

Summary
Richtlinien für die computergestützte strukturelle und funktionelle Charakterisierung von Proteinen unter Verwendung der I-Tasser Pipeline beschrieben. Ab Abfrage Proteinsequenz sind 3D-Modelle generiert mit mehreren Threading Ausrichtungen und iterative strukturellen Aufbau-Simulationen. Funktionelle Konsequenzen werden anschließend auf der Grundlage Übereinstimmungen mit Proteinen mit bekannter Struktur und Funktionen gezeichnet.
Abstract
Genome Sequencing Projekte haben Millionen von Protein-Sequenz, die Kenntnis ihrer Struktur und Funktion erfordern, um das Verständnis ihrer biologischen Rolle zu verbessern verschlüsselt. Obwohl experimentelle Methoden können detaillierte Informationen für einen Bruchteil dieser Proteine bieten, ist Computermodelle für die Mehrheit der Eiweißmoleküle, die experimentell charakterisierten benötigt werden. Die I-Tasser Server ist ein on-line-Workbench für hochauflösende Modellierung von Proteinstruktur und-funktion. Bei einer Proteinsequenz enthält eine typische Ausgabe aus dem I-Tasser Server Sekundärstruktur Vorhersage prognostiziert Lösungsmittel Zugänglichkeit der einzelnen Rückstand, homologe Proteine durch Vorlage Threading und Struktur Ausrichtungen erkannt, bis zu fünf abendfüllenden tertiäre strukturelle Modelle und Struktur-basierte funktionelle Annotationen für Enzymklassifikation, Gene Ontology Bedingungen und Protein-Ligand-Bindungsstellen. Alle Vorhersagen sind mit einer Zuversicht Partitur markiert dieerzählt, wie genau die Vorhersagen ohne Kenntnis der experimentellen Daten. Zur Erleichterung der speziellen Anforderungen der Endanwender, bietet der Server-Kanäle, um vom Benutzer festgelegten inter-Rückstandes Abstand nehmen und Kontakt-Karten zur interaktiven Veränderung der I-Tasser Modellierung, sondern auch ermöglicht es Benutzern, alle Proteine als Vorlage angeben, oder jede Vorlage auszuschließen Proteine während die Struktur der Montage-Simulationen. Die strukturellen Informationen können von den Nutzern auf experimentelle Beweise oder biologische Erkenntnisse mit dem Ziel der Verbesserung der Qualität von I-Tasser Vorhersagen basieren gesammelt werden. Der Server wurde als die besten Programme für Proteinstruktur und-funktion Vorhersagen in der jüngsten Gemeinde-weiten CASP Experimente ausgewertet. Es gibt derzeit> 20.000 registrierten Wissenschaftlern aus über 100 Ländern, die mit dem on-line I-Tasser Server.
Protocol
Method Überblick
Nach der Sequenz-to-Struktur-to-Funktion Paradigma beinhaltet das I-Tasser Verfahren 1-4 für Struktur und Funktion Modellierung vier aufeinander folgenden Schritten: (a) Vorlage Identifizierung von LOMETS 5; (b)-Fragment Struktur Zusammenbau von Replika- Austausch Monte-Carlo-Simulationen 6; (c) atomarer Ebene Strukturverfeinerung mit REMO 7 und FG-MD 8, und (d) Struktur-basierte Funktion Interpretationen mit Cofaktor 9.
Template Identifikation: Bei einer Abfrage Sequenz, die durch den Benutzer übermittelt, wird die Sequenz zunächst durch einen Vertreter PDB Struktur-Bibliothek durch eine lokal installierte LOMETS meta-Threading-Server eingefädelt. Threading ist eine Sequenz-Struktur-Alignment Verfahren zur Identifizierung von Proteinen, die Vorlage ähnliche Struktur haben oder enthalten ähnliche Strukturmotiv wie der Abfrage-Protein verwendet. Um die Abdeckung der homologen templ erhöhenaß Erkennungen verbindet LOMETS mehrere state-of-the-art Algorithmen, die verschiedene Methoden Threading. Da unterschiedliche Threading-Programme verschiedene Scoring-Systeme und Ausrichtung Empfindlichkeiten haben, ist die Qualität der erzeugten Gewinde Ausrichtungen von jedem threading Programm normalisiert Z-Score, die wie folgt definiert wird bewertet: 
wo Z-Score ist das Ergebnis in Einheiten der Standardabweichung bezogen auf den statistischen Mittelwert aller Ausrichtungen vom Programm generiert und Z 0 ist ein Programm-spezifische Z-score-Cutoff bestimmt basierend auf umfangreichen Threading Benchmark-Tests 5 bis differenzieren "gute 'und' schlechte 'Vorlagen. Eine Vorlage mit einem hohen Z-Score bedeutet, dass die Top-Vorlagen eine Ausrichtung der Gäste deutlich höher als die meisten anderen Vorlagen, die in der Regel bedeutet, dass die Ausrichtung auf ein gutes Modell entspricht haben. Wenn die meisten der Top-Threading-Vorlagen haben hallogh normalisiert Z-Werte, ist die Genauigkeit der endgültigen I-Tasser Modell in der Regel hoch. Allerdings, wenn das Protein groß ist und die Berichterstattung über das Einfädeln Ausrichtungen ist es, einen kleinen Bereich des Abfrage-Protein beschränkt, hat eine hohe normierte Z-Score nicht unbedingt eine hohe Genauigkeit bei der Modellierung der full-length-Modell. Top zwei Threading-Ausrichtungen von jedem threading-Programm werden gesammelt und für den nächsten Schritt der Struktur der Montage eingesetzt.
Iterative Struktur der Montage-Simulation: Nach dem Einfädeln Prozedur, Abfrage-Sequenz wird in threading ausgerichtet und nicht ausgerichteten Regionen aufgeteilt. Continuous-Fragmente in threading Ausrichtung sind aus Vorlagen ausgeschnitten und direkt für die Struktur der Montage verwendet, während die unaligned Loop-Regionen durch Ab-initio Modellierung gebaut werden. Die Struktur der Montage Verfahren basiert auf einem Gitter-System durch die Replik Austausch Monte-Carlo-Simulationen 6 geführte durchgeführt. Die I-Tasser Kraftfeld umfasst Wasserstoff-bonding Interaktionen 10, wissensbasierten statistische energetisch von bekannten Protein-Strukturen in der PDB 11, Sequenz-basierte Kontakt Vorhersagen aus SVMSEQ 12 und räumlichen Beschränkungen aus LOMETS 5 Threading-Vorlagen gesammelt werden. Die konformative Lockvögel in der Tieftemperatur-Repliken in den Simulationen erzeugt werden durch Spicker 13 Cluster auf Strukturen der niedrigen freien Energie Zustände zu identifizieren. Cluster Schwerpunkte der Top-Cluster sind durch Mittelung der 3D-Koordinaten aller Cluster-Struktur-Attrappen und für das endgültige Modell-Generation erhalten. Die Simulations-und Clustering-Verfahren werden zweimal zur Entfernung von sterischen Zusammenstöße und die weitere Verfeinerung der globalen Topologie wiederholt.
