

Summary
Richtlijnen voor de computer op basis van structurele en functionele karakterisering van eiwitten met behulp van de I-TASSER pijplijn is beschreven. Vanaf vraag eiwitsequentie, zijn 3D-modellen gegenereerd met behulp van meerdere threading uitlijningen en iteratief structurele assemblage simulaties. Functionele gevolgtrekkingen worden daarna getrokken op basis van wedstrijden om eiwitten met bekende structuur en functies.
Abstract
Genoom sequencing projecten zijn versleuteld miljoenen eiwitsequentie, die de kennis van hun structuur en functie nodig hebben om het begrip van hun biologische rol te verbeteren. Hoewel de experimentele methoden kunnen gedetailleerde informatie te verstrekken voor een klein deel van deze eiwitten, is computationele modellering nodig is voor het merendeel van de eiwit-moleculen die experimenteel zijn ongekarakteriseerde. De I-TASSER server is een on-line werkbank voor hoge-resolutie modellering van eiwit structuur en functie. Gegeven een eiwitsequentie, een typisch uitgang van de I-TASSER server bevat secundaire structuur voorspelling, voorspelde solvent toegankelijkheid van elk residu, homologe template eiwitten aangetroffen door te rijgen en structuur uitlijningen, tot vijf full-length tertiaire structurele modellen, en structure-based functionele annotaties voor enzym classificatie, Gene Ontology termen en eiwit-ligand binding sites. Alle voorspellingen zijn gemarkeerd met een score die het vertrouwenvertelt hoe nauwkeurig de voorspellingen zijn zonder het te weten de experimentele data. Ter vergemakkelijking van de speciale wensen van de eindgebruikers, de server geeft kanalen naar de gebruiker opgegeven inter-residu op afstand te accepteren en kaarten contact met interactief veranderen de I-TASSER modelleren, maar ook kunnen gebruikers met vermelding van eventuele eiwitten als sjabloon of uit te sluiten elke template eiwitten tijdens de structuur van de montage simulaties. De structurele informatie kan worden verzameld door de gebruikers op basis van experimentele bewijzen of biologische inzichten met als doel het verbeteren van de kwaliteit van de I-TASSER voorspellingen. De server werd geëvalueerd als de beste programma's voor proteïne structuur en functie voorspellingen in het recente community-brede CASP experimenten. Er zijn momenteel> 20.000 geregistreerde wetenschappers uit meer dan 100 landen die gebruik maken van de on-line I-TASSER server.
Protocol
Methode overzicht
Naar aanleiding van de sequentie-to-structuur-to-paradigma, de I-TASSER procedure 1-4 voor de structuur en functie van modellen bestaat uit vier opeenvolgende stappen van: (a) template identificatie door LOMETS 5, (b) fragment structuur montage door replica- uitwisseling Monte Carlo simulaties 6, (c) atomair niveau de structuur verfijning met behulp van REMO 7 en FG-MD 8, en (d) structure-based functie interpretaties met behulp van co-factor 9.
Sjabloon identificatie: Voor een query-sequentie die door de gebruiker, is de volgorde eerste schroefdraad door middel van een vertegenwoordiger van VOB structuur bibliotheek door een lokaal geïnstalleerde LOMETS meta-threading server. Threading is een sequentie-structuur alignment procedure die wordt gebruikt voor het identificeren van eiwitten die template vergelijkbare structuur hebben of bevatten vergelijkbare structurele motief als de query eiwit. Om de dekking van de homologe templ vergrotenaten detecties, LOMETS combineert meerdere state-of-the-art algoritmen die verschillende threading methodieken. Aangezien verschillende threading-programma's hebben verschillende scoringssystemen en uitlijning gevoeligheden, is de kwaliteit van de gegenereerde threading uitlijningen van elkaar threading programma beoordeeld door genormaliseerde Z-score, die wordt gedefinieerd als: 
waar de Z-score is de score in standaarddeviatie-eenheden ten opzichte van het statistische gemiddelde van alle uitlijningen gegenereerd door het programma, en Z 0 is een programma-specifiek Z-score cutoff bepaald op basis van grootschalige threading benchmark-tests 5 om een onderscheid te 'goed 'en' slechte 'templates. Een sjabloon met een hoge Z-score betekent dat de top templates een afstemming scoren significant hoger dan de meeste van de andere sjablonen, die meestal betekent dat de uitlijning correspondeert met een goed model te hebben. Als de meeste van de top threading templates hebben high genormaliseerde Z-scores, de nauwkeurigheid van de uiteindelijke I-TASSER model is meestal hoog. Echter, als het eiwit groot is en de dekking van de threading uitlijningen is beperkt tot een klein gebied van de query eiwit, heeft een hoge genormaliseerde Z-score niet per se een hoog model nauwkeurigheid voor de full-length model. Top twee threading alignementen van elk threading programma zijn verzameld en gebruikt voor de volgende stap van structuur montage.
Iteratieve structuur van de montage-simulatie: Na de threading procedure, query volgorde is opgesplitst in threading uitgelijnd en niet aangepaste regio's. Continue fragmenten in te rijgen lijn zijn uitgesneden uit sjablonen en direct gebruikt voor de structuur montage, terwijl de niet aangepaste lus regio's zijn gebouwd door ab initio modellering. De structuur assemblage procedure wordt uitgevoerd op een rooster systeem geleid door de replica uitwisseling van Monte Carlo simulaties 6. De I-TASSER krachtveld bestaat uit waterstof-bonding interacties 10, op kennis gebaseerde statistische termen van energie afgeleid van bekende eiwitstructuren in het VOB 11, sequentie-gebaseerde contact voorspellingen van SVMSEQ 12, en ruimtelijke beperkingen verzameld van LOMETS 5 threading templates. De conformationele lokvogels gegenereerd in de lage-temperatuur replica's tijdens de simulaties zijn geclusterd door Spicker 13 tot en met structuren van een lage vrije-energie-staten te identificeren. Cluster hartlijnen van de top clusters worden verkregen door het gemiddelde van de 3D-coördinaten van alle geclusterde structurele lokvogels en gebruikt voor het uiteindelijke model generatie. De simulatie en clustering procedure twee keer herhaald voor het verwijderen van sterische botsingen en verdere verfijning van het globale topologie.
