

Summary
הנחיות מחשב מבוסס אפיון מבניים ותפקודיים של חלבון באמצעות צינור I-TASSER מתואר. החל מ רצף חלבון השאילתה, מודלים 3D נוצרות באמצעות מערכים השחלה מרובים איטרטיבי סימולציות הרכבה המבני. מסקנות פונקציונלית נמשכים לאחר מכן מבוסס על התאמות לחלבונים עם מבנה ופונקציות ידוע.
Abstract
פרויקטים לקביעת רצף הגנום מוצפנת יש מיליונים של רצף חלבון, אשר דורשים ידע של מבנה ותפקוד שלהם כדי לשפר את ההבנה של תפקידם הביולוגי שלהם. למרות שיטות ניסיוניות יכול לספק מידע מפורט על חלק קטן של חלבונים אלה, מודלים חישוביים דרוש רוב מולקולות חלבון אשר uncharacterized בניסוי. שרת I-TASSER הוא שולחן העבודה on-line דוגמנות ברזולוציה גבוהה של מבנה החלבון ותפקודו. בהינתן רצף החלבון, פלט טיפוסי משרת I-TASSER כולל חיזוי מבנה משני, ניבא נגישות ממס שאריות של כל אחד, חלבונים הומולוגיים תבנית זוהה על ידי השחלה ועל יישור המבנה, עד חמש באורך מלא, מודלים מבניים שלישוני, מבנה מבוסס הסברים פונקציונליים לסיווג האנזים, אונטולוגיה ג'ין התנאים חלבון ליגנד אתרי הקישור. כל התחזיות מתויגים עם ציון האמון אשרמספר כיצד מדויק התחזיות הן מבלי לדעת את נתוני הניסוי. כדי להקל על בקשות מיוחדות של משתמשי הקצה, השרת מספק ערוצי לקבל את המשתמש שצוין בין שאריות מרחק וקשר מפות אינטראקטיבי לשנות את הדוגמנות I-TASSER, זה גם מאפשר למשתמשים לציין כל החלבונים כתבנית, או להוציא כל תבנית חלבונים במהלך סימולציות מבנה הרכבה. המידע מבניים יכול להיות שנאספו על ידי המשתמשים בהתבסס על עדויות ניסיוניות או תובנות ביולוגיות במטרה לשפר את איכות I-TASSER התחזיות. השרת הוערך כמו התוכניות הטובות ביותר עבור מבנה החלבון ותחזיות לתפקד הקהילה רחב הניסויים האחרונים CASP. כרגע יש> 20,000 מדענים רשומים מ -100 מדינות המשתמשים on-line I-TASSER השרת.
Protocol
שיטת סקירה
בעקבות הפרדיגמה רצף אל המבנה אל הפונקציה, ההליך I-TASSER 1-4 למבנה דוגמנות פונקציה כוללת ארבעה שלבים רצופים של: (א) על ידי זיהוי תבנית LOMETS 5 (ב) מבנה שבר והרכבה מחדש על ידי-העתק חילופי סימולציות מונטה קרלו 6 (ג) ברמה האטומית באמצעות שכלול מבנה רמו 7 ו FG-MD 8; (ד) מבנה המבוסס על פרשנויות פונקציה באמצעות cofactor 9.