Atomic-Ebenen-Modell Konstruktion und Verfeinerung: Die Cluster-Schwerpunkte nach Spicker Clustering erhaltenen Protein-Modelle (jeweils Rückstand durch seine C α-und Seitenketten-Zentrum der Masse vertreten) und h reduziertave begrenzte biologische Anwendung. Der Bau von Voll-Atom-Modell aus der reduzierten Modelle erfolgt in zwei Schritten durchgeführt. Im ersten Schritt wird REMO 7 verwendet, um Full-Atom-Modelle von C-alpha Spuren konstruieren durch die Optimierung der H-Brücken-Netzwerken. Im zweiten Schritt, REMO full-Atom-Modelle weiter von FG-MD 14, die das Rückgrat-Torsionswinkel, Bindungslängen und-Seitenkette Rotamer Orientierungen verbessert raffiniert, durch molekulare Simulationen, wie die strukturelle Fragmente aus dem gesuchten geführte PDB-Strukturen von TM-align. Die FG-MD raffinierten Modelle sind als die endgültige Modelle für Tertiärstruktur Vorhersagen durch I-Tasser verwendet.
Die Qualität der erzeugten Modelle basieren auf einem Vertrauen des Gastes (C-Score), die basierend auf der Z-Score von LOMETS Threading Ausrichtungen und die Konvergenz der I-Tasser Simulationen, mathematisch formuliert definiert wird geschätzt: 
wo
Die C-Score hat eine starke Korrelation mit der Qualität der I-Tasser Modelle. Durch die Kombination von C-score-und Protein-Länge kann die Genauigkeit der ersten I-Tasser Modelle mit einem mittleren Fehler von 0,08 für die TM-Score und 2 Å für die RMSD 15 geschätzt werden. In der Regel Modelle mit C-Score> - sind 1,5 erwartet, dass eine korrekte Faltung zu haben. Hier werden RMSD und TM-Score sowohl bekannte Maßnahmen der topologischen Ähnlichkeit zwischen dem Modell und nativen Struktur. TM-score wertvolleES-Serie in [0, 1], wobei eine höhere Punktzahl deutet auf eine bessere Struktur entsprechen 16,17. Doch für niedrigeren Rangs Modelle (dh 2 nd -5 th-Modelle), ist die Korrelation von C-Partitur mit TM-Score und RMSD viel schwächer (~ 0,5), und kann nicht für eine zuverlässige Schätzung der absolute Modell Qualität eingesetzt werden.
Ist erste Modell immer das beste Modell in I-Tasser Simulationen? Die Antwort auf diese Frage hängt von der Ziel-Typ. Für leichte Ziele, ist das erste Modell in der Regel das beste Modell und seine C-Score ist in der Regel viel höher als der Rest der Modelle. Doch für harte Ziele, wo Threading keinen signifikanten Vorlage trifft, ist das erste Modell nicht unbedingt das beste Modell und I-Tasser tatsächlich hat Schwierigkeiten bei der Auswahl der besten Vorlage und Modelle. Es wird daher empfohlen, alle 5 Modelle für harte Ziele zu analysieren und markieren Sie diese auf den experimentellen Daten und biologischen Kenntnisse.
Funktion predictions: In den letzten Schritt, sind endgültig 3D-Modelle von FG-MD generiert verwendet werden, um drei Aspekte der Funktion von Proteinen vorherzusagen, und zwar: a) Enzyme Commission (EC)-Nummern 18 und (b) Gene Ontology (GO) 19 Begriffe und ( c) Bindungsstellen für kleine Moleküle Liganden. Für alle drei Aspekte sind funktionelle Interpretationen generiert mit Cofaktor, der einen neuen Ansatz zur Protein die Funktion auf globale und lokale Ähnlichkeit mit Vorlage Proteine in PDB mit bekannter Struktur und Funktionen vorherzusagen ist. Erstens ist die globale Topologie der vorhergesagten Modelle gegen funktionale Template-Bibliotheken abgestimmt mit strukturellen Ausrichtung Programm TM-align 20. Anschließend werden eine Reihe von Proteinen am ähnlichsten dem Ziel Modelle aus der Bibliothek auf ihre globale Struktur Ähnlichkeit basiert ausgewählt, und eine umfangreiche lokale Suche wird durchgeführt, um Struktur und Sequenz-Ähnlichkeit in der Nähe des aktiven / Bindungsstelle Region zu identifizieren. Die daraus resultierenden globalen und lokalen Ähnlichkeit Scores werden verwendet, um den RangVorlage Proteine (funktionelle Homologe) und übertragen Sie die Annotation (EG-Nummern und Gene Ontology 19 Begriffe) auf der Oberseite Scoring Hits basieren. Ebenso sind Ligand-Bindungsstelle Rückstände und die Ligandenbindung-Modus basiert auf dem lokalen Ausrichtung der Abfrage mit bekannten Liganden-Bindungsstelle Rückstände in den Top Scoring-Funktion Templates 9 abgeleitet.