Atomaire niveau van modelbouw en verfijning: Het cluster hartlijnen verkregen na Spicker clustering worden verlaagd eiwit modellen (elk residu vertegenwoordigd door haar C α en side-keten centrum van de massa) en have beperkte biologische toepassing. De bouw van de full-atomaire model van het verminderde modellen gebeurt in twee stappen. In de eerste stap wordt REMO 7 gebruikt voor de volledige atomaire modellen te construeren van C-alfa sporen door het optimaliseren van de H-band netwerken. In de tweede stap, REMO full-atomaire-modellen verder worden verfijnd door FG-MD 14, die de ruggengraat torsie hoeken, obligatie lengtes, en de side-keten rotameer oriëntaties verbetert, door moleculaire dynamische simulaties, zoals geleid door de structurele fragmenten doorzocht van de VOB structuren door TM-lijnen. De FG-MD verfijnde modellen worden gebruikt als de uiteindelijke modellen voor tertiaire structuur voorspellingen van I-TASSER.
De kwaliteit van de gegenereerde modellen zijn geschat op basis van een vertrouwen score (C-score), die is gedefinieerd op basis van de Z-score van LOMETS threading uitlijningen en de convergentie van de I-TASSER simulaties, wiskundig geformuleerd als: 
waar
De C-score heeft een sterke correlatie met de kwaliteit van de I-TASSER modellen. Door het combineren van C-score en eiwit lengte, kan de nauwkeurigheid van de eerste I-TASSER modellen worden geschat met een gemiddelde fout van 0,08 voor de TM-score en 2 A voor de RMSD 15. In het algemeen, modellen met een C-score> - zijn 1,5 zal naar verwachting een juiste plooi te hebben. Hier RMSD en TM-score zijn beide bekende maatregelen van de topologische gelijkenis tussen het model en de natieve structuur. TM-score waardevollees variëren in [0, 1], waarbij een hogere score duidt op een betere structuur overeenkomen met 16,17. Maar voor lagere-ranked modellen (dat wil zeggen 2 e -5 e-modellen), de correlatie van de C-score met TM-score en RMSD is veel zwakker (~ 0.5), en kan niet worden gebruikt voor een betrouwbare schatting van absolute model kwaliteit.
Is het eerste model altijd het beste model in I-TASSER simulaties? Het antwoord op deze vraag hangt af van de doelgroep type. Voor een eenvoudige doelwitten, het eerste model is meestal de beste model en de C-score is meestal veel hoger dan de rest van de modellen. Echter, voor harde doelen, waar de draad niet significant template hits, het eerste model is niet per se de beste model en I-TASSER eigenlijk heeft moeite bij het selecteren van de beste template en modellen. Het is daarom aan te raden om alle vijf modellen te analyseren voor harde doelen en hen op basis van de experimentele informatie en de biologische kennis te selecteren.
Functie Predictions: In de laatste stap, zijn definitief 3D-modellen gegenereerd op basis van FG-MD gebruikt om drie aspecten van eiwitten functie te voorspellen, te weten: a) Enzyme Commissie (EC) nummers 18 en (b) Gene Ontology (GO) 19 termen en ( c) bindingsplaatsen voor kleine molecule liganden. Voor alle drie aspecten, zijn functionele interpretaties gegenereerd met behulp van co-factor, dat is een nieuwe benadering van eiwit functie op basis van globale en lokale gelijkenis met template eiwitten in het VOB met bekende structuur en functies te voorspellen. Ten eerste is de globale topologie van de voorspelde modellen vergeleken met functionele template libraries met behulp van structurele afstemming programma TM-lijn 20. Vervolgens wordt een reeks van eiwitten het meest lijkt op de doelgroep modellen geselecteerd uit de bibliotheek op basis van hun globale structuur gelijkenis, en een uitgebreide lokale zoekopdracht wordt uitgevoerd om de structuur en sequentie-overeenkomst in de buurt van het actieve / bindingsplaats regio te identificeren. De resulterende globale en lokale gelijkenis scores worden gebruikt om de rangtemplate eiwitten (functionele homologen) en breng de annotatie (EG nummers en Gene Ontology 19 voorwaarden) op basis van de hoogst scorende hits. Evenzo worden ligand bindingsplaats residuen en de ligand binding modus afgeleid op basis van de lokale afstemming van de vraag met bekende ligand bindingsplaats residuen in de hoogst scorende functie templates 9.
De kwaliteit van de functie (EG en GO termijn) voorspelling in I-TASSER is geëvalueerd op basis van functionele homologie score (FH-score), die een maat is van de mondiale en lokale overeenkomst tussen de query en de template, en wordt gedefinieerd als: 
waar C-score is een schatting van de kwaliteit van de voorspelde model als omschreven in Eq. (2), TM-score meet de globale structurele gelijkenis tussen het model en de template eiwitten; RMSD ali is de RMSD tussen het model en de template structuur in de regio structureel lijn van TM-align 20; Cov vertegenwoordigt de dekking van de structurele uitlijning (dwz de verhouding van de structureel gericht residuen gedeeld door de query lengte); ID-ali is de sequentie-identiteit in de TM-align uitlijning. De geschatte vertrouwen score voor EG-nummer voorspellingen bevat ook een term voor het beoordelen van actieve site match (ACM) tussen de query en sjabloon in een bepaalde streek, berekend als: 
waarbij N t het aantal sjabloon residuen aanwezig binnen de lokale gebied vertegenwoordigt, N ali is het nummer van de aangepaste query-template residu paren, D II is de C α afstand tussen de i-de twee uitgelijnde residuen, d 0 = 3,0 Å is de afstand cutoff, M ii is de BLOSUM scores tussen et paar uitgelijnd residuen. In het algemeen, de Fh-score is in het bereik [0, 5] en ACM score tussen [0, 2], Waarbij hogere scores geeft meer zelfvertrouwen functionele opdrachten. ACM score wordt ook gebruikt voor het evalueren van de lokale structuur en sequentie overeenkomst de buurt van de ligand-bindende sites, die wordt aangeduid als BS-score.