זיהוי תבנית: עבור רצף שאילתה שהוגשה על ידי המשתמש, הרצף הוא מושחל הראשון LOMETS מותקנת באופן מקומי meta-threading שרת דרך הספרייה מבנה נציג PDB. Threading היא הליך רצף מבנה יישור המשמש לזיהוי חלבונים תבנית אשר ייתכן מבנה דומה או להכיל מוטיב מבניים דומים כמו חלבון השאילתה. כדי להגדיל את הכיסוי של templ הומולוגייםלתגליות אכל, LOMETS משלב מספר רב של המדינה-of-the-art אלגוריתמים כיסוי מתודולוגיות השחלה שונה. מאז תוכניות השחלה שונים יש מערכות ניקוד שונה ורגישויות היישור, האיכות של מערכים השחלה שנוצר כל תוכנית השחלה נבחנת על ידי ציון-Z מנורמל, אשר מוגדר כ: 
איפה Z-ציון הוא הציון ביחידות סטיית התקן ביחס לממוצע סטטיסטי של כל המערך שנוצר על ידי תוכנית, ו-Z 0 היא תוכנית ספציפית Z-הציון הקובע נקבע על בסיס בקנה מידה גדול מבחני ביצועים השחלה 5 להבדיל "טוב 'ו' רע 'תבניות. תבנית עם ציון-Z גבוה משמעות הדבר היא כי תבניות למעלה יש ציון התאמה גבוה משמעותית ביותר של תבניות אחרות, אשר בדרך כלל מרמז כי יישור מתאים מודל טוב. אם רוב תבניות השחלה העליון יש הייGH מנורמל-Z ציוני, את הדיוק של הדגם I-TASSER הסופית היא בדרך כלל גבוהה. עם זאת, אם החלבון הוא גדול הכיסוי של מערכים השחלה מוגבל לאזור קטן של חלבון השאילתה, גבוהה מנורמל Z-ציון לא בהכרח אומר דוגמנות דיוק גבוה עבור דגם באורך מלא. הדף שני מערכים השחלה מתוכנית כל השחלה נאספים ומשמשים לשלב הבא של מבנה הרכבה.
איטרטיבי הרכבה סימולציה מבנה: בעקבות ההליך השחלה, רצף שאילתה מחולק לאזורים השחלה בציר המעוות. שברי רציף יישור השחלה הם נכרת מתוך תבניות בשימוש ישירות להרכבה המבנה, בעוד אזורים לולאה המעוות נבנים על ידי הדוגמנות מלכתחילה. הליך ההרכבה המבנה מבוצע על מערכת סריג בהדרכת החליפין העתק סימולציות מונטה קרלו 6. השדה I-TASSER כוח כולל מימן בוnding אינטראקציות 10, מבוססי ידע מבחינת אנרגיה סטטיסטי הנגזר מבנים חלבוניים ידוע PDB 11, המבוססת על רצף של קשר תחזיות SVMSEQ 12, מעצורים מרחבי שנאספו LOMETS 5 תבניות השחלה. פתיונות קונפורמציה שנוצר בטמפרטורה נמוכה העתקים במהלך סימולציות מקובצים על ידי SPICKER 13 לזהות מבנים של אנרגיה חופשית מדינות נמוכה. Centroids Cluster של אשכולות העליון מתקבלים על ידי חישוב ממוצע של כל הקואורדינטות 3D פיתיונות מבניים אשכולות משמש לייצור הדגם הסופי. סימולציה וההליך באשכולות חוזרים פעמיים להסרת עימותים סטרית ועוד הזיקוק טופולוגיה העולמי.
ברמה האטומית מודל הבנייה עידון: centroids אשכול המתקבל לאחר באשכולות SPICKER מופחתים מודלים חלבון (כל שאריות מיוצג על ידי α שלה C, בצד שרשרת של מרכז המסה) ו hשד מוגבל ביולוגי היישום. בניית מודל מלא אטומי ממודלים הקטינה מתבצע בשני שלבים. בשלב הראשון, רמו 7 משמש לבניית מלא האטום מודלים מ C-alpha עקבות על ידי אופטימיזציה של רשתות H-האג"ח. בשלב השני, רמו מלא אטומי הדגמים מעודן עוד יותר על ידי FG-MD 14, אשר משפר את פיתול עמוד השדרה זוויות, אורכים איגרות החוב, לצד שרשרת אוריינטציות rotamer, על ידי סימולציות דינמיות מולקולרית, מודרך על ידי שברי מבניים חיפש מן מבנים PDB על ידי TM-align. FG-MD מודלים מעודן משמשים הדגמים הסופיים עבור תחזיות מבנה שלישוני על ידי I-TASSER.
