

Summary
Retningslinjer for computerbaseret strukturelle og funktionelle karakterisering af protein ved hjælp af I-TASSER rørledningen er beskrevet. Fra forespørgsel protein sekvens, er 3D-modeller genereret ved hjælp af flere tråde alignments og iterativ strukturelle samling simuleringer. Funktionelle slutninger er derefter trukket baseret på kampe til proteiner med kendt struktur og funktioner.
Abstract
Genomsekvensering projekter har kryptograferede millioner af protein sekvens, som kræver kendskab til deres struktur og funktion for at forbedre forståelsen af deres biologiske rolle. Selvom eksperimentelle metoder kan give detaljerede oplysninger for en lille brøkdel af disse proteiner er datamodellering behov for hovedparten af protein molekyler, der er eksperimentelt uncharacterized. I-TASSER server er en on-line workbench til høj opløsning modellering af protein struktur og funktion. Givet et protein sekvens, indeholder en typisk output fra I-TASSER server sekundær struktur forudsigelse, forudsagde opløsningsmiddel tilgængeligheden af de enkelte restkoncentrationer, homologe skabelon proteiner opdaget ved gevindskæring og struktur justeringer, op til fem i fuld længde tertiær strukturelle modeller, og struktur-baserede funktionelle annotationer for enzym klassificering, Gene ontologi vilkår og protein-ligand bindingssteder. Alle forudsigelser er kodet med en tillid score, somfortæller, hvordan præcis de forudsigelser er uden at kende de eksperimentelle data. For at lette de særlige anmodninger fra slutbrugere, serveren giver kanaler til at acceptere bruger-specificerede inter-rester afstand og kontakt kort til interaktivt ændre I-TASSER modellering, og det giver også brugerne mulighed for at angive eventuelle proteiner som skabelon, eller at udelukke enhver skabelon proteiner i løbet af strukturen samling simuleringer. De strukturelle oplysninger kan indsamles af brugere baseret på eksperimentelle beviser eller biologiske indsigter med det formål at forbedre kvaliteten af I-TASSER forudsigelser. Serveren blev vurderet som det bedste programmer for proteiners struktur og funktion forudsigelser i den nylige EU-dækkende CASP eksperimenter. Der er i øjeblikket> 20.000 registrerede forskere fra over 100 lande, der bruger online-I-TASSER server.
Protocol
Metode overblik
Efter sekvens-til-struktur-til-funktion paradigme, indebærer jeg-TASSER procedure 1-4 for struktur og funktion modellering fire på hinanden følgende trin af: (a) skabelon identifikation ved LOMETS 5, (b) fragment struktur samling af replika- udveksling af Monte Carlo simuleringer 6; (c) atomare niveau struktur raffinement bruger REMO 7 og FG-MD-8, og (d) struktur-baserede funktion fortolkninger hjælp cofaktor 9.
Skabelon identifikation: For en forespørgsel sekvens fremlagt af brugeren, er rækkefølgen first gevind gennem en repræsentant FBF struktur bibliotek ved en lokalt installeret LOMETS meta-threading server. Threading er en sekvens-struktur tilpasning procedure, der anvendes til at identificere skabelon proteiner, som kan have lignende struktur eller indeholder samme strukturelle motiv som forespørgslen protein. For at øge dækningen af homologe Templspiste registreringer, LOMETS kombinerer flere state-of-the-art algoritmer, der dækker forskellige tråde metoder. Siden andet gevind programmer har forskellige pointsystemer og tilpasning følsomheder, er kvaliteten af den genererede gevindskæring alignments fra hver gevindskæring program vurderes af normaliseret Z-score, som er defineret som: 
hvor Z-score er den score i standardafvigelsen enheder i forhold til den statistiske gennemsnit af alle alignments genereret af programmet, og Z 0 er et program-specifikke Z-score cutoff opgøres på grundlag af store threading benchmark-test 5 for at skelne gode 'og' dårlige 'skabeloner. En skabelon med en høj Z-score betyder, at de øverste skabeloner er en tilpasning scorer væsentligt højere end de fleste andre skabeloner, som normalt indebærer, at tilpasning svarer til en god model. Hvis de fleste af de øverste gevind skabeloner har high normaliserede Z-scores, nøjagtigheden af den endelige I-TASSER model er som regel høj. Men hvis proteinet er stor og dækningen af gevindskæring justeringer er begrænset til et lille område af forespørgslen protein, giver en høj normaliseret Z-score ikke nødvendigvis ensbetydende med en høj modellering nøjagtighed for fuld længde model. To bedste threading alignments fra hver threading program er indsamlet og anvendt til det næste trin i strukturen forsamling.
Iterativ struktur samling simulering: Efter gevindskæring procedure, er query sekvens opdelt i gevindskæring justeret og unaligned regioner. Kontinuerlig fragmenter i trådning tilpasning er skåret fra skabeloner og anvendes direkte til strukturen forsamling, mens unaligned løkken regioner er bygget af ab initio modellering. Strukturen samling proceduren er udført på et gitter system, styret af replika udveksling Monte Carlo simuleringer 6. I-TASSER kraftfelt omfatter brint-Bonde interaktioner 10, videnbaserede statistiske hensyn til energi stammer fra kendte protein strukturer i FBF 11, sekvens-baserede kontakt forudsigelser fra SVMSEQ 12 og rumlige begrænsninger indsamlet fra LOMETS 5 gevindskæring skabeloner. Den konformationelle lokkefugle genereret i den lave temperatur kopier i løbet af simuleringer er grupperet efter SPICKER 13 til at kortlægge strukturer lave fri-energi stater. Cluster centroids af de øverste klynger ved at tage gennemsnittet 3D-koordinater på alle klynger strukturelle lokkefugle og anvendes til den endelige model generation. Simuleringen og klyngedannelse procedure gentages to gange for at fjerne steriske sammenstød og yderligere raffinering den globale topologi.