1. Indiening van eiwitsequentie
- Bezoek de I-TASSER webpagina op http://zhanglab.ccmb.med.umich.edu/I-TASSER om te beginnen met structuur en functie modelleren experiment.
- Kopieer en plak de aminozuursequentie in de verstrekte formulier of direct te uploaden vanaf uw computer door te klikken op de knop "Bladeren". I-TASSER server accepteert momenteel sequenties met tot 1500 residuen. Eiwitten langer dan 1500 resten zijn meestal multi-domein eiwitten, en worden aanbevolen om te worden opgesplitst in afzonderlijke domeinen voordat zij naar de I-TASSER.
- Geef uw e-mail adres (verplicht) en een naam voor de job (optioneel).
- Gebruikers kunnen naar keuze opgeven externe inter-residue contact / afstand beperkingen, add-in een extra sjabloon of template uit te sluiten sommige eiwitten tijdens de structuur modelleren. Meer informatie over het gebruik van deze opties in de "Discussion" sectie.
- Voor het indienen van de reeks, klik op de "Run I-TASSER" knop. De browser zal worden gericht op een bevestigingspagina met de gebruiker opgegeven informatie, job identificatie (Job ID) nummer en een link naar een webpagina waar de resultaten zullen worden gestort na voltooiing van de baan. Gebruikers kunnen bookmark deze link of noteer de baan identificatienummer voor toekomstig gebruik.
2. Beschikbaarheid van de resultaten
- Controleer de status van uw ingediende werk door een bezoek aan de I-TASSER wachtrij pagina op http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php . Klik op het tabblad Zoeken en gebruik de Job ID-nummer of de query volgorde om uw ingediende zoeken naar een baan.
- Na de structuur en functie modeling is voltooid, wordt een notificatie e-mail met daarin beeld van de voorspelde structuren en een web-link naar u worden verzonden. Klik op deze link of open de link bookmark bij stap 1.5 te bekijken en te downloaden van de resultaten.
3. Secundaire structuur en oplosmiddel toegankelijkheid voorspellingen
- Controleer de FASTA geformatteerde zoeksequentie weergegeven op de bovenkant van het resultaat pagina. Als er extra terughoudendheid / template werd opgegeven tijdens de volgorde indiening, een link naar de webpagina weergeven door de gebruiker opgegeven informatie kan ook worden gezien (Figuur 1A).
- Onderzoek van de secundaire structuur voorspelling weergegeven als: alfa helix (H), beta-streng (S) of spoel (C) en het vertrouwen score van voorspelling (0 = laag, 9 = hoog) voor elk residu. Kijk voor regio met lange stukken van regelmatige secundaire structuur (H of S) voorspellingen, tot de kern-regio schatten in het eiwit. Structurele klasse van eiwitten kunnen ook geanalyseerd worden op basis van de verdeling van secundaire structuren elementen. Alzo, lang regio's van de spoel elementen in het eiwit wijzen gewoonlijk op ongestructureerde / wanordelijke gebieden.
- Bekijk de voorspelde solvent toegankelijkheid (figuur 1C) om na te gaan begraven en oplosmiddel belichte gebieden in de query. Waarden van de voorspelde solvent toegankelijkheid variëren van 0 (begraven residu) tot 9 (blootgestelde residu). Regio die voornamelijk begraven residuen kunnen worden gebruikt om de kern regio af te bakenen in het eiwit, terwijl de regio's met een oplosmiddel worden blootgesteld en hydrofiele residuen zijn potentiële hydratatie / functionele sites.
4. Tertiaire structuur voorspellingen
- Scroll naar beneden naar de voorspelde tertiaire structuren van eiwitten query's, weergegeven in een interactief Jmol applet (figuur 2) te bekijken. Klik met de linkermuisknop op de applet om het uiterlijk van de weergegeven structuur te veranderen, zoom in specifieke regio, selecteert u specifieke residu types in de voorspelde model of de berekening van inter-residu afstanden.
- Analyseer de modellen voor de aanwezigheid van lange ongestructureerde regio's. Deze rREGIO 'S meestal overeen met ongeordende regio's in eiwit of wijzen op een gebrek van de sjabloon uitlijning. Deze regio's hebben over het algemeen een lage modellering nauwkeurigheid en het verwijderen van deze regio's tijdens het modelleren van N & C-terminus regio zal het modelleren nauwkeurigheid te verbeteren.
- Download het VOB opgemaakte bestanden structuur van het model door te klikken op de "Download Model" links. U kunt deze bestanden in een moleculaire visualisatie software (bijv. pymol, Rasmol enz.) voor verdere analyse van de structurele kenmerken.
- Analyseer het vertrouwen score (C-score) van de structuur van modellering om de kwaliteit van de voorspelde structuren te schatten. C-score (Vgl. 2) waarden zijn meestal in het bereik [-5, 2], waarin een hogere score wijst op een model van betere kwaliteit. De geschatte TM-score en RMSD van het eerste model is weergegeven als "Geschatte nauwkeurigheid van Model 1". Voor lange eiwitten, is het raadzaam om het model kwaliteit op basis van TM-score te evalueren, zoals TM-score is gevoeliger voor de topologische veranderingen dan RMSD. < li> Klik op "meer informatie over C-score" link naar C-score te analyseren, cluster grootte en de cluster dichtheid van alle modellen. Geschatte TM-score en RMSD zijn alleen gepresenteerd voor de eerste I-TASSER model, omdat de C-score van lager gerangschikt modellen is niet sterk gecorreleerd met TM-score of RMSD. De kwaliteit van lagere-gerangschikte modellen kunnen gedeeltelijk worden beoordeeld op basis van hun cluster dichtheid en cluster grootte ten opzichte van het eerste model, waarin modellen van grotere cluster en een hogere dichtheid zijn gemiddeld dichter bij de natieve structuur.