איכות הדגמים שנוצר נאמדים על בסיס ניקוד ביטחון (C-ציון), המוגדרת על בסיס ניקוד-Z של מערכים LOMETS השחלה ואת ההתכנסות של I-TASSER סימולציות, מתמטי כמו: 
איפה
C-ציון יש מתאם גבוה עם איכות המודלים I-TASSER. על ידי שילוב של C-ציון ואורך החלבון, ברמת דיוק של I-TASSER הדגמים הראשונים ניתן להעריך עם שגיאה ממוצעת של 0.08 עבור התפלגות-TM ו 2 עבור 15 RMSD. באופן כללי, מודלים עם ציון C-> - 1.5 צפויים יש לקפל הנכון. כאן, RMSD ו-TM התוצאה הן אמצעי מוכר של דמיון טופולוגי בין המודל והמבנה הילידים. TM-ציון valuמגוון es ב [0, 1], שם ניקוד גבוה יותר מצביע על המבנה טוב יותר להתאים 16,17. אולם עבור נמוך בדירוג מודלים (כלומר 2 nd דגמי ה -5), המתאם של הציון-C עם ניקוד-TM ו RMSD הוא הרבה יותר חלש (~ 0.5), ולא יכול לשמש אומדן אמין של איכות המודל מוחלט.
האם המודל הראשון תמיד את המודל הטוב ביותר I-TASSER סימולציות? התשובה לשאלה זו תלויה בסוג מטרה. עבור מטרות קלות, הדגם הראשון הוא בדרך כלל את המודל הטוב ביותר שלה C-התוצאה היא בדרך כלל הרבה יותר גבוה מאשר שאר המודלים. עם זאת, עבור מטרות קשות, בו השחלה אינה פוגעת תבנית משמעותית, הדגם הראשון אינו בהכרח המודל הטוב ביותר ואני-TASSER למעשה מתקשה בבחירת תבנית מיטב הדגמים. לכן מומלץ לנתח את כל 5 מודלים מטרות קשה לבחור אותם על סמך המידע הניסויי ואת הידע הביולוגי.
הפונקציה predictions: בשלב האחרון, הסופי 3D-מודלים שנוצר FG-MD משמשים כדי לחזות שלושה היבטים של תפקוד החלבון, כלומר: א) אנזימים הנציבות (EC) המספרים 18 ו (ב) אונטולוגיה ג'ין (GO) 19 התנאים ( ג) אתרי קישור ligands מולקולה קטנה. במשך כל שלושת ההיבטים, פרשנויות פונקציונלי נוצרות באמצעות cofactor, אשר היא גישה חדשה כדי לחזות פונקציה של חלבון על סמך הדמיון גלובליים ומקומיים חלבונים תבנית PDB עם מבנה ופונקציות ידוע. ראשית, טופולוגיה הגלובלית של המודלים ניבאו משתווה נגד ספריות תבנית תפקודית באמצעות התוכנית יישור מבניים TM-ליישר 20. לאחר מכן, סדרה של חלבונים דומים ביותר הדגמים יעד נבחרים מהספריה על סמך הדמיון במבנה העולמי שלהם, חיפוש מקומי נרחב מתבצע כדי לזהות את מבנה רצף הדמיון ליד אזור פעיל / מחייב את האתר. עשרות כתוצאה דמיון גלובליים ומקומיים משמשים לדרגתתבנית של חלבונים (homologues תפקודית) ולהעביר את ביאור (מספרים EC ו אונטולוגיה ג'ין 19 מונחים) מבוסס על להיטים הבקיע העליון. כמו כן, מחייב ליגנד באתר שאריות מצב ליגנד המחייבים הינם להסיק בהתבסס על יישור המקומי של שאילתה עם ידוע מחייב ליגנד שאריות אתר בפונקציה תבניות העליון הבקיע 9.