Die Qualität der Funktion (EG und GO Begriff) Vorhersage in I-Tasser basiert auf funktionelle Homologie Score (Fh-Score), die ein Maß für die globale und lokale Ähnlichkeit zwischen der Anfrage und Vorlage ausgewertet und ist definiert als: 
wo C-Score ist eine Schätzung der Qualität der vorhergesagten Modell wie in Gl. (2); TM-Score misst die globale strukturelle Ähnlichkeit zwischen dem Modell und Vorlage Proteine; RMSD ali ist die RMS-Abweichung zwischen dem Modell und der Template-Struktur in der strukturell ausgerichteten Region von TM-align 20; Cov stellt die Berichterstattung über die strukturelle Ausrichtung (dh das Verhältnis der strukturell ausgerichtet Rückstände von der Abfrage Länge unterteilt); ID ali ist die Sequenzidentität in der TM-align Ausrichtung. Die geschätzte Vertrauen Punktzahl für EG-Nummer Voraussagen auch ein Begriff für die Bewertung aktiven Zentrum match (ACM) zwischen Abfrage und Vorlage innerhalb einer definierten Region, wie folgt berechnet: 
wobei N t die Anzahl der Vorlage vorhandenen Rückstände innerhalb der lokalen Bereich stellt, N ali die Anzahl der ausgerichteten Abfrage-template-Rest-Paare ist, d ii der C α Abstand zwischen i-ten Paar ausgerichteter Rückstände, ist d 0 = 3,0 Å der Abstand Cutoff, ist M ii der BLOSUM Scores zwischen ith Paar ausgerichteter Rückstände. Im Allgemeinen ist der FH-Score im Bereich [0, 5] und ACM-Score ist zwischen [0, 2], Wobei höhere Werte eine mehr Selbstvertrauen funktionale Aufgaben. ACM-Score ist auch für die Beurteilung der lokalen Struktur und Sequenz-Ähnlichkeit in der Nähe des Liganden-Bindungsstellen, die als BS-Score bezeichnet wird.
1. Einreichung der Proteinsequenz
- Besuchen Sie die I-Tasser Webseite unter http://zhanglab.ccmb.med.umich.edu/I-TASSER mit Struktur und Funktion Modellierung Versuch zu starten.
- Kopieren Sie den Aminosäure-Sequenz in das dafür vorgesehene Formular oder direkt hochladen von Ihrem Computer, indem Sie die Schaltfläche "Durchsuchen". I-Tasser Server akzeptiert derzeit Sequenzen mit bis zu 1500 Rückstände. Proteine mehr als 1500 Reste sind in der Regel Multi-Domain-Proteine und wird empfohlen, bei einzelnen Domänen vor dem Absenden an die I-Tasser aufgeteilt werden.
- Geben Sie Ihre E-Mail-Adresse (obligatorisch) und einen Namen für den Job (optional).
- Benutzer können optional externe inter-residue Kontakt / Abstandsrestraints, Add-In ein zusätzliches Template oder ausschließen einige Vorlage Proteine während die Struktur Modellierung. Erfahren Sie mehr über die Verwendung dieser Optionen in der "Diskussion" Abschnitt.
- So senden Sie die Sequenz, auf der "Run I-Tasser" klicken. Der Browser wird auf eine Bestätigungsseite angezeigte Benutzer bestimmte Informationen, Auftrags-ID (Job-ID) und einem Link zu einer Webseite, wo die Ergebnisse nach Abschluss der Arbeit abgelegt werden gerichtet werden. Nutzer können Lesezeichen auf diesen Link oder notieren Sie sich den Job-Identifikationsnummer für die Zukunft.
2. Verfügbarkeit der Ergebnisse
- Überprüfen Sie den Status Ihrer eingereichten Arbeit durch den Besuch der I-Tasser Warteschlange Seite bei http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php . Klicken Sie auf die Registerkarte Suchen und nutzen Sie die Job-ID-Nummer oder die Abfrage-Sequenz zu Ihrem eingereichten Jobsuche.
- Nach der Struktur und Funktion moDeling fertig ist, wird eine Benachrichtigung per E-Mail mit Bild von den vorhergesagten Strukturen und einem Web-Link an Sie gesendet werden. Klicken Sie auf diesen Link oder öffnen Sie den Link Lesezeichen bei Schritt 1.5 zum Ansehen und Herunterladen der Ergebnisse.
3. Sekundäre Struktur und Lösungsmittel Zugänglichkeit Prognosen
- Überprüfen Sie die FASTA formatierten Abfrage-Sequenz auf der Oberseite der Ergebnisseite angezeigt. Wenn keine zusätzlichen Zurückhaltung / template während Reihenfolge Vorlage, einen Link zu der Webseite angezeigt werden vom Benutzer angegebenen Informationen angegeben wurde auch ersichtlich (Abbildung 1A) werden.
- Untersuchen Sie die Sekundärstruktur Vorhersage angezeigt: alpha-Helix (H), beta-Strang (S) oder Spule (C) und das Vertrauen der Gäste der Vorhersage (0 = niedrig, 9 = hoch) für jeden Rückstand. Suchen Sie nach Region mit langen Strecken regelmäßige Sekundärstruktur (H oder S) Voraussagen, um die Kern-Region des Proteins zu schätzen. Strukturelle Klasse von Protein kann auch analysiert über die Verteilung der Sekundärstrukturen Elementen basieren. Also lange Regionen Spulenelemente in das Protein zeigen normalerweise unstrukturierten / ungeordnete Bereiche.
- Sehen Sie sich die prognostizierte Lösungsmittelzugänglichkeit (Abbildung 1C) zu prüfen, begraben und Lösungsmittel exponierten Regionen in der Abfrage. Werte von vorhergesagten Lösungsmittelzugänglichkeit Bereich von 0 (begraben Rückstand) bis 9 (ausgesetzt Rückstand). Region, die hauptsächlich begraben Rückstände können verwendet werden, um den Kernbereich des Proteins beschreiben werden, während Regionen mit Lösungsmittel ausgesetzt und hydrophile Reste Potenzial Hydratation / funktionellen Stellen sind.
4. Tertiärstruktur Prognosen
- Blättern Sie nach unten, um die vorhergesagte Tertiärstrukturen Abfrage Protein, in einem interaktiven Jmol Applet (Abbildung 2) angezeigt zu sehen. Ein Linksklick auf das Applet, um das Aussehen der angezeigten Struktur zu verändern, zoomen Sie in bestimmte Region, wählen spezifische Rest-Typen in der vorhergesagten Modell oder berechnen inter-Rest distanziert.
- Analysieren Sie die Modelle für das Vorhandensein von langen unstrukturierte Regionen. Diese rEGIONEN meist ungeordneten Bereiche im Protein entsprechen oder zeigen mangelnde Vorlage Ausrichtung. Diese Regionen haben in der Regel niedriger Modellierung Genauigkeit und Entfernen dieser Regionen bei der Modellierung von N & C-Terminus Region die Modellierung Genauigkeit zu verbessern.
- Laden Sie die PDB-Dateien formatiert Struktur des Modells, indem Sie auf den "Download-Modell" Links. Sie können diese Dateien in einem beliebigen molekularen Visualisierungs-Software (z. B. Pymol, Rasmol etc.) zur weiteren Analyse der strukturellen Merkmale zu öffnen.