- Lage C-score voorspellingen meestal duiden op een lage nauwkeurigheid voorspelling. In de meeste dergelijke gevallen, de query-eiwit ontbreekt een goede template in de bibliotheek en heeft een afmeting buiten het bereik van ab initio modellering (dwz> 120 residuen). In deze gevallen kunnen gebruikers zoeken voor extra ruimtelijke beperkingen en gebruik ze om de I-TASSER modellering (zie Overleg sectie) te verbeteren. Het is ook aangemoedigd om de sequenties te leggen aan onze QUARK server (QUARK / "> http://zhanglab.ccmb.med.umich.edu/QUARK/) voor een zuivere ab initio modellering als het eiwit grootte is onder de 200 residuen.
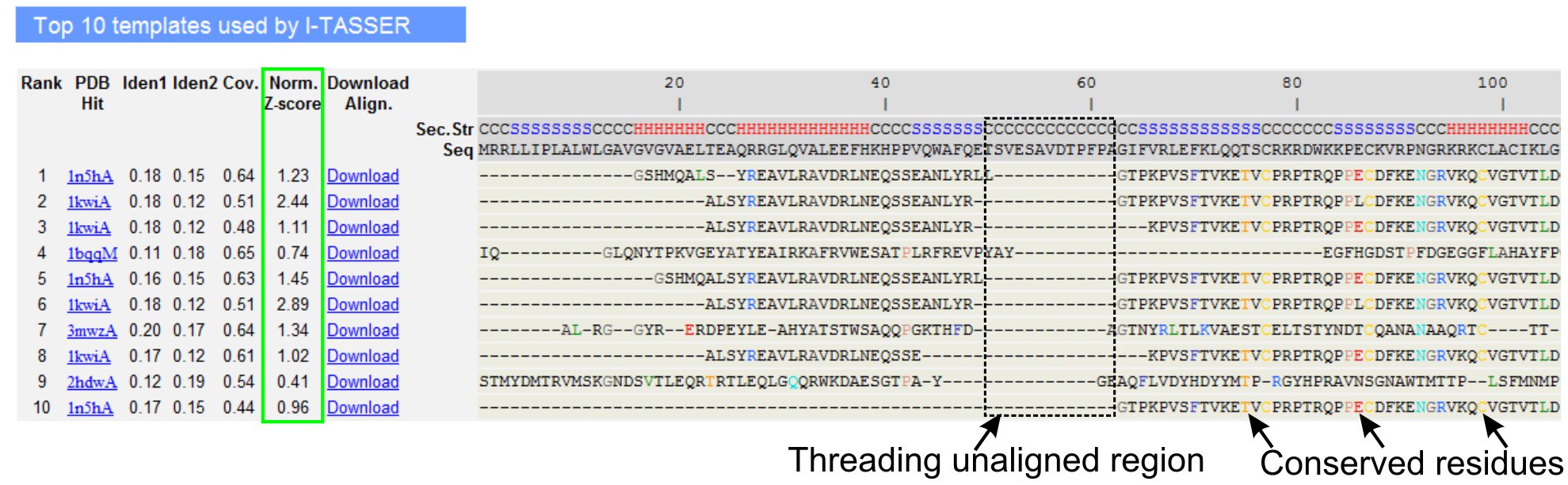
5. LOMETS doelgroep template uitlijning
- Scroll naar beneden naar de top tien threading templates van de query eiwit, zoals vastgesteld door LOMETS threading-programma's (Figuur 3) te analyseren. Bekijk de genormaliseerde Z-score (Vgl. 1), te zien in 'Norm. Z-score 'kolom, om de kwaliteit van threading uitlijningen te analyseren. Afstemmingen met een genormaliseerde Z-score> 1 geeft een zelfverzekerd afstemming en waarschijnlijk hebben dezelfde plooi als de query eiwit.
- Analyseer de sequentie-identiteit in de threading-gebonden regio (kolom 'Iden. 1') en voor de gehele keten (kolom 'Iden. 2') naar de homologie tussen de query en de sjabloon eiwitten te beoordelen. Hoge sequentie-identiteit is een indicator van de evolutionaire verwantschap tussen de query en template eiwitten.
- Bekijk de threading uitgelijnd residuen weergegeven in gekleurde om visueel te identificeren tegenserved residuen / motieven in de query en het sjabloon eiwitten. Een hogere sequentie-identiteit in te rijgen uitgelijnd regio, in vergelijking met hele keten alignment wijst ook op de aanwezigheid van geconserveerde structurele motief / domains in de query.
- Beoordelen van de dekking van de threading uitlijning door het bekijken van de 'Cov.' kolom en het controleren van de uitlijning. Als de dekking van de top uitlijningen is laag en beperkt tot slechts een klein deel van de query eiwit of afwezig zijn voor een lange segment van query-sequentie, wordt de query eiwit bevat meestal meer dan een domein en het wordt aanbevolen om de volgorde en het model split de domeinen individueel (figuur 3).
- Download het VOB geformatteerde sequentie-structuur alignment bestanden door te klikken op de "Download Align" links. Deze uitlijning bestand kan worden geopend in een moleculaire visualisatie-programma opgenomen in de Materialen sectie, en kan ook worden gebruikt voor het toevoegen van extra beperkingen in de structuur modelleren (Stap 1.4).