Atomic-model for konstruktion og justering: Klyngen centroids opnået efter SPICKER clustering er nedsat protein-modeller (hver rester repræsenteret ved sin C α og side-chain tyngdepunkt) og have begrænsede biologiske ansøgning. Opførelsen af fuld atommodel fra den reducerede modeller er udført i to trin. I det første trin, er REMO 7 bruges til at konstruere fuld-atomare modeller fra C-alfa-spor ved at optimere H-obligation netværk. I det andet trin er REMO fuld-atomare modeller yderligere forfinet af FG-MD 14, som forbedrer rygraden torsion vinkler, obligations-længder, og side-chain rotamer retningslinjer, som molekylære dynamiske simuleringer, styret som af den strukturelle fragmenter søges fra FBF strukturer af TM-align. Den FG-MD raffinerede modeller bruges som den endelige modeller for tertiær struktur forudsigelser af I-TASSER.
Kvaliteten af de genererede modeller er estimeret baseret på en tillid score (C-score), som er defineret baseret på Z-score på LOMETS gevindskæring alignments og konvergensen af I-TASSER simuleringer, matematisk formuleret som: 
hvor
C-score har en stærk korrelation med kvaliteten af I-TASSER modeller. Ved at kombinere C-score og protein længde, kan nøjagtigheden af de første I-TASSER modeller estimeres med en gennemsnitlig fejl på 0,08 for den TM-score og 2 A for RMSD 15. Generelt modeller med C-score> - er 1,5 forventes at have et korrekt fold. Her RMSD og TM-score er begge velkendte foranstaltninger af topologiske lighed mellem modellen og indfødte struktur. TM-score værdifuldees sortiment i [0, 1], hvor en højere score indikerer en bedre struktur matcher 16,17. Men for lavere rangeret modeller (dvs. 2 nd -5 th modeller), korrelationen mellem C-score med TM-score, og RMSD er meget svagere (~ 0,5), og kan ikke bruges til pålidelige skøn over absolut model kvalitet.
Er første model altid den bedste model i I-TASSER simuleringer? Svaret på dette spørgsmål afhænger af målet type. For lette mål, er den første model som regel den bedste model og dens C-score er normalt meget højere end resten af modellerne. Men for hårde mål, hvor gevindskæring ikke har væsentlig skabelon hits, er den første model ikke nødvendigvis den bedste model, og jeg-TASSER faktisk har svært ved at udvælge de bedste skabelonen og modeller. Det anbefales derfor at analysere alle de 5 modeller for hårde mål og vælge dem baseret på de eksperimentelle oplysninger og biologisk viden.
Funktion Gætictions: I det sidste trin, er endelige 3D-modeller genereret fra FG-MD brugt til at forudsige tre aspekter af protein funktion, nemlig: a) Enzym Kommissionen (EF) nummer 18 og (b), Gene Ontology (GO) 19 termer og ( c) bindingssteder for småmolekyle ligander. For alle de tre aspekter, er funktionelle fortolkninger genereres ved hjælp af co-faktor, som er en ny tilgang til at forudsige proteinets funktion er baseret på globale og lokale lighed med skabelon proteiner i FBF med kendt struktur og funktioner. Først er det globale topologi af de forudsagte modeller matchet mod funktionelle skabelon biblioteker ved hjælp af strukturelle tilpasning programmet TM-align 20. Dernæst er et sæt af proteiner mest ligner målet modeller udvalgt fra biblioteket på basis af deres global struktur lighed, og en omfattende lokal søgning er udført for at identificere struktur og rækkefølge ligheden nær den aktive / bindingssted region. Den resulterende globale og lokale lighed scores bruges til at rangordneskabelon proteiner (funktionel homologer) og overføre annotation (EF-numre og Gene Ontology 19 termer) baseret på den øverste scoring hits. Tilsvarende er ligand bindingssted rester og ligandbindende tilstand udledes baseret på den lokale tilpasning af forespørgsel med kendte ligand bindingssted rester i øverste scoring funktionen skabeloner 9.
Kvaliteten af funktion (EF og GO sigt) forudsigelse i I-TASSER vurderes baseret på funktionel homologi score (Fh-score), som er et mål for globale og lokale lighed mellem forespørgslen og skabelon, og er defineret som: 
hvor C-score er et estimat af kvaliteten af de forudsagte model, som defineret i Eq. (2), TM-score måler den samlede strukturelle lighed mellem modellen og skabelon proteiner; RMSD ali er RMSD mellem modellen og den skabelon struktur i strukturelt afstemt regionen fra TM-align 20; Cov repræsenterer dækningen af den strukturelle tilpasning (dvs. forholdet mellem de strukturelt tilpasset rester divideret med forespørgslen længde); ID ali er rækkefølgen identitet i TM-align tilpasning. Den estimerede tillid score for EF-nummeret forudsigelser omfatter også et udtryk for vurdering af aktive site match (ACM) mellem forespørgsel og skabelon i et afgrænset lokalområde, beregnes som: 
hvor N t er antallet af skabelonen rester til stede i lokalområdet, N ali er antallet af den tilpassede query-skabelon rester par, D II er C α afstanden mellem I th par tilpasset restprodukter, d 0 = 3,0 Å er afstanden cutoff, M ii er BLOSUM score mellem ed par tilpasset rester. Generelt er Fh-score i intervallet [0, 5] og Acm score er mellem [0, 2], Hvor højere score indikerer mere sikker funktionelle opgaver. ACM score er også brugt til vurdering af den lokale struktur og rækkefølge ligheden nær ligand-bindingssteder, der er benævnt BS-score.