- Analysieren Sie das Vertrauen des Gastes (C-Wert) der Struktur-Modellierung, um die Qualität der vorhergesagten Strukturen zu schätzen. C-score (Gl. 2)-Werte sind in der Regel im Bereich [-5, 2], wobei eine höhere Punktzahl spiegelt ein Modell von besserer Qualität. Die geschätzten TM-Score und RMSD der ersten Modell wird als "Geschätzte Genauigkeit von Modell 1" gezeigt. Für lange Proteine, ist es empfehlenswert, das Modell-Qualität auf TM-Score basiert auswerten, wie TM-Score ist empfindlicher auf die topologische Änderungen als RMSD. < li> auf "mehr über C-score"-Link, um C-score Analyse, Cluster-Größe und Cluster-Dichte aller Modelle klicken. Geschätzte TM-Score und RMSD nur für die erste I-Tasser Modell vorgestellt werden, denn C-Score von niedrigeren Rang Modelle ist nicht stark mit TM-Score oder RMSD korreliert. Quality of niedrigeren Rangs Modelle können teilweise auf der Grundlage ihrer Cluster-Dichte und Cluster-Größe gegenüber dem ersten Modell beurteilt werden, wobei Modelle von größeren Cluster und eine höhere Dichte im Durchschnitt näher an der nativen Struktur.
- Low C-score Prognosen zeigen normalerweise eine geringe Genauigkeit Vorhersage. In den meisten solchen Fällen fehlt die Abfrage Protein eine gute Vorlage in die Bibliothek und hat eine Größe außerhalb der Reichweite von ab-initio Modellierung (dh> 120 Reste). In diesen Fällen kann der Benutzer für zusätzliche räumliche Beschränkungen suchen und nutzen sie, um die I-Tasser Modellierung (siehe Diskussion Abschnitt) zu verbessern. Es wird ebenfalls gefördert, um die Sequenzen zu unserem QUARK-Server senden (QUARK / "> http://zhanglab.ccmb.med.umich.edu/QUARK/) für eine reine ab initio Modellierung, wenn die Größe des Proteins ist unter 200 Rückstände.
5. LOMETS Ziel Vorlage Ausrichtung
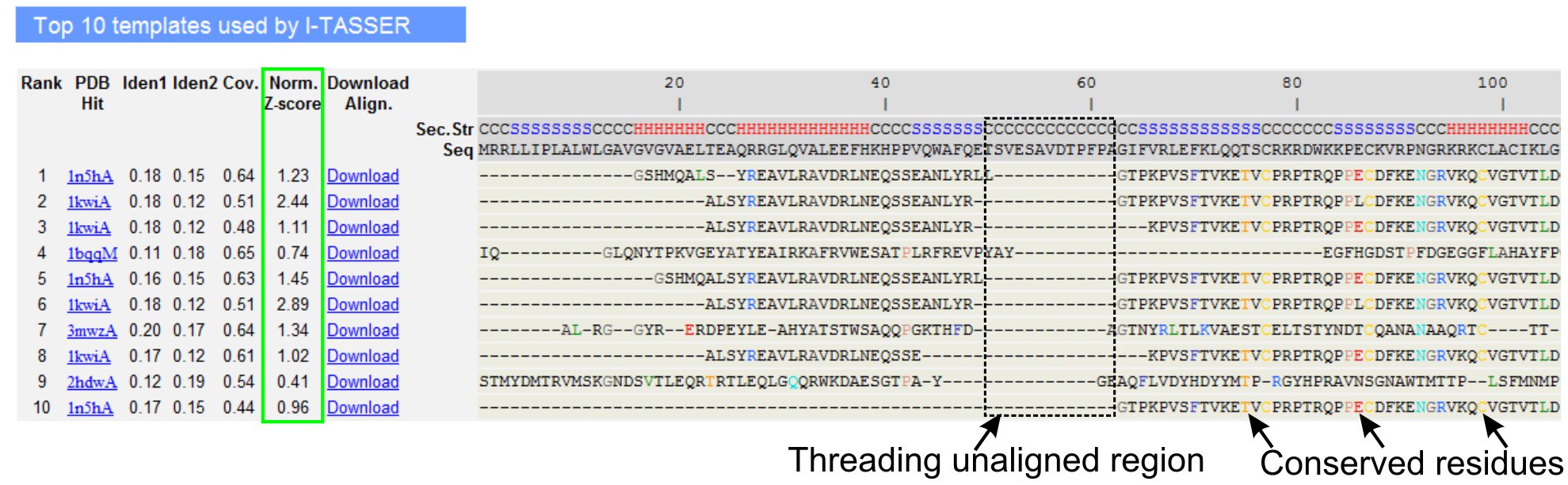
- Scrollen Sie zu den Top Ten Threading Vorlagen der Abfrage-Protein, wie LOMETS Threading-Programme (Abbildung 3) identifiziert zu analysieren. Sehen Sie sich die normierte Z-score (Gl. 1), in "Norm dargestellt. Z-score "-Spalte, um die Qualität der Gewinde Ausrichtungen zu analysieren. Alignments mit einem normierten Z-Score> 1 spiegelt eine selbstbewusste Ausrichtung und wahrscheinlich haben die gleiche Falte, wie die Abfrage Protein.
- Analysieren Sie die Sequenz-Identität in der Threading-aligned Region (Spalte 'Iden. 1') und für die gesamte Kette (Spalte 'Iden. 2') auf die Homologie zwischen der Abfrage und der Vorlage Proteine zu beurteilen. Hohe Sequenzidentität ist ein Indikator für evolutionäre Verwandtschaft zwischen der Abfrage und Vorlage Proteine.
- Sehen Sie sich die Gewinde ausgerichtet Rückstände in farbigen gezeigt, visuell zu identifizieren Nachteileerved Rückstände / Motive in der Abfrage und die Vorlage Proteine. Eine höhere Sequenzidentität in Threading-aligned Region, im Vergleich zu ganzen Kettenflucht auch zeigt das Vorhandensein von konservierten Strukturmotiv / domains in der Abfrage.
- Bewerten Sie die Abdeckung des Threading Ausrichtung, indem Sie die 'Cov. Spalte und die Kontrolle der Ausrichtung. Wenn die Abdeckung der oberen Ausrichtungen ist gering und beschränkt sich auf nur ein kleiner Bereich der Abfrage-Protein oder fehlen für eine lange Segment der Abfrage-Sequenz, dann die Abfrage Protein enthält in der Regel mehr als eine Domain und es wird empfohlen, die Reihenfolge und das Modell Split die Domänen einzeln (Abbildung 3).