6.Structurele analogen in het VOB
- Bekijk de volgende tabel (figuur 4) van het resultaat pagina om de top tien structurele analogen van de eerste voorspelde model, zoals die is bepaald door het structurele afstemming programma TM-lijn 20. Een TM-score> 0,5 geeft aan dat de gedetecteerde analoge en het model een soortgelijke topologie hebben en kan worden gebruikt om de structurele klasse / eiwit familie van de query eiwit 16 vast te stellen, terwijl die met TM-score <0,3 betekent dat een willekeurige structuur gelijkenis.
- Analyseer de sequentie-identiteit en RMSD in de structureel uitgelijnd regio getoond in 'IDEN een' en 'RMSD een' kolommen aan het behoud van de ruimtelijke motieven in het model en de structurele analoge beoordelen. Inspecteer de gekleurde en uitgelijnd residu paren in de aanpassing aan deze structureel geconserveerde residuen en motieven te identificeren.
- Klik op het VOB-code vermeld in de kolom 'VOB Hit' om RCSB website te bezoeken en meer te leren over hun structurele classificatie (SCOP, Rk en PFAM) en functionele informatie (EG-nummer, gekoppeld GO voorwaarden en gebonden ligand).
7. Functie voorspellen met behulp van co-factor
- Scroll naar beneden in het resultaat pagina om functionele interpretaties voor de zoekopdracht eiwitten te analyseren. Eiwit functies worden opgesomd in drie context tabellen, het weergeven van: Enzyme Commissie (EC) cijfers, Gene Ontology (GO) termen, en het ligand bindingsplaatsen.
- Bekijk de 'TM-score', 'RMSD een', 'IDEN een' en 'Ver.' kolommen in elke tabel de parameters van de globale structuur gelijkenis en het behoud van ruimtelijke patronen tussen model en geïdentificeerd functionele homologen (templates) te analyseren.
8. Enzym Commissie aantal voorspelling
- Bekijk de top vijf potentiële enzym homologen van query's eiwit getoond in de "voorspelde EG-nummer" table (figuur 5). De betrouwbaarheid van de EG-nummer voorspellen met behulp van deze templates is weergegeven in de kolom "EG-Score '. Op basis van benchmarking analyse 23, functionele overeenkomst (de eerste drie cijfers van het EG-nummer) tussen de query en de template eiwitten betrouwbare wijze kunnen worden geïnterpreteerd met behulp van EG-score> 1,1.
- Kijk voor consensus van de functie (EG-nummer) bij de templates, die de gelijkaardige vouw (dat wil zeggen TM-score> 0,5) hebben als de query eiwit. Als er meerdere templates hebben dezelfde EG-nummer en EG-score> 1,1, de betrouwbaarheid van de voorspelling is zeer hoog. Echter, als de EG-Score hoog is, maar er is een gebrek aan consensus onder de geïdentificeerde hits, dan is de voorspelling wordt minder betrouwbaar en de gebruikers wordt aangeraden de GO-termijn voorspellingen te raadplegen.
- Klik op de link die op de EG-nummers aan de ExPASy Enzym-database te bezoeken en de functie analyseren, inclusief de reactie gekatalyseerd, co-factor-eisen en de metabole route, van de template eiwit in detail.
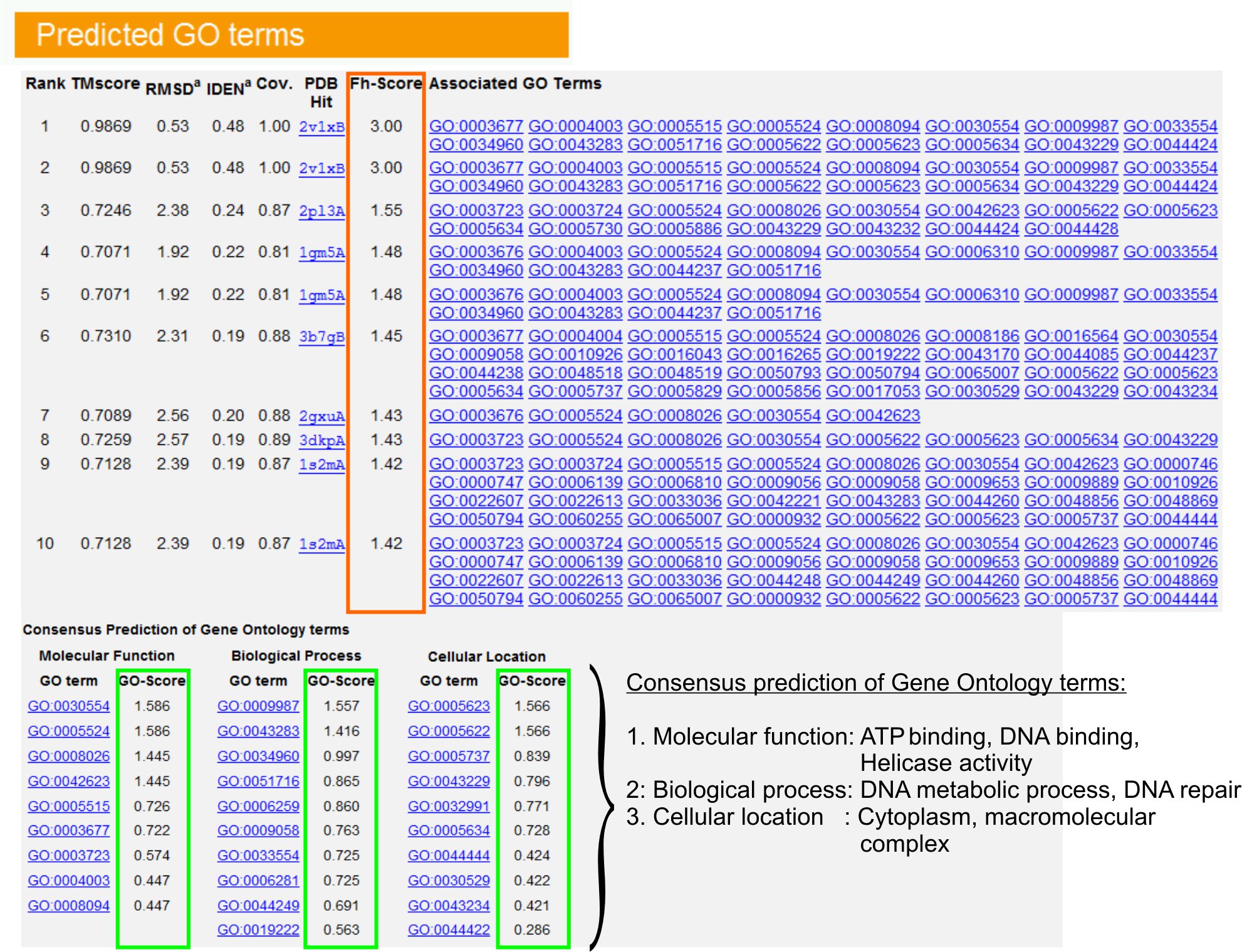
9. Gene Ontology (GO) termijn voorspellingen
- Bekijk de "voorspelde GO termen" tabel (Figure 6) naar boven tien homologen van query eiwit in het VOB bibliotheek, geannoteerd met Gene Ontology (GO) termen te identificeren. Elk eiwit wordt meestal geassocieerd met meerdere GO termen, beschrijven de moleculaire functies (MF), biologische processen (BP) en cellulaire component (CC). Klik op een term om de Amigo website te bezoeken en de definitie en afstamming te analyseren.