1. Indsendelse af protein sekvens
- Besøg I-TASSER webside på http://zhanglab.ccmb.med.umich.edu/I-TASSER til at starte med struktur og funktion modellering eksperiment.
- Kopier og indsæt aminosyresekvens ind på ansøgningsskemaet eller direkte uploade det fra din computer ved at klikke på "Gennemse"-knappen. I-TASSER server i øjeblikket accepterer sekvenser med op til 1500 rester. Proteiner længere end 1500 rester er normalt multi-domæne proteiner, og anbefales at blive opdelt i de enkelte domæner, før du sender til I-TASSER.
- Giv din e-mail-adresse (obligatoriske) og et navn til jobbet (valgfrit).
- Brugere kan eventuelt angive eksterne inter-residue kontakt / distance begrænsninger, tilføjelsesprogrammet en ekstra skabelon eller udelukke nogle skabelon proteiner i løbet af strukturen modelleringsprocessen. Lær mere om at bruge disse indstillinger i "Debat" sektionen.
- For at indsende sekvensen, skal du klikke på "Run I-TASSER" knappen. Browseren vil blive dirigeret til en bekræftelses side, der viser brugeren bestemte oplysninger, job identifikation (Job ID) nummer og et link til en webside, hvor resultaterne vil blive deponeret efter endt job. Brugere kan bogmærke dette link eller notere jobbet identifikationsnummer for fremtidig reference.
2. Tilgængelighed af resultater
- Kontroller status for dine indsendt job ved at besøge I-TASSER kø side på http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php . Klik på fanen Søg, og brug Job ID-nummer eller forespørgslen sekvens til at søge dine sendte jobbet.
- Efter den struktur og funktion modeling er færdig, vil en meddelelse e-mail med billede af den forventede strukturer og et web-link blive sendt til dig. Klik på dette link eller åbne linket bogmærket på Trin 1,5 til se og downloade resultaterne.
3. Sekundær struktur og solvens tilgængelighed forudsigelser
- Kontroller FASTA formaterede forespørgsel sekvens vises øverst på resultatsiden. Hvis nogen yderligere tilbageholdenhed / skabelon blev angivet under sekvens fremlæggelse, et link til den webside, vise brugerdefinerede informationer kan også ses (figur 1A).
- Undersøg sekundær struktur forudsigelse vises som: alpha helix (H), beta Strand (S) eller spole (C) og tillid score på forudsigelse (0 = lav, 9 = høj) for de enkelte restkoncentrationer. Kig efter region med lange strækninger af regelmæssige sekundær struktur (H eller S) forudsigelser, at vurdere den kerne-region i protein. Strukturelle klasse af protein kan også analyseres baseret på distribution af sekundære strukturer elementer. Alså, længe regioner coil elementer i proteinet normalt indikerer ustruktureret / rodet regioner.
- Se forudsagt opløsningsmiddel tilgængelighed (Figur 1C) for at fastslå begravet og solvens udsatte regioner i forespørgslen. Værdier for forudsagde opløsningsmiddel tilgængelighed varierer fra 0 (begravet rest) til 9 (udsat rester). Region indeholder det meste begravet rester kan bruges til at afgrænse de centrale region i protein, mens regioner med opløsningsmiddel eksponeret og hydrofile rester er potentielle hydrering / funktionelle sites.
4. Tertiær struktur forudsigelser
- Rul ned for at se den forudsagte tertiære strukturer forespørgsel protein, der vises i en interaktiv Jmol applet (Figur 2). Venstre klik på applet til at ændre udseendet af viste struktur, zoome ind bestemt region, vælg bestemt rester typer i den forudsagte modellen eller beregne inter-rester afstande.
- Analysere modeller for tilstedeværelsen af lange ustruktureret regioner. Disse rEGIONSUDVALGET normalt svarer til forstyrret regioner i protein eller angiver manglende skabelon justering. Disse regioner har generelt lav modellering nøjagtighed og fjerne disse regioner i løbet af modellering fra N & C-terminus region vil forbedre modellerne nøjagtighed.
- Download FBF formaterede struktur-filer af modellen ved at klikke på "Download Model" links. Du kan åbne disse filer i en vilkårlig molekylær visualisering software (f.eks Pymol, Rasmol osv.) for yderligere analyse af de strukturelle træk.
- Analyser den tillid score (C-score) af struktur modellering til at vurdere kvaliteten af de forudsagte strukturer. C-score (Eq. 2) værdier er typisk i intervallet [-5, 2], hvor en højere score afspejler en model af bedre kvalitet. Den anslåede TM-score og RMSD af første model er vist som "Anslået nøjagtigheden af model 1". For lange proteiner, anbefales det at evaluere modellen kvalitet baseret på TM-score, som TM-score er mere følsom over for de topologiske ændringer end RMSD. < li> Klik på "mere om C-score" for at analysere C-score, cluster størrelse og klynge tæthed af alle modeller. Anslået TM-score og RMSD præsenteres kun for den første I-TASSER model, fordi C-score på lavere rangerende modeller ikke er stærkt korreleret med TM-score eller RMSD. Kvaliteten af lavere rangerede modeller kan delvist vurderes ud fra deres klynge tæthed og cluster størrelse i forhold til den første model, hvori modeller fra større klynge og højere tæthed i gennemsnit er tættere på den oprindelige struktur.