- Laden Sie die PDB formatiert Sequenz-Struktur-Alignment-Dateien mit einem Klick auf den "Download Align" Links. Diese Ausrichtung kann in einem beliebigen molekularen Visualisierungsprogramm in den Materialien aufgeführt geöffnet werden, und kann auch für das Hinzufügen von zusätzlichen Beschränkungen bei der Struktur-Modellierung (Schritt 1.4) verwendet werden.
6.Strukturelle Analoga in PDB
- Sehen Sie sich die folgende Tabelle (Abbildung 4) der Ergebnis-Seite zu den Top Ten der strukturelle Analoga der ersten vorhergesagt Modell, wie identifiziert durch die strukturelle Ausrichtung Programm bestimmen 20 TM-align. Ein TM-Score> 0,5 zeigt, dass die erfasste analoge und Modell eine ähnliche Topologie und kann verwendet werden, um die strukturelle Klasse / Protein-Familie der Abfrage Protein 16 zu bestimmen, während diejenigen mit TM-Score <0,3 bedeutet eine zufällige Struktur Ähnlichkeit.
- Analysieren Sie die Sequenz-Identität und RMSD in der strukturell ausgerichteten Region gezeigt "IDEN a 'und' RMSD a 'Spalten, um die Erhaltung der räumlichen Motive in das Modell und die strukturelle analog zu beurteilen. Sichtprüfung der farbigen und ausgerichtet Rest-Paare in der Ausrichtung auf diese strukturell konservierte Reste und Motive zu identifizieren.
- Klicken Sie auf den PDB-Code in der "PDB Hit 'Spalte zur RCSB Website zu besuchen und erfahren Sie mehr gezeigt, über deren strukturelle Klassifizierung (SCOP, CATH und PFAM) und funktionelle Informationen (EG-Nummer, verbunden GO Bedingungen und gebundenen Liganden).
7. Function Vorhersage mit Cofaktor
- Blättern Sie in der Ergebnis-Seite, um funktionale Interpretationen für die Abfrage Protein zu analysieren. Protein-Funktionen sind in drei Zusammenhang Tabellen aufgelistet, die Anzeige: Enzyme Commission (EC) Zahlen, Gene Ontology (GO) Begriffe und Ligand-Bindungsstellen.
- Sehen Sie sich die "TM-Score ',' RMSD a ',' IDEN a 'und' Cov. Spalten in jeder Tabelle zu analysieren Parameter der globalen Struktur Ähnlichkeit und Erhaltung der räumlichen Muster zwischen Modell und identifiziert funktionalen Homologen (Templates).
8. Enzyme Commission Anzahl Voraussage
- Sehen Sie sich die Top fünf potenziellen Enzym Homologen der Abfrage Protein in der gezeigten "Predicted EG-Nummern" Tabelle (Abbildung 5). Das Konfidenzniveau von EG-Nummer Vorhersage mit Hilfe dieser Vorlagen ist in "EG-Score"-Spalte angezeigt. Basierend auf benchmarking Analyse 23, funktionelle Ähnlichkeit (ersten 3 Ziffern der EG-Nummer) zwischen der Abfrage und Vorlage Protein zuverlässig interpretiert werden mittels EC-Score> 1,1.
- Suchen Sie nach Konsens-Funktion (EC-Nummern) zu den Vorlagen, die ähnlich fach (dh TM-Score> 0,5) haben, wie die Abfrage Protein. Wenn mehrere Vorlagen gleichzeitig EG-Nummer und EC-Wert> 1,1 haben, ist das Konfidenzniveau der Vorhersage sehr hoch. Wenn jedoch das EG-Score hoch ist, aber es gibt einen Mangel an Konsens zwischen den identifizierten Hits, dann der Vorhersage wird weniger zuverlässig und Benutzern wird empfohlen, die GO längerfristige Voraussagen zu konsultieren.
- Klicken Sie auf den Link auf der EC-Nummern angeboten werden, die ExPASy Enzyme Datenbank zu besuchen und analysieren die Funktion, einschließlich der Reaktion katalysiert, Co-Faktor Anforderungen und den Stoffwechselweg, der die Vorlage Protein im Detail.
9. Gene Ontology (GO) und längerfristige Voraussagen
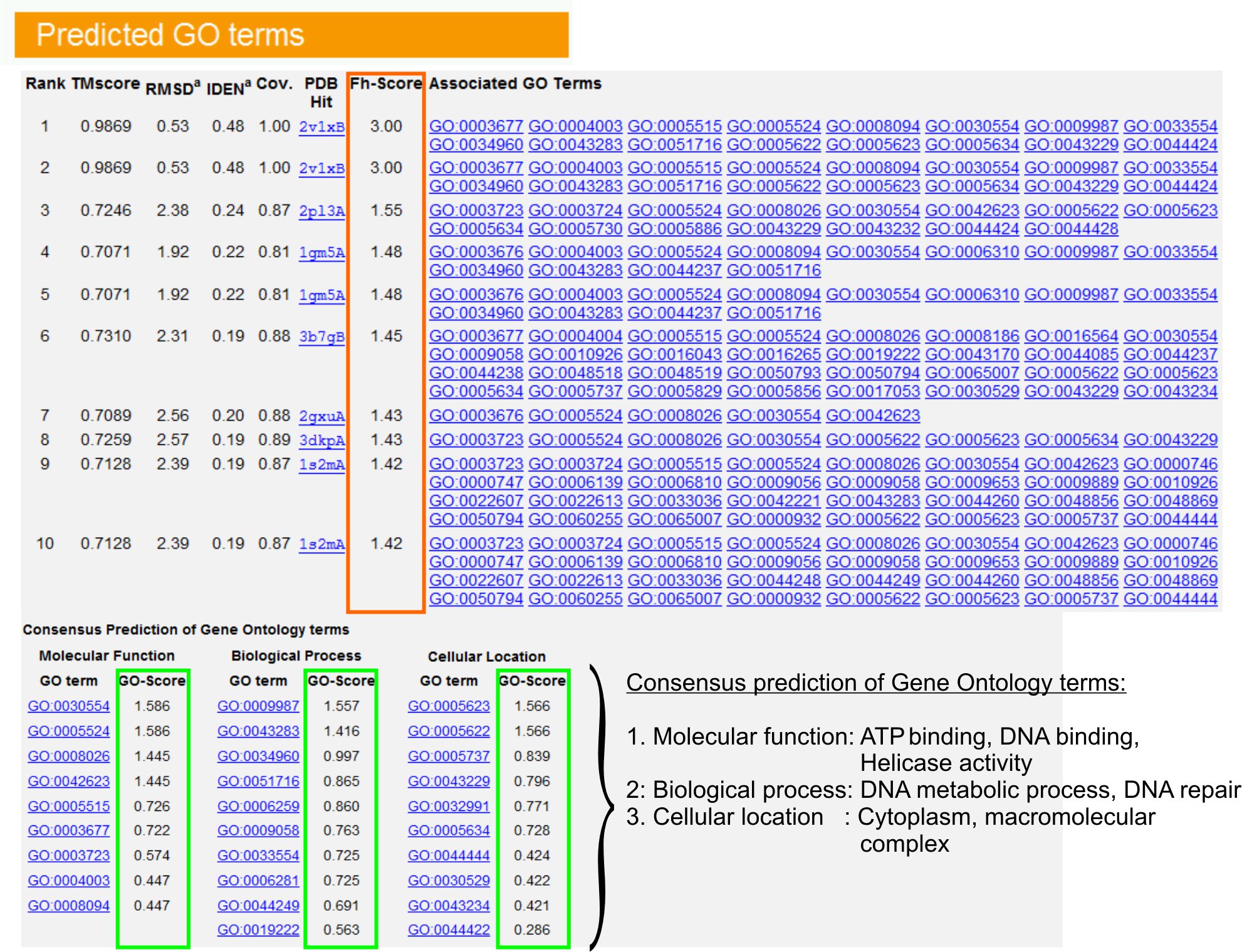
- Sehen Sie sich die "Predicted GO Begriffe" Tabelle (Abb.Abbildung 6) zu identifizieren Top-Ten-Homologe von Abfrage-Protein in der PDB-Bibliothek, versehen mit Gene Ontology (GO) Bezug. Jedes Protein ist in der Regel mit mehreren GO Begriffe verbunden sind, beschreiben ihre molekularen Funktionen (MF), biologische Prozesse (BP) und zellulären Komponente (CC). Klicken Sie auf jedes Glied der Amigo Webseite zu besuchen und zu analysieren, ihre Definition und Herkunft.