- Analyseer de Fh-score (Functional homologie score) kolom om de functionele overeenkomst tussen de query en de template eiwitten toegang tot en het betrouwbaarheidsniveau van de overdracht van functionele annotatie van deze eiwitten te schatten. In onze benchmark-studie 23, zou 50% van de inheemse GO voorwaarden correct worden geïdentificeerd vanaf de eerste geïdentificeerde template met behulp van een Fh-score cutoff van 0,8, met een totale nauwkeurigheid van 56%.
- Bekijk de 'Consensus voorspelling van GO termen "tabel om de samenloop van de functie tussen de sjablonen te analyseren. Deze gemeenschappelijke functies worden gebruikt voor het voorspellen van de GO termen (MF, BP en CC) van de queryeiwit en beoordelen van de betrouwbaarheid (GO-score) van GO termijn voorspellingen. Op basis van de benchmarking-test 23, zijn de beste vals positieve en vals negatieve uitslagen verkregen voor voorspellingen met een GO-score cutoff = 0,5, met afnemende dekking van de voorspelling op diepere niveaus ontologie.
10. Eiwit-ligand bindingsplaats voorspellingen
- Scroll naar beneden naar de onderkant van de pagina om top tien ligand binding site voorspellingen voor de query-eiwit te bekijken. Voorspelde bindingsplaatsen zijn gerangschikt op basis van het aantal voorspelde ligand conformaties die gemeenschappelijke bindingsplaats delen. De beste geïdentificeerde bindingsplaats is al weergegeven in de Jmol applet. Klik op de radio knoppen om andere voorspellingen te analyseren en de ligand interactie residuen te visualiseren.
- Analyseer de BS-score kolom om de plaatselijke overeenkomst tussen het model en sjabloon bindingsplaats evalueren. Op basis van de benchmark 9, BS-score> 1.1 geeft een hoge volgorde en structuur similarity de buurt van de voorspelde bindingsplaats in model en bekend bindingsplaats in de sjabloon.
- Download het VOB bestand geformatteerd structuur van het complex door te klikken op de "Download" link. Gebruikers kunnen deze bestanden in een moleculaire visualisatie-programma en interactief bekijk de voorspelde bindingsplaats en ligand-eiwit interacties op hun lokale computer.
11. Representatieve resultaten

Figuur 1 Een uittreksel van de I-TASSER resultaat pagina met (A) FASTA geformatteerd zoeksequentie;. (B) voorspelde secundaire structuur en de bijbehorende zelfvertrouwen scores, en (C) voorspelde solvent toegankelijkheid van de residuen. Geanalyseerd kerngebied en het potentieel hydratatie site in de query worden gemarkeerd in cyaan en rood rechthoeken, respectievelijk.

Figuur 2.

Figuur 3. Een voorbeeld van I-TASSER resultaat pagina met top tien geïdentificeerd threading templates en uitlijning door LOMETS 5 threading-programma's. De kwaliteit van de threading uitlijningen is geëvalueerd op basis van genormaliseerde Z-score (groen gemarkeerd), waar een waarde> 1 geeft een zelfverzekerde lijn. Uitgelijnd residuen in de template die identiek zijn aan de overeenkomstige vraag residuen zijn gemarkeerd in kleur op de aanwezigheid van geconserveerde residu / motief te geven, terwijl een gebrek aan afstemming in de meeste top templates geeft de aanwezigheid van meerdere domeinen in de query eiwit en de niet aangepaste residuen corresponderen met domeinnaam linker regio's. Klik hier om de full-sized versie van figuur 3 te bekijken.

Figuur 4. Een voorbeeld van een resultaatpagina, waarbij de top tien geïdentificeerde structurele analogen en structurele uitlijning, geïdentificeerd door TM-align 20 structureel alignment programma. De rangschikking van de analogen die in is gebaseerd op de TM-score (blauw gemarkeerd) van de structurele aanpassing. Een TM-score> 0,5 geeft aan dat de twee vergeleken structuren een soortgelijke topologie hebben, terwijl een TM-score <0,3 betekent dat een overeenkomst tussen twee willekeurige structuren. Structureel lijn residu paren zijn gemarkeerd in kleur op basis van hun amino-acid eigenschap heeft, terwijl de niet aangepaste regio's worden aangeduid met "-".ove.com/files/ftp_upload/3259/3259fig4large.jpg "> Klik hier om de full-sized versie van figuur 4 te bekijken.