- Lavt C-score forudsigelser normalt indikerer en lav nøjagtighed forudsigelse. I de fleste sådanne tilfælde mangler forespørgslen proteinet en god skabelon på biblioteket og har en størrelse ud over området for ab initio modellering (dvs.> 120 rester). I disse tilfælde kan brugerne søge yderligere rumlige begrænsninger og bruge dem til at forbedre den I-TASSER modellering (se diskussion afsnit). Det er også opfordres til at indsende sekvenser til vores QUARK server (QUARK / "> http://zhanglab.ccmb.med.umich.edu/QUARK/) for en ren ab initio modellering, hvis proteinet størrelse er under 200 rester.
5. LOMETS mål skabelon tilpasning
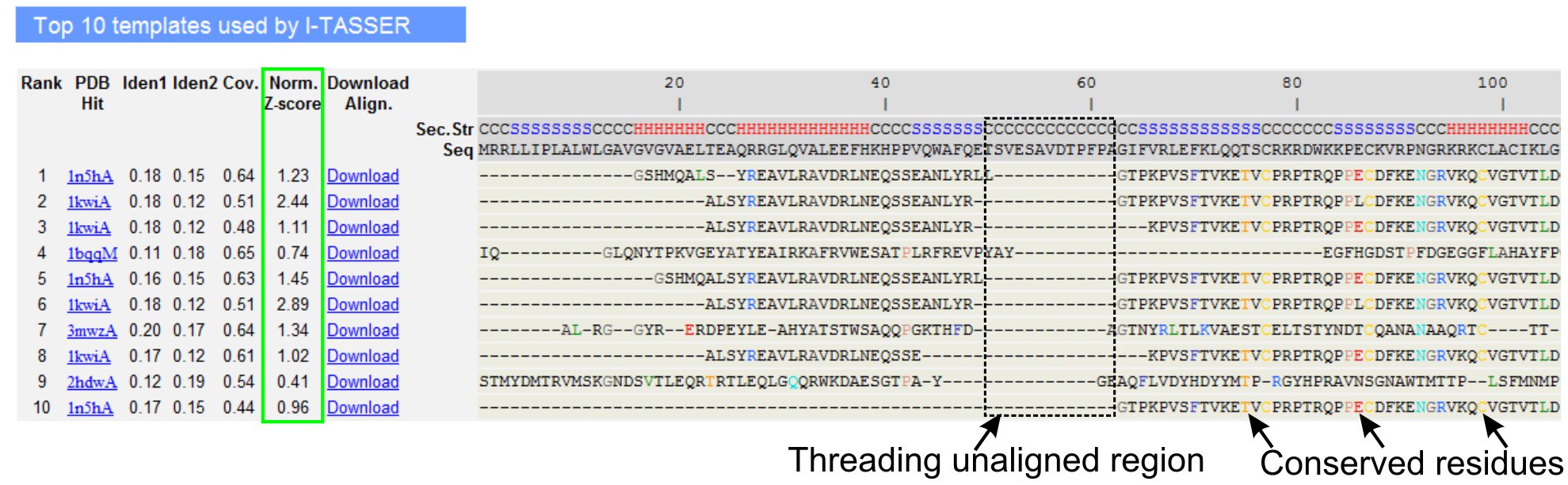
- Rul ned for at analysere de ti bedste trådning skabeloner af forespørgslen protein, som er identificeret af LOMETS gevindskæring programmer (Figur 3). Se normaliseret Z-score (Eq. 1), der er vist i 'Norm. Z-score 'kolonnen, til at analysere kvaliteten af gevindskæring alignments. Alignments med et normaliseret Z-score> 1 er udtryk for en sikker tilpasning og sandsynligvis har samme fold som forespørgslen protein.
- Analyser rækkefølgen identitet i trådning alliancefrie regionen (kolonnen »Iden. 1 ') og for hele kæden (kolonnen» Iden. 2 «) til at vurdere homologi mellem forespørgslen og skabelonen proteiner. Høj sekvens identitet er en indikator for evolutionære slægtskab mellem forespørgslen og skabelon proteiner.
- Se trådning afstemt rester vist i farvet til visuelt at identificere ulempererved rester / motiver i forespørgslen og skabelonen proteiner. En højere sekvens identitet i gevind-tilpasset regionen, i forhold til hel-kæde tilpasning også indikerer tilstedeværelsen af velbevarede strukturelle motiv / domæner i forespørgslen.
- Vurdere dækning af gevindskæring tilpasning ved at se på 'Cov.' kolonne og inspektion af justeringen. Hvis dækningen af de bedste alignments er lav og begrænset til kun et lille område af forespørgslen protein eller fraværende i en længere segment af query sekvens, så forespørgslen protein normalt indeholder mere end ét domæne, og det anbefales at opdele sekvensen og model domænerne individuelt (Figur 3).
- Download FBF formateret sekvens-struktur tilpasning filer ved at klikke på "Download Juster" links. Disse tilpasning fil kan åbnes i enhver molekylær visualisering program, der er opført i materialer afsnittet, og kan også bruges til at tilføje yderligere begrænsninger i strukturen modellering (Trin 1,4).