- Analysieren Sie die Fh-Score (Functional Homologie score) Spalte, um die funktionelle Ähnlichkeit zwischen der Anfrage und Vorlage Proteine Zugang und schätzen das Vertrauen Ebene zu übertragen funktionellen Annotation von diesen Proteinen. In unserer Benchmarking-Studie 23, konnten 50% der einheimischen GO Begriffe richtig aus dem ersten identifizierten Vorlage mit einem Fh-score-Cutoff von 0,8, mit einer Genauigkeit von 56% identifiziert werden.
- Sehen Sie sich die "Consensus Prognose von GO Begriffe" Tabelle, um die Zustimmung der Funktion zwischen den Vorlagen zu analysieren. Diese gemeinsamen Funktionen sind für die Vorhersage der GO Begriffe (MF, BP und CC) von der Abfrage verwendetProtein und bewerten das Konfidenzniveau (GO-Score) von GO längerfristige Voraussagen. Basierend auf der Benchmarking-Test 23, sind die beste falsch positive und falsch negative Raten für Vorhersagen mit GO-score cutoff = 0,5 erhalten, mit abnehmender Reichweite der Vorhersage auf tieferen Ebenen Ontologie.
10. Protein-Ligand-Bindungsstelle Prognosen
- Scrollen Sie zum unteren Rand der Seite, um Top-Ten-Ligand-Bindungsstelle Vorhersagen für die Abfrage Protein zu sehen. Predicted Bindungsstellen sind basierend auf der Anzahl der vorhergesagten Ligand Konformationen, die gemeinsame Bindungstasche Aktie rangiert. Die besten identifizierten Bindungsstelle ist bereits in der Jmol Applet angezeigt. Klicken Sie auf die Radio-Buttons zu anderen Prognosen analysieren und visualisieren den Liganden interagieren Rückstände.
- Analysieren Sie die BS-score Spalte zur lokalen Ähnlichkeit zwischen dem Modell und Vorlage Bindungsstelle zu bewerten. Basierend auf den Benchmark-9, BS-Score> 1,1 deutet auf eine hohe Sequenz und Struktur similarity in der Nähe der vorhergesagten Bindungsstellen in Modell und bekannte Bindungsstelle in der Vorlage.
- Laden Sie die PDB-Datei formatiert Struktur des Komplexes durch einen Klick auf den Link "Download". Benutzer können diese Dateien in einem beliebigen molekularen Visualisierung Programm zu öffnen und interaktiv Blick die vorhergesagte Bindungsstelle und Ligand-Protein-Interaktionen auf ihrem lokalen Computer.
11. Repräsentative Ergebnisse

Abbildung 1 Ein Auszug aus I-Tasser Ergebnisseite zeigt (A) FASTA formatierten Abfrage-Sequenz;. (B) vorhergesagt sekundäre Struktur und die damit verbundenen Vertrauen Partituren, und (C) vorhergesagt Lösungsmittel Zugänglichkeit der Rückstände. Analysiert Kernregion und potenziellen Hydratation Ort in der Abfrage werden in cyan und rote Rechtecke markiert, beziehungsweise.

Abbildung 2.

Abbildung 3. Ein Beispiel für I-Tasser Ergebnisseite zeigt Top Ten identifiziert Threading-Vorlagen und Ausrichtungen von LOMETS 5 Threading-Programme. Die Qualität der Gewinde Ausrichtungen wird auf der Basis normalisierter Z-Score (grün markiert), wobei ein Wert> 1 zeigt eine selbstbewusste Ausrichtung ausgewertet. Ausgerichtet Rückstände in der Vorlage, die identisch mit den entsprechenden Abfrage-Reste werden farblich hervorgehoben, um Präsenz von konservierten Rest / Motiv zeigen, während ein Mangel an Übereinstimmung in den meisten Top-Vorlagen zeigt das Vorhandensein von mehreren Domains in der Abfrage-Protein und die unaligned Rückstände Domain Linker Regionen entsprechen. Klicken Sie hier, um die Full-Size-Version von Abbildung 3 zu sehen.

Abbildung 4. Ein Beispiel für Ergebnis-Seite mit Top-Ten festgestellten strukturellen Analoga und strukturellen Ausrichtung, identifiziert durch TM-align 20 Struktureinheiten Alignment-Programm. Das Ranking der Analoga gezeigt in ist auf dem TM-Score (blau) der strukturellen Ausrichtung basiert. Ein TM-Score> 0,5 zeigt an, dass die beiden verglichenen Strukturen eine ähnliche Topologie haben, während ein TM-Score <0,3 bedeutet eine Ähnlichkeit zwischen zwei beliebigen Strukturen. Strukturell ausgerichtet Rest-Paare sind in Farbe auf ihre Aminosäure-Eigenschaft basiert hervorgehoben, während die unaligned Regionen angegeben "-".ove.com/files/ftp_upload/3259/3259fig4large.jpg "> Klicken Sie hier, um die volle Größe Version von Abbildung 4 zu sehen.