Figuur 5. Een voorbeeld van I-TASSER resultaat pagina met geïdentificeerd enzym homologen van de query eiwit in het VOB bibliotheek. De betrouwbaarheid van de EG-nummer voorspelling is geanalyseerd op basis van EC-score (groen gemarkeerd), waar EG-score> 1.1 geeft functionele gelijkenis (dezelfde eerste 3 cijfers van het EG-nummer), tussen de query en template eiwit.

Figuur 6. Een voorbeeld van I-TASSER resultaat pagina met GO termijn voorspellingen voor de query eiwit. Functionele homologen voor de query eiwit in de Gene Ontology template bibliotheek zijn gerangschikt op basis van hun Fh-score (in oranje rechthoek). Gemeenschappelijke functionele kenmerken van deze top-hits scoren zijn afgeleid te alge at de laatste GO termijn voorspellingen voor de query eiwit. De kwaliteit van de voorspelde GO voorwaarden is geschat op basis van GO-score (aangegeven in groen), waar een GO-score> 0.5 geeft een betrouwbare voorspelling. Klik hier om de full-sized versie van figuur 6 te bekijken.

Figuur 7. Een voorbeeld van I-TASSER resultaat pagina met top tien eiwit ligand-bindingsplaats voorspellingen met behulp van de cofactor 9 algoritme. De rangschikking van de voorspelde bindingsplaatsen is gebaseerd op het aantal van de voorspelde ligand conformaties dat gemeenschappelijke binding pocket aandeel in de query. BS-score (rood gemarkeerd) is een maat van de lokale volgorde en structuur overeenkomst tussen de voorspelde en sjabloon bindingsplaats, en is nuttig voor het analyseren van het behoud van de bindingsplaats zakken.
les/ftp_upload/3259/3259fig8.jpg "/>
Figuur 8. Een voorbeeld van externe dwang bestanden die gebruikt worden om voor het opgeven van residu-residu contact / afstand beperkingen.

Figuur 9. Voorbeeld van terughoudendheid bestanden die gebruikt worden voor het specificeren van een template eiwit aan de I-TASSER server. Gebruiker kan de query-template alignment hetzij in (A) FASTA-formaat, of (B) 3D-formaat.

Figuur 10. Een voorbeeld bestand dat wordt gebruikt voor de uitsluiting van template tijdens de I-TASSER structuur modelleren procedure. De eerste kolom bevat het VOB ID van het sjabloon eiwitten worden uitgesloten. De tweede kolom wordt gebruikt om de sequentie-identiteit cutoff die voor andere, soortgelijke sjablonen worden gebruikt in de template bibliotheek te geven.
Discussion
Het protocol hierboven gepresenteerde is een algemene richtlijn voor de structuur en functie van het modelleren met behulp van de I-TASSER server. Hoewel deze geautomatiseerde procedure werkt zeer goed voor het grootste deel van de eiwitten, menselijk ingrijpen vaak helpen om een aanzienlijke verbetering van de modellering nauwkeurigheid, met name voor de eiwitten die nauw templates gebrek in het VOB bibliotheek. Gebruikers kunnen ingrijpen tijdens de I-TASSER model op de volgende manieren: (a) splitsing van multi-domein eiwitten, (b) het verstrekken van externe beperkingen om de structuur montage verbeteren, en (c) het verwijderen van templates in de modellering.
Splitsen van multi-domein eiwit:
Veel lange eiwit-sequenties bevatten vaak meerdere domeinen gebonden door flexibele linker regio's, waardoor hun structuuropheldering moeilijk met behulp van zowel experimentele en computationele technieken. Niettemin, zoals domeinen zijn onafhankelijk van elkaar vouwen entiteiten en kunnen verschillende moleculaire functie uit te voeren, het iswenselijk om lange multi-domein eiwitten en model split elk domein afzonderlijk. Modelleren van domeinen individueel zal niet alleen maar versnellen de voorspelling proces, maar verhoogt ook de kwaliteit van de query-template alignment, wat resulteert in meer betrouwbare structuur en functie voorspellingen.
Domeingrenzen in eiwit-sequenties kunnen worden voorspeld behulp van vrij verkrijgbare externe online programma's zoals NCBI CDD 24, 25 of PFAM InterProScan 26. Ook als LOMETS draadsnijden uitlijningen zijn beschikbaar voor de query eiwit, kan domeingrenzen worden gelokaliseerd door visueel te identificeren lange stukken van niet aangepaste residuen in de top-threading sjablonen (zie stap 5.4). Deze niet aangepaste regio's, hoofdzakelijk overeen met domeinnaam linker regio's. Als multi-domein templates zijn reeds beschikbaar in de sjabloon VOB bibliotheek met alle uitgelijnd vraag domeinen, wordt de query eiwit kan worden gemodelleerd als volledige lengte.
Zorg voor externe beperkingen
A. Geef de contact / afstand beperkingen
Experimenteel gekenmerkt inter-residu contacten / afstanden, bijvoorbeeld een MRI-ofcross-linking experimenten, kan worden gespecificeerd door het uploaden van een bestand terughoudendheid. Een voorbeeld bestand wordt weergegeven in figuur 8, waar een kolom geeft het type van de beperking, dat wil zeggen "DIST" of "contact". Voor afstand hoofdsteun (DIST), kolommen 2 en 4 bevatten residu posities (i, j), kolommen 3 en 5 bevatten de atoom-types in het residu en kolom 6 specificeert de afstand tussen de twee opgegeven atomen. Voor contact beperkingen (CONTACT), kolommen 2 en 3 bevatten de posities (i, j) van residuen die moeten worden in het contact. De afstand tussen de zijketens centrum van deze contact op te nemen residu paren wordt beslist op basis van waargenomen afstanden in bekende structuren in het VOB. I-TASSER zal proberen om deze atoom paren dicht bij de opgegeven afstand tijdens de structuur verfijning simulaties te trekken.