6.Strukturelle analoger i FBF
- Vis det næste tabel (Figur 4) af resultatet side til at bestemme top ti strukturelle analoger af det første forudsagde modellen, som er identificeret af den strukturelle tilpasning programmet TM-align 20. En TM-score> 0,5 tyder på, at det fundne analoge og model har en lignende topologi og kan bruges til at bestemme de strukturelle klassen / protein familie af forespørgslen protein 16, mens dem med TM-score <0,3 betyder en tilfældig struktur lighed.
- Analyser rækkefølgen identitet og RMSD i strukturelt tilpasset regionen vist i "Iden a« og »RMSD en 'kolonner til at vurdere bevarelse af rumlige motiver i modellen og de strukturelle analog. Efterse de farvede og afstemt rester par i tilpasningen til at identificere disse strukturelt bevarede rester og motiver.
- Klik på FBF kode, som vises i 'FBF Hit' kolonne for at besøge RCSB hjemmeside og lære mere om deres strukturelle klassificering (SCOP, Cath og PFAM) og funktionelle oplysninger (EF-nummer, tilknyttede GO vilkår og bundne ligand).
7. Funktion forudsigelse ved hjælp af cofaktor
- Rul ned i resultatet side til at analysere funktionelle fortolkninger for forespørgslen protein. Protein funktioner er opregnet i tre led tabeller, der viser: Enzym-Kommissionen (EF) numre, Gene Ontology (GO) vilkår, og ligand bindingssteder.
- Se 'TM-score', 'RMSD a', 'identi a «og» Cov.' kolonner i hver tabel til at analysere parametrene for global struktur lighed og bevarelse af rumlige mønstre mellem model og identificeret funktionelle homologe (skabeloner).
8. Enzym Kommissionen nummer forudsigelse
- Se top fem mulige enzym homologe af forespørgsel protein vist i "Predicted EF-numre" tabel (Figur 5). Konfidensniveauet af EF nummer forudsigelse ved hjælp af disse skabeloner er vist i »EF-Score 'kolonne. Baseret på benchmarking analyse 23, funktionel lighed (3 første cifre i EF-nummer) mellem forespørgslen og skabelon protein kan opgøres pålideligt fortolkes ved hjælp af EF-score> 1.1.
- Kig efter konsensus af funktion (EF-numre) blandt de skabeloner, som har de samme fold (dvs. TM-score> 0,5), som forespørgslen protein. Hvis flere skabeloner har samme EF-nummer og EF-score> 1.1, konfidensniveau forudsigelse er meget høj. Men hvis EF-Score er høj, men der er en manglende konsensus blandt de identificerede hits, så forudsigelsen bliver mindre pålidelige og brugerne anbefales at konsultere GO sigt forudsigelser.
- Klik på linket på EF-numre til at besøge ExPASy Enzym-databasen og analysere den funktion, herunder reaktionen katalyseret, co-faktor krav og metaboliseringsvej, af skabelonen protein i detaljer.
9. Gene Ontology (GO) sigt forudsigelser
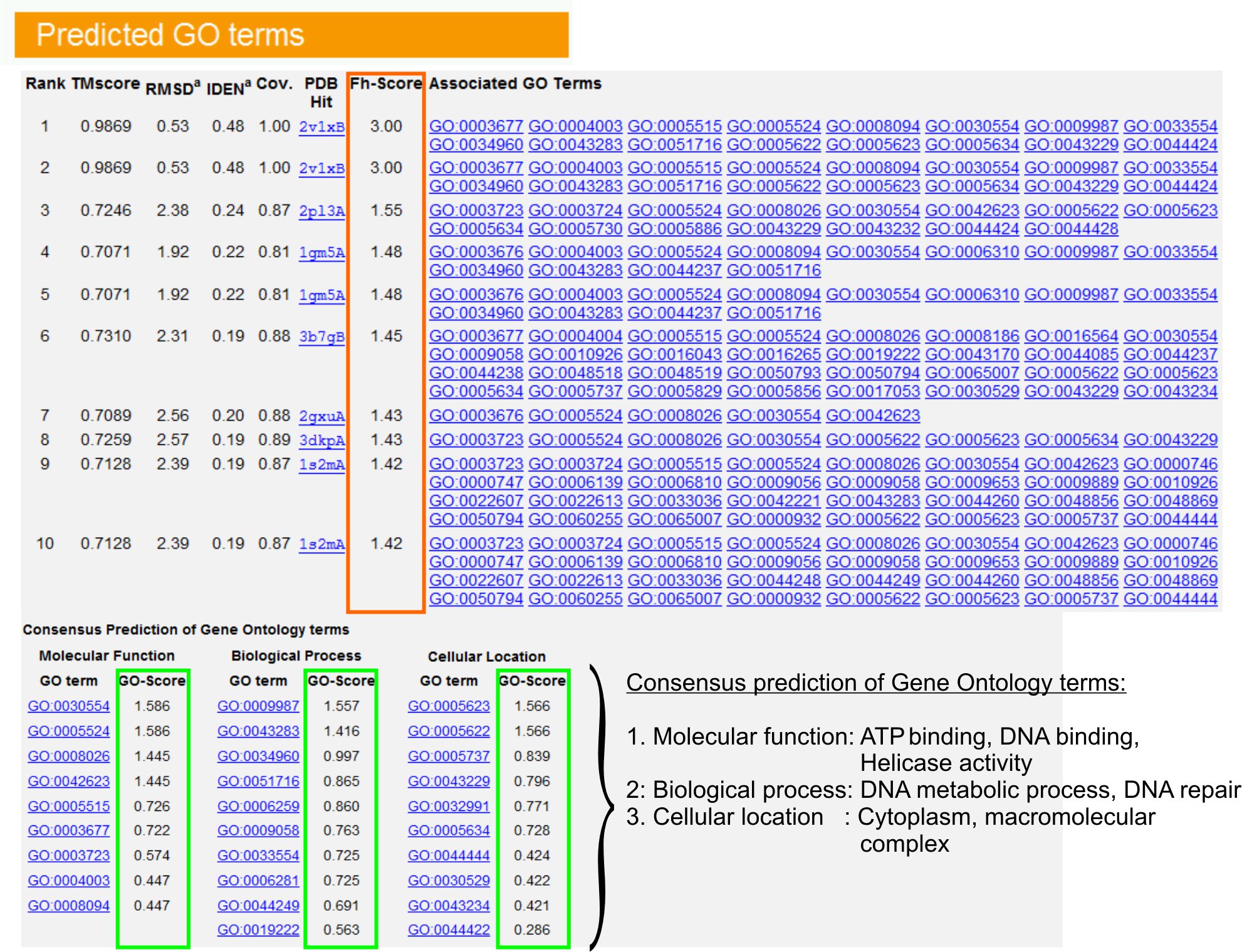
- Se "forudsagde GO ordene" tabel (FigURE 6) for at identificere top ti homologe af forespørgsel protein i FBF biblioteket, kommenteret med Gene Ontology (GO) vilkår. Hvert protein er normalt forbundet med flere GO vilkår, der beskriver dens molekylære funktioner (MF), biologiske processer (BP) og cellulære komponent (CC). Klik på hvert sigt at besøge Amigo hjemmeside og analysere dens definition og afstamning.
- Analyser Fh-score (Functional homologi score) kolonne for at få adgang til den funktionelle lighed mellem forespørgslen og skabelon proteiner og et skøn over konfidensniveau at overføre funktionelle annotation fra disse proteiner. I vores benchmarking-undersøgelse 23, kunne 50% af den indfødte GO vilkårene være korrekt identificeret fra den første identificerede skabelon ved hjælp af en Fh-score cutoff på 0,8, med en samlet nøjagtighed på 56%.