Abbildung 5. Ein Beispiel für I-Tasser Ergebnisseite zeigt identifizierten Enzym Homologen der Abfrage Protein in der PDB-Bibliothek. Das Konfidenzniveau von EG-Nummer Vorhersage basierend auf EC-Score (grün markiert), in denen die EG-Score> 1,1 zeigt funktionelle Ähnlichkeit (gleicher ersten 3 Ziffern der EG-Nummer) zwischen Abfrage und Vorlage Protein analysiert.

Abbildung 6. Ein Beispiel für I-Tasser Ergebnisseite zeigt GO Begriff Voraussagen für die Abfrage Protein. Funktionelle Homologe für die Abfrage Protein in der Gene Ontology-Template-Bibliothek sind auf der Grundlage ihrer Fh-score (in orange Rechteck) eingestuft. Gemeinsame funktionellen Merkmale dieser Top-Scoring-Hits sind auf Gener abgeleitet aßen die letzten GO Begriff Voraussagen für die Abfrage Protein. Die Qualität der vorhergesagten GO Begriffe wird geschätzt auf GO-Score (grün dargestellt), wo eine GO-Score> 0,5 deutet auf eine zuverlässige Vorhersage. Basierte Klicken Sie hier, um die Full-Size-Version von Abbildung 6 zu sehen.

Abbildung 7. Ein Beispiel für I-Tasser Ergebnisseite zeigt Top-Ten-Protein-Ligand-Bindungsstelle Vorhersagen mit dem Cofaktor 9-Algorithmus. Das Ranking der vorhergesagten Bindungsstellen auf der Anzahl der vorhergesagten Ligand Konformationen, die gemeinsame Bindungstasche Anteil an der Abfrage basiert. BS-Score (rot hervorgehoben) ist ein Maß für lokale Sequenz und Struktur Ähnlichkeit zwischen den vorhergesagten und Vorlage Bindungsstelle, und ist nützlich für die Analyse der Erhaltung der Bindungsstelle Taschen.
les/ftp_upload/3259/3259fig8.jpg "/>
Abbildung 8. Ein Beispiel für externe Zurückhaltung Dateien verwendet werden, um für die Angabe Rest-Rest-contact / Abstandsrestraints.

Abbildung 9. Beispiel der Zurückhaltung Dateien für die Angabe einer Vorlage Proteins an die I-Tasser-Server verwendet. Der Benutzer kann die Abfrage-template Ausrichtung entweder im (A) FASTA-Format, oder (B) 3D-Format.

Abbildung 10 dargestellt. Ein Beispiel-Datei für den Ausschluss der Vorlage bei der I-Tasser Strukturmodellierung Verfahren verwendet. Die erste Spalte enthält die PDB ID der Vorlage Proteine ausgeschlossen werden. In der zweiten Spalte wird verwendet, um die Sequenzidentität Cutoff, die für andere ähnliche Vorlagen in der Vorlagen-Bibliothek verwendet werden soll.
Discussion
Das Protokoll oben dargestellten ist eine allgemeine Richtlinie für die Struktur und Funktion Modellierung mit der I-Tasser Server. Obwohl, dieses automatisierte Verfahren funktioniert sehr gut für die meisten Proteine, menschliche Eingriffe oft zu einer erheblichen Verbesserung der Modellierung Genauigkeit, vor allem für die Proteine, die in der Nähe Vorlagen Mangel in der PDB-Bibliothek. Benutzer können während der I-Tasser Modellierung in der folgenden Weise eingreifen: (a) Spaltung von Multi-Domain-Proteine, (b) Bereitstellung von äußeren Zwängen, um die Struktur der Montage zu verbessern, und (c) Entfernen von Vorlagen bei der Modellierung.
Splitting Multi-Domänen-Protein:
Viele lange Protein-Sequenzen enthalten häufig mehrere Domains durch flexible Linker Regionen, die ihre Strukturaufklärung schwer mit beiden experimentellen und theoretischen Techniken macht angebunden. Dennoch, als Domänen unabhängig sind Falt-Einheiten und können unterschiedliche molekulare Funktion zu erfüllen, es istwünschenswert, lange Multi-Domain-Proteine und Modell aufgeteilt jede Domain einzeln. Modeling-Domains einzeln nicht nur beschleunigen, die Vorhersage-Prozess, sondern erhöht auch die Qualität der Abfrage-template Ausrichtung, die sich in zuverlässiger Struktur und Funktion Vorhersagen.
Domain Grenzen in Proteinsequenzen vorhergesagt mit frei verfügbaren externen Online-Programme wie NCBI CDD 24, 25 oder PFAM InterProScan 26 sein. Auch, wenn LOMETS Threading Ausrichtungen für die Abfrage Protein sind, können Domain-Grenzen durch visuellen Erkennung weite Strecken unaligned Rückstände in den Top Threading-Vorlagen (siehe Schritt 5.4) befinden. Diese unaligned Regionen, vor allem, um Domain-Linker Regionen entsprechen. Wenn Multi-Domain-Templates sind bereits in der Vorlage PDB-Bibliothek mit allen Abfrage Domänen ausgerichtet verfügbar ist, dann die Abfrage Protein kann als voller Länge modelliert werden.
Geben äußeren Zwängen
A. Geben Sie contact / Abstandsrestraints
Experimentell charakterisiert inter-Rückstandes Kontakte / Distanzen, zum Beispiel aus NMR-oderVernetzung Experimente können durch Hochladen einer Zurückhaltung angegeben werden. Ein Beispiel-Datei in Abbildung 8, wo Spalte 1 gibt die Art der Zurückhaltung ist angezeigt, dh "DIST" oder "Kontakt". Für Abstand Zurückhaltung (DIST), Spalten 2 und 4 Rückstände enthalten Positionen (i, j), enthalten Spalten 3 und 5 das Atom-Typen in den Rückstand und Spalte 6 gibt den Abstand zwischen den beiden angegebenen Atome. Für den Kontakt Beschränkungen (CONTACT), enthalten Spalten 2 und 3 Positionen (i, j) von Rückständen, die bei Berührung sein sollte. Der Abstand zwischen den Seitenketten Zentrum dieser Kontaktaufnahme Rest-Paaren ist auf Grund der beobachteten Distanzen in bekannten Strukturen in PDB entschieden. I-Tasser wird versuchen, diese Atompaare in der Nähe der angegebenen Entfernung während der Strukturverfeinerung Simulationen zu ziehen.