B. Geef een eiwitstructuur sjabloon
LOMETS threading programma's gebruiken een vertegenwoordiger van het VOB bibliotheek om plausibele plooien te vinden voor de query protein. Hoewel het gebruik van een representatieve structuur bibliotheek helpt het verminderen van de tijd die nodig is om de sequentie-structuur uitlijningen te berekenen, is het mogelijk dat een goed sjabloon eiwit wordt gemist in de bibliotheek of het sjabloon kan niet zijn geïdentificeerd door middel LOMETS threading-programma's, ook al is het aanwezig in de bibliotheek. In deze gevallen dient de gebruiker de gewenste eiwit structuur als de sjabloon.
Om aan te geven eiwitstructuur als een extra sjabloon, kunnen gebruikers zowel uploaden een PDB geformatteerd structuur bestand of geef het VOB ID van een gedeponeerd eiwitstructuur in het VOB bibliotheek. De I-TASSER genereert de query-template uitlijning met behulp van Muster-programma 23 en zal ruimtelijke beperkingen te verzamelen van zowel de gebruiker opgegeven template en LOMETS sjablonen om de structuur montage simulatie gids. Omdat de nauwkeurigheid van de LOMETS beperkingen is verschillend voor verschillende doelen, het gewicht van de LOMETS beperkingen is sterker in eenvoudig (homologe) targets dan die harde (niet-homologe) doelen, die systematisch zijn afgestemd op onze benchmark training.
Gebruikers kunnen ook aangeven hun eigen query-template uitlijningen. De server accepteert de aanpassing in twee formaten: de FASTA formaat (figuur 9A) en de 3D-formaat (figuur 9B). De FASTA formaat is standaard en beschreven op http://zhanglab. ccmb.med.umich.edu / FASTA / . De 3D-formaat is vergelijkbaar met de standaard VOB-formaat ( http://www.wwpdb.org/documentation/format32/sect9.html ), maar twee extra kolommen afgeleid van de sjablonen zijn toegevoegd aan het ATOM records (zie figuur 9B):
Kolommen 1-30: Atom (C-alpha only) en residu namen voor de query sequentie.
Kolommen 31-54: Coördinaten van de C-alfa-atomen van de query gekopieerd van de overeenkomstige atomen in de sjabloon.
Kolommen 55-59: Overeenkomstige residu nummer in de sjabloon op basis van afstemming
Kolommen 60-64: Overeenkomstige residu naam in de sjabloon
Uit te sluiten templates eiwitten
Eiwitten zijn flexibel moleculen en kan aannemen meerdere conformationele staten om hun biologische activiteit te veranderen. Zo hebben de structuren van veel proteïne kinases en membraaneiwitten opgelost in zowel actieve als inactieve conformatie. Ook de aanwezigheid of afwezigheid van gebonden ligand kan leiden tot grote structurele bewegingen. Terwijl alle conformationele toestanden van de template zijn gelijk voor de threading-programma's, is het wenselijk om de query met behulp van sjablonen in slechts een bepaalde toestand model. Een nieuwe optie op de server kan de gebruiker template eiwitten uit te sluiten tijdens de structuur van het modelleren. Deze functie zou ook kan de gebruiker de homologie niveau van templates worden gebruikt voor het modelleren te kiezen. Gebruikers kunnen sluiten sjabloon eiwitten frOM de I-TASSER bibliotheek door:
A. Het specificeren van een sequentie-identiteit cutoff
Gebruikers kunnen deze optie gebruiken om homologe eiwitten uit te sluiten van de I-TASSER template bibliotheek. De homologie niveau is ingesteld op basis van de sequentie-identiteit cutoff, dat wil zeggen het aantal identieke residu tussen de query en de sjabloon eiwit gedeeld door de volgorde lengte van de query sequentie. Bijvoorbeeld als de gebruiker typt in "70%" in de meegeleverde vorm, alle sjablonen eiwitten die een sequentie-identiteit hebben> 70% aan de query eiwit I-zal worden uitgesloten van de I-TASSER template bibliotheek.
B. uitsluiten specifieke sjabloon eiwitten
Specifieke sjabloon eiwitten kunnen worden uitgesloten van de I-TASSER template library door het uploaden van een lijst met VOB-ID's van de structuren uit te sluiten. Een voorbeeld bestand wordt weergegeven in Figuur 10. Als het hetzelfde eiwit kan bestaan als meerdere vermeldingen in het VOB bibliotheek, I-TASSER server zal uitsluiten door de opgegeven standaard templates (in Kolom1), alsmede alle andere sjablonen uit de bibliotheek die een identiteit hebben> 90% aan de opgegeven templates. Gebruikers kunnen ook aangeven een andere identiteit cutoff, bijvoorbeeld 70%, waarbij alle sjablonen met identiteit> 70% op bepaalde template eiwitten worden uitgesloten.
Disclosures
Geen belangenconflicten verklaard.
Acknowledgments
Het project wordt gedeeltelijk ondersteund door de Alfred P. Sloan Foundation, NSF Career Award (DBI 1.027.394) en het Nationale Instituut van Algemene Medische Wetenschappen (GM083107, GM084222).
Materials
| Name | Company | Catalog Number | Comments |
| Material Name | Type | Company | Catalogue Number |
| FASTA formatted amino acid sequence of the protein to be modeled (see, http://www.ncbi.nlm.nih.gov/BLAST/fasta.shtml). | |||
| A personal computer with access to the internet and a web browser. | |||
| Molecular visualizing software, e.g. RASMOL or PYMOL, for analyzing the predicted tertiary structure and functional sites. |
References
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H. CD-Search: protein domain annotations on the fly. Nucleic. Acids. Res. 32, W327-W331 (2004).
- Finn, R. D. The Pfam protein families database. Nucleic. Acids. Res. 38, D211-D222 (2010).
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).

{kind=link}
{kind=link}