- Se "Konsensus forudsigelse af GO udtrykkene" tabel til at analysere sammenfald af funktion mellem skabeloner. Disse fælles funktioner benyttes til at forudsige GO vilkår (MF, BP og CC) af forespørgslenprotein og vurdere konfidensniveau (GO-score) af GO sigt forudsigelser. Baseret på benchmarking-test 23, er de bedste falsk positive og falsk negative vækstrater opnået for forudsigelser med GO-score cutoff = 0,5, med faldende dækning af forudsigelse på dybere ontologi niveauer.
10. Protein-ligand bindingssted forudsigelser
- Rul ned til bunden af siden for at se top ti ligand bindingssted forudsigelser for forespørgslen protein. Predicted bindingssteder er rangeret baseret på antallet af forudsagde ligand konformationer, der deler fælles bindende lomme. Den bedste identificerede bindingssted allerede vises i Jmol applet. Klik på radioknapper til at analysere andre forudsigelser og visualisere ligand interagerer rester.
- Analyser BS-score kolonne for at vurdere de lokale lighed mellem modellen og skabelonens bindingssted. Baseret på den toneangivende 9,> 1,1 BS-score indikerer høj sekvens og struktur similarity nær det forudsagte bindingssted i model og kendt bindingssted i skabelonen.
- Download FBF formateret struktur-fil af komplekset ved at klikke på "Download" linket. Brugere kan åbne disse filer i en vilkårlig molekylær visualisering program og interaktivt se forudsagt bindingssted og ligand-protein interaktioner på deres lokale computer.
11. Repræsentative resultater

Figur 1 viser et uddrag af I-TASSER resultat side viser (A) FASTA formateret query sekvens;. (B) forudsagt sekundær struktur og tilhørende tillid scores, og (C) forudsagt opløsningsmiddel tilgængelighed rester. Analyseret kerne region og potentielle hydrering websted i forespørgslen er fremhævet med cyan og røde rektangler, hhv.

Figur 2.

Figur 3. Et eksempel på I-TASSER resultat side viser top ti identificeret gevindskæring skabeloner og alignments af LOMETS 5 gevindskæring programmer. Kvaliteten af gevindskæring justeringer vurderes baseret på normaliserede Z-score (markeret med grønt), hvor en værdi> 1 afspejler en sikker tilpasning. Alliancefri rester i den skabelon, der er identiske med de tilsvarende forespørgsel rester er fremhævet i farve for at indikere tilstedeværelsen af velbevarede rester / motiv, mens en manglende tilpasning i de fleste af Top skabeloner indikerer tilstedeværelsen af flere domæner i forespørgslen protein og unaligned restprodukter svarer til domæne linkeren regioner. Klik her for at se fuld størrelse version af figur 3.

Figur 4. Et eksempel på resultatet side viser top ti identificerede strukturelle analoger og strukturelle justeringer, identificeret ved TM-align 20 strukturelle tilpasning program. Rangordningen af analoger vist i er baseret på TM-score (fremhævet med blåt) af den strukturelle tilpasning. En TM-score> 0,5 tyder på, at de to forhold strukturer har en lignende topologi, mens en TM-score <0,3 betyder en lighed mellem to tilfældige strukturer. Strukturelt tilpasset rester par er fremhævet i farver baseret på deres amino-syre ejendom, mens unaligned regioner er angivet med "-".ove.com/files/ftp_upload/3259/3259fig4large.jpg "> Klik her for at se fuld størrelse version af figur 4.