B. Geben Sie einen Protein-Struktur-Vorlage
LOMETS Threading-Programme verwenden eine repräsentative PDB Bibliothek plausible Falten für die Abfrage prot findenEin. Obwohl die Verwendung einer repräsentativen Struktur-Bibliothek hilft, die Zeit benötigt, um die Sequenz-Struktur-Alignments berechnen zu reduzieren, ist es möglich, dass eine gute Vorlage Protein in der Bibliothek ausgelassen wird oder die Vorlage kann nicht durch LOMETS Threading-Programme identifiziert wurden, obwohl es in der Bibliothek vorhanden. In diesen Fällen sollte der Benutzer die gewünschte Protein Struktur wie die Vorlage.
So legen Sie Proteinstruktur als zusätzliche Vorlage kann der Anwender entweder laden Sie eine PDB-Datei formatiert Struktur oder geben Sie die PDB ID einer abgeschiedenen Proteinstruktur in PDB-Bibliothek. Die I-Tasser erzeugt die Abfrage-template Ausrichtung mit MUSTER Programm 23 und räumlichen Beschränkungen sowohl vom Benutzer angegebene Vorlage und LOMETS Vorlagen, um die Struktur der Montage-Simulation Guide zu sammeln. Da die Genauigkeit der LOMETS Beschränkungen ist für verschiedene Ziele, ist das Gewicht des LOMETS Beschränkungen stärker in leicht (homologen) targets als in hart (nicht-homologe) Ziele, die systematisch in unsere Benchmark-Training abgestimmt.
Benutzer können auch ihre eigenen Abfrage-template Ausrichtungen. Der Server akzeptiert die Ausrichtung in zwei Formaten erhältlich: das FASTA-Format (Abbildung 9A) und das 3D-Format (Abbildung 9B). Das FASTA-Format als Standard beschrieben und auf http://zhanglab. ccmb.med.umich.edu / FASTA / . Die 3D-Format ist vergleichbar mit dem Standard-PDB-Format ( http://www.wwpdb.org/documentation/format32/sect9.html ), aber zwei zusätzliche Spalten aus den Vorlagen abgeleitet werden, um das Atom Aufzeichnungen (siehe Abbildung 9B) angefügt:
Spalten 1-30: Atom (C-alpha nur) und Rück-Namen für die Abfrage-Sequenz.
Spalten 31-54: Koordinaten von C-alpha-Atome der Abfrage aus der entsprechenden Atome in der Vorlage kopiert.
Spalten 55-59: Entsprechende Rückstände in der Vorlage auf die Ausrichtung auf
Spalten 60-64: Entsprechende Rückstände im Template
Ausschließen Vorlagen Proteine
Proteine sind flexible Moleküle und können mehrere Konformationen anzunehmen, um ihre biologische Aktivität zu verändern. Zum Beispiel haben Strukturen vieler Proteinkinasen und Membranproteine in aktive und inaktive Konformation gelöst. Auch An-oder Abwesenheit des gebundenen Liganden kann großen strukturellen Bewegungen. Während alle Konformationen der Vorlage gleichermaßen für das Einfädeln Programme, ist es wünschenswert, um die Abfrage mithilfe von Vorlagen in nur einem bestimmten Zustand Modell. Eine neue Option auf dem Server ermöglicht dem Benutzer die Vorlage Proteinen während der Struktur-Modellierung auszuschließen. Dieses Feature würde es auch ermöglichen dem Benutzer, die Homologie Niveau der Vorlagen für die Modellierung verwendet werden soll. Benutzer können ausgeschlossen Vorlage Proteine from der I-Tasser Bibliothek durch:
A. Angeben einer Sequenzidentität cutoff
Benutzer können diese Option verwenden, um homologe Proteine aus dem I-Tasser Template-Bibliothek ausgeschlossen werden. Die Homologie-Ebene auf der Grundlage der Sequenzidentität Cutoff eingestellt ist, dh die Anzahl der identischen Rückstand zwischen der Abfrage und der Vorlage Protein durch die Sequenz Länge der Abfrage Reihenfolge aufgeteilt. Zum Beispiel, wenn der Benutzer in "70%" in das dafür vorgesehene Formular, alle Vorlagen Proteine, die eine Sequenzidentität haben> 70%, um die Abfrage Protein I-wird aus dem I-Tasser Template-Bibliothek ausgeschlossen werden.
B. Ausschließen bestimmter Vorlage Proteine
Spezielle Vorlage Proteine können aus dem I-Tasser Template-Bibliothek durch das Hochladen einer Liste mit PDB IDs der Strukturen ausgeschlossen werden ausgeschlossen. Ein Beispiel-Datei ist in Abbildung 10 dargestellt. Da das gleiche Protein kann als mehrere Einträge in der PDB-Bibliothek, I-Tasser se existierenRVer wird standardmäßig ausgeschlossen angegebenen Vorlagen (in Spalte1) sowie alle anderen Vorlagen aus der Bibliothek, die eine Identität zu haben> 90% auf die angegebenen Vorlagen. Benutzer können auch eine andere Identität Cutoff, zB 70%, in der alle Vorlagen mit Identität> 70% auf angegebenen Vorlage Proteine ausgeschlossen werden.
Disclosures
Keine Interessenskonflikte erklärt.
Acknowledgments
Das Projekt wird zum Teil durch Alfred P. Sloan Foundation, NSF Career Award (DBI 1.027.394) und das National Institute of General Medical Sciences (GM083107, GM084222) unterstützt.
Materials
| Name | Company | Catalog Number | Comments |
| Material Name | Type | Company | Catalogue Number |
| FASTA formatted amino acid sequence of the protein to be modeled (see, http://www.ncbi.nlm.nih.gov/BLAST/fasta.shtml). | |||
| A personal computer with access to the internet and a web browser. | |||
| Molecular visualizing software, e.g. RASMOL or PYMOL, for analyzing the predicted tertiary structure and functional sites. |
References
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H. CD-Search: protein domain annotations on the fly. Nucleic. Acids. Res. 32, W327-W331 (2004).
- Finn, R. D. The Pfam protein families database. Nucleic. Acids. Res. 38, D211-D222 (2010).
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).

{kind=link}
{kind=link}