 6 وتحدد المسافة بين الذرات المحددين. لقيود الاتصال (الاتصال) ، والأعمدة 2 و 3 تحتوي على مواقف (ط ، ي) من مخلفات التي ينبغي أن تكون على اتصال. تقرر المسافة بين الجانب سلاسل وسط بقايا هذه الأزواج بالاتصال على أساس المسافات التي لوحظت في الهياكل المعروفة في PDB. I - TASSER سوف احاول ان اضع هذه الأزواج ذرة بالقرب من المسافة المحددة خلال المحاكاة صقل الهيكل.
باء تحديد قالب بنية البروتين
LOMETS برامج الترابط استخدام المكتبة للعثور على ممثل PDB طيات المعقول البروتوكول الاضافي للاستعلامعين. على الرغم من استخدام مكتبة هيكل ممثل يساعد على تقليل الوقت اللازم لحساب التحالفات تسلسل الهيكل ، فمن الممكن أن يكون غاب عن البروتين قالب جيدة في المكتبة أو قد لا القالب تم تحديدها من قبل برامج LOMETS الترابط ، حتى ولو كان موجودة في المكتبة. في هذه الحالات ، يجب على المستخدم تحديد بنية البروتين المطلوب كقالب.
لتحديد بنية البروتين بمثابة قالب إضافية ، يمكن للمستخدمين تحميل إما ملف PDB هيكل تنسيق أو تحديد هوية PDB من بنية البروتين المودعة في مكتبة PDB. سوف I - TASSER توليد محاذاة الاستعلام القالب باستخدام برنامج حشد 23 و سيجمع القيود المكانية من المستخدم على حد سواء قالب محدد وLOMETS قوالب للاسترشاد بها في محاكاة هيكل التجمع. لأن دقة القيود LOMETS يختلف عن أهداف مختلفة ، وثقل القيود LOMETS أقوى في تا (مثلي) سهلrgets من ذلك في الأهداف الثابتة (غير المتماثلة) ، والتي تم ضبطها بشكل منهجي في تدريبنا القياسي.
يمكن للمستخدمين أيضا تحديد الخاصة بهم الاستعلام قالب التحالفات. الخادم يقبل المحاذاة في شكلين : الشكل FASTA (الشكل 9A) وشكل 3D (الشكل 9B). تنسيق FASTA هو معيار وصفها في http://zhanglab. ccmb.med.umich.edu / FASTA / . شكل 3D مشابه لتنسيق PDB القياسية ( http://www.wwpdb.org/documentation/format32/sect9.html ) ، ولكن تضاف عمودين إضافية مشتقة من قوالب لسجلات ATOM (انظر الشكل 9B) :
أعمدة 1-30 : اتوم (C - ألفا فقط) وبقايا أسماء لتسلسل الاستعلام.
الأعمدة 31-54 : إحداثيات C - ذرات ألفا من الاستعلام نسخ من ذرات المناظرة في القالب.
الأعمدة 55-59 : عدد بقايا الموافق في القالب على أساس التوافق
الأعمدة 60-64 : اسم بقايا الموافق في القالب
استبعاد القوالب البروتينات
البروتينات هي جزيئات مرنة ويمكن أن تتبنى الدول متعلق بتكوين عدة لتغيير نشاطهم البيولوجي. على سبيل المثال ، قد حلت هياكل تحركات البروتينات وكثير من البروتينات في الغشاء التشكل على حد سواء نشطة وغير نشطة. كما يمكن وجود أو غياب الالتزام يجند تسبب تحركات هيكلية كبيرة. في حين أن جميع الدول بتكوين جزئي من القالب على حد سواء لبرامج الترابط ، فمن المستحسن أن نموذج الاستعلام باستخدام القوالب في دولة واحدة فقط خاصة. خيار جديد على الخادم يسمح للمستخدم لاستبعاد البروتينات قالب خلال النمذجة هيكل. وهذه الميزة تسمح للمستخدم أيضا اختيار مستوى التماثل من القوالب لاستخدامها للنمذجة. يمكن للمستخدمين قالب استبعاد البروتينات الابام المكتبة I - TASSER من قبل :
أ تحديد هوية قطع تسلسل
يمكن للمستخدمين استخدام هذا الخيار لاستبعاد البروتينات مثلي من مكتبة قالب I - TASSER. يتم تعيين مستوى التماثل على أساس تسلسل قطع الهوية ، أي عدد من بقايا متطابقة بين الاستعلام والبروتين قالب مقسوما على طول تسلسل تسلسل الاستعلام. على سبيل المثال ، إذا كان المستخدم في أنواع "70 ٪" في شكل المقدمة ، قوالب جميع البروتينات التي لها هوية تسلسل> 70 ٪ من البروتين الاستعلام I - سيتم استبعادها من مكتبة قالب I - TASSER.
باء استبعاد البروتينات قالب معين
ويمكن استبعاد البروتينات قالب معين من مكتبة قالب I - TASSER عن طريق تحميل قائمة تحتوي على معرفات PDB الهياكل سيتم استبعادها. ويظهر ملف سبيل المثال في الشكل 10. كما يمكن للبروتين موجود نفس كإدخالات متعددة في مكتبة PDB ، I - TASSER حد ذاتهاسوف rver افتراضيا استبعاد قوالب معينة (في Column1) ، فضلا عن جميع القوالب الأخرى من المكتبة التي تحتوي على هوية> 90 ٪ لقوالب معينة. يمكن للمستخدمين أيضا تحديد قطع هوية مختلفة ، على سبيل المثال 70 ٪ ، حيث سيتم استبعاد كافة القوالب مع الحفاظ على الهوية> 70 ٪ من البروتينات قالب محدد.
Disclosures
الإعلان عن أي تضارب في المصالح.
Acknowledgments
ويدعم هذا المشروع جزئيا من قبل ألفريد سلون مؤسسة P. ، جائزة شهادة NSF (DBI 1027394) ، والمعهد الوطني للعلوم الطبية العامة (GM083107 ، GM084222).
Materials
| Name | Company | Catalog Number | Comments |
| Material Name | Type | Company | Catalogue Number |
| FASTA formatted amino acid sequence of the protein to be modeled (see, http://www.ncbi.nlm.nih.gov/BLAST/fasta.shtml). | |||
| A personal computer with access to the internet and a web browser. | |||
| Molecular visualizing software, e.g. RASMOL or PYMOL, for analyzing the predicted tertiary structure and functional sites. |
References
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H. CD-Search: protein domain annotations on the fly. Nucleic. Acids. Res. 32, W327-W331 (2004).
- Finn, R. D. The Pfam protein families database. Nucleic. Acids. Res. 38, D211-D222 (2010).
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).

{kind=link}
{kind=link}