Figur 5. Et eksempel på I-TASSER resultat side viser identificeret enzym homologe af forespørgslen protein i FBF biblioteket. Konfidensniveauet af EF nummer forudsigelse er analyseret baseret på EF-score (markeret med grønt), hvor EF-score> 1,1 indikerer funktionel lighed (samme første 3 cifre i EF-nummer) mellem forespørgsel og skabelon protein.

Figur 6. Et eksempel på I-TASSER resultat side viser GO sigt forudsigelser for forespørgslen protein. Funktionel homologer for forespørgslen protein i Gene Ontology Template Library er rangordnet ud fra deres Fh-score (i orange firkant). Fælles funktionelle egenskaber fra disse top-scoring hits er udledt til gene spiste den endelige GO sigt forudsigelser for forespørgslen protein. Kvaliteten af den forudsagte GO begreber er estimeret baseret på GO-score (vist med grønt), hvor en GO-score> 0,5 indikerer en pålidelig forudsigelse. Klik her for at se fuld størrelse version af figur 6.

Figur 7. Et eksempel på I-TASSER resultat side viser top ti protein liganden bindingssted forudsigelser ved hjælp af cofaktor 9 algoritme. Placeringen af den forudsagte bindingssteder er baseret på antallet af forudsagde ligand konformationer, der deler fælles bindende lomme i forespørgslen. BS-score (markeret med rødt) er et mål for lokal sekvens og struktur lighed mellem det forudsagte og skabelonens bindingssted, og er nyttig til at analysere bevarelse af bindingssted lommer.
les/ftp_upload/3259/3259fig8.jpg "/>
Figur 8. Et eksempel på ekstern tilbageholdenhed filer, der bruges til at til at angive rester rester kontakt / distance begrænsninger.

Figur 9. Eksempel på tilbageholdenhed filer, der bruges til at angive en skabelon protein til I-TASSER server. Brugeren kan angive forespørgslen-skabelonen tilpasning enten (A) FASTA format, eller (B) 3D-format.

Figur 10. Et eksempel fil, der bruges til at udelukke skabelon under I-TASSER struktur modellering procedure. Den første kolonne indeholder det foreløbige budgetforslag ID af skabelonen proteiner, der skal udelukkes. Den anden kolonne bruges til at angive den rækkefølge identitet cutoff, som vil blive anvendt til andre lignende skabeloner i skabelonen biblioteket.
Discussion
Protokollen præsenteret ovenfor er en generel retningslinje for struktur og funktion modellering ved hjælp af I-TASSER server. Selv om denne automatiske procedure virker meget godt for de fleste af de proteiner, menneskelige indgreb ofte hjælpe i væsentlig grad at forbedre modellerne nøjagtighed, især for de proteiner, som mangler tæt skabeloner i FBF biblioteket. Brugere kan gribe ind i løbet af I-TASSER modellering på følgende måder: (a) opdeling af multi-domæne proteiner (b) at give eksterne begrænsninger for at forbedre strukturen forsamling, og (c) At fjerne skabeloner i løbet af modellering.
Opdeling af multi-domæne protein:
Mange lange protein-sekvenser ofte indeholder flere domæner tøjret ved hjælp af fleksible linker regioner, hvilket gør deres struktur opklaringen vanskelig ved hjælp af både eksperimentelle og beregningsmæssige teknikker. Ikke desto mindre, som domæner er uafhængigt folde enheder og kan udføre forskellige molekylære funktion, det erønskeligt at opdele lange multi-domæne proteiner og model hvert enkelt domæne. Modeling domæner individuelt vil ikke kun fremskynde forudsigelsen processen, men også øger kvaliteten af query-skabelon tilpasning, hvilket resulterer i mere pålidelig struktur og funktion forudsigelser.
Domæne grænser i protein-sekvenser kan forudsiges ved hjælp af frit tilgængelige eksterne online-programmer som NCBI CDD 24, PFAM 25 eller InterProScan 26. Også, hvis LOMETS gevindskæring justeringer er tilgængelige for forespørgslen protein, kan domænenavn grænser være placeret ved visuelt at identificere lange strækninger af unaligned rester i øverste trådning skabeloner (se trin 5.4). Disse unaligned regioner overvejende svarer til domæne linkeren regioner. Hvis multi-domæne skabeloner er allerede tilgængelige i skabelonen FBF bibliotek med alle forespørgslen justeret domæner, så forespørgslen protein kan modelleres som fuld længde.
Sørg for eksterne begrænsninger
A. Angiv kontakt / distance begrænsninger
Eksperimentelt præget inter-rester kontakter / afstande, for eksempel fra NMR ellertværbindende eksperimenter, kan specificeret ved at uploade et tilbageholdenhed fil. Et eksempel fil er vist i figur 8, hvor kolonne 1 angiver den type frihedsberøvelse, nemlig "DIST" eller "kontakt". Ved fjernsalg tilbageholdenhed (DIST), kolonne 2 og 4 indeholder rester positioner (i, j), kolonne 3 og 5 indeholder de atom-typer i den rest, og kolonne 6 angiver afstanden mellem de to angivne atomer. For kontakt begrænsninger (kontakt), kolonne 2 og 3 indeholder de positioner (i, j), restprodukter, som skal være i kontakt. Afstanden mellem sidekæder centrum af disse kontakter rester par afgøres baseret på observerede afstande i kendte strukturer i FBF. I-TASSER vil forsøge at tegne disse atom par tæt på den angivne afstand i løbet af strukturen raffinement simuleringer.
B. Angiv et protein struktur skabelon
LOMETS threading programmer bruger en repræsentant FBF biblioteket for at finde en plausibel folder for forespørgslen protein. Selv om der ved hjælp af en repræsentativ struktur bibliotek hjælper til at reducere den tid, der kræves for at beregne den sekvens-struktur alignments, er det muligt, at en god skabelon protein er savnet i biblioteket eller skabelon kan ikke have været identificeret ved LOMETS gevindskæring programmer, selvom det er til stede i biblioteket. I disse tilfælde skal brugeren angive den ønskede protein struktur som skabelon.
Hvis du vil angive protein-struktur som en ekstra skabelon, kan brugere enten uploade et FBF formateret struktur fil eller angive FBF-ID for en deponeret protein struktur i FBF bibliotek. I-TASSER vil generere forespørgslen-skabelonen tilpasning ved hjælp af Muster-programmet 23, og vil indsamle rumlige begrænsninger fra både den angivne bruger skabelonen, og LOMETS skabeloner til at guide strukturen forsamling simulering. Fordi nøjagtigheden af LOMETS begrænsninger er forskellig for forskellige mål, vægten af LOMETS begrænsninger er stærkere i let (homologe) targets end den hårde (ikke-homologe) mål, som er blevet systematisk tunet i vores benchmark træning.
Brugere kan også angive deres egne query-skabelon alignments. Serveren accepterer tilpasning i to formater: FASTA format (Figur 9A) og 3D-format (Figur 9B). Den FASTA format er standard og beskrevet på http://zhanglab. ccmb.med.umich.edu / FASTA / . 3D-formatet svarer til den standard FBF format ( http://www.wwpdb.org/documentation/format32/sect9.html ), men yderligere to kolonner er afledt af de skabeloner føjes til ATOM poster (se figur 9B):
Kolonne 1-30: Atom (C-alfa-only) og rester navne til forespørgslen sekvens.
Kolonne 31-54: Koordinater af C-alfa atomer af forespørgslen kopieret fra den tilsvarende atomer i skabelonen.
Kolonne 55-59: tilsvarende restkoncentrationer nummer i den skabelon baseret på tilpasning
Kolonne 60-64: tilsvarende restkoncentrationer navn i skabelonen
Udelad skabeloner proteiner
Proteiner er fleksible molekyler og kan vedtage flere konformationelle stater til at ændre deres biologiske aktivitet. For eksempel har strukturer i mange protein kinaser og membranproteiner blevet løst i både aktive og inaktive kropsbygning. Også tilstedeværelsen eller fraværet af bundne ligand kan forårsage store strukturelle bevægelser. Mens alle de konformationelle stater i skabelonen er ens for trådning programmer, er det ønskeligt at modellere forespørgsel ved hjælp af skabeloner i kun én bestemt tilstand. En ny mulighed på serveren giver brugeren mulighed for at udelukke skabelon proteiner i løbet struktur modellering. Denne funktion vil også gøre det muligt for brugeren at vælge homologi niveau af skabeloner, der kan anvendes til modellering. Brugere kan udelukke skabelon proteiner frOM I-TASSER bibliotek ved at:
A. Angivelse af en sekvens identitet cutoff
Brugere kan bruge denne mulighed for at udelukke homologe proteiner fra I-TASSER skabelon bibliotek. Det homologi er indstillet baseret på rækkefølgen identitet cutoff, det vil sige antallet af identiske rest mellem forespørgslen og skabelonen protein divideret med sekvens længden af forespørgslen sekvens. For eksempel, hvis brugeren typer i "70%" i den viste form, alle skabeloner proteiner, der har en sekvens identitet> 70% til forespørgslen protein jeg-vil blive udelukket fra I-TASSER skabelon bibliotek.
B. Udeluk bestemt skabelon proteiner
Bestemt skabelon proteiner, kan udelukkes fra I-TASSER Template Library ved at uploade en liste med FBF-id'er for de strukturer, der skal udelukkes. Et eksempel fil er vist i figur 10. Da de samme protein kan eksistere som flere poster i FBF bibliotek, I-TASSER se-floden vil som standard udelukke den angivne skabeloner (i Kolonne1) samt alle andre skabeloner fra det bibliotek, der har en identitet> 90% til den angivne skabeloner. Brugere kan også angive en anden identitet cutoff, fx 70%, hvor alle skabeloner med identitet> 70% til specifikke skabelon proteiner vil blive udelukket.
Disclosures
Ingen interessekonflikter erklæret.
Acknowledgments
Projektet er delvist understøttet af Alfred P. Sloan Foundation, NSF Career Award (DBI 1.027.394), og National Institute of General Medical Sciences (GM083107, GM084222).
Materials
| Name | Company | Catalog Number | Comments |
| Material Name | Type | Company | Catalogue Number |
| FASTA formatted amino acid sequence of the protein to be modeled (see, http://www.ncbi.nlm.nih.gov/BLAST/fasta.shtml). | |||
| A personal computer with access to the internet and a web browser. | |||
| Molecular visualizing software, e.g. RASMOL or PYMOL, for analyzing the predicted tertiary structure and functional sites. |
References
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H. CD-Search: protein domain annotations on the fly. Nucleic. Acids. Res. 32, W327-W331 (2004).
- Finn, R. D. The Pfam protein families database. Nucleic. Acids. Res. 38, D211-D222 (2010).
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).

{kind=link}
{kind=link}