איכות של הפונקציה (EC ו GO טווח) התחזית ב-I TASSER מוערך על בסיס ציון הומולוגיה תפקודית (FH-ניקוד) שהיא מדד הדמיון גלובליים ומקומיים בין השאילתה בתבנית, והוא מוגדר כ: 
שם C-ציון הוא אומדן של איכות המודל החזוי כהגדרתו Eq. (2); TM-הציון מודד את הדמיון המבני בין המודל הגלובלי וחלבונים תבנית: RMSD עלי הוא RMSD בין המודל והמבנה תבנית באזור מיושר מבנית מ-TM ליישר 20; Cov מייצג את הכיסוי של יישור המבני (כלומר היחס בין המשקעים מיושר מבנית חלקי אורך השאילתה); מזהה עלי הוא זהות רצף יישור TM-ליישר את. הציון ביטחון מוערך לתחזיות EC מספר כולל גם לטווח להערכת להתאים אתר פעיל (ACM) בין שאילתה תבנית בתוך אזור המוגדר המקומית, מחושב: 
כאשר N לא מייצג את מספר שאריות התבנית הנוכחית בתחום המקומי, עלי N הוא מספר את השאילתה, תבנית זוגות מיושר שאריות, ד ii הוא המרחק בין זוג α C i ה שאריות מיושר, ד 0 = 3.0 הוא הפסקת מרחק, M ii הוא ציוני BLOSUM בין זוג ith שאריות מיושר. באופן כללי, FH-הציון הוא בטווח [0, 5] ציון ACM הוא בין [0, 2], שם ציונים גבוהים המעידים מטלות פונקציונלי יותר ביטחון. ציון ACM משמש גם להערכת מבנה המקומית דמיון רצף ליד ליגנד מחייב אתרים, המכונה התפלגות-BS.
1. הגשת רצף החלבון
- בקר בדף I-TASSER האינטרנט בכתובת http://zhanglab.ccmb.med.umich.edu/I-TASSER להתחיל עם מבנה דוגמנות הניסוי לתפקד.
- העתק והדבק את רצף החומצות האמיניות אל תוך בטופס המסופק או להעלות אותו ישירות מהמחשב על ידי לחיצה על הלחצן "עיון". I-TASSER השרת כרגע מקבל עם רצפים עד 1500 שאריות. חלבונים יותר מאשר 1500 הם בדרך כלל שאריות רב תחום חלבונים, מומלץ להיות מחולק תחומים בודדים לפני הגשת אל I-TASSER.
- ספקו את כתובת הדואר האלקטרוני שלך (חובה) ואת שם העבודה (לא חובה).
- המשתמשים יכולים לציין חיצוני אופציונלי בין מילקשר idue / מרחק מעצורים, התוספת תבנית נוספת או להוציא כמה חלבונים תבנית במהלך תהליך דוגמנות המבנה. למידע נוסף על אפשרויות אלה באמצעות בסעיף "דיון".
- כדי להגיש את הרצף, ללחוץ על כפתור "הפעל I-TASSER". הדפדפן יופנה לדף אישור הצגת מידע המשתמש שצוין, הזדהות עבודה (ID איוב) מספר וקישור לדף אינטרנט שבו תוצאות יופקדו לאחר השלמת העבודה. משתמשים יכולים סימניה על קישור זה או לרשום את מספר הזיהוי עבודה לשימוש עתידי.
2. זמינות של תוצאות
- בדוק את מצב העבודה שהוגשה שלך על ידי ביקור בדף I-TASSER תור http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php . לחץ על הכרטיסייה חיפוש ושימוש מזהה מספר איוב או את רצף השאילתה חיפוש עבודה שהוגשה שלך.
- לאחר המבנה והתפקוד מוdeling נגמר, הדואר האלקטרוני הודעה ובה תמונה של המבנים חזה אינטרנט קישור תישלח אליך. לחץ על קישור זה או לפתוח את הקישור בסימניה בכל שלב 1.5 כדי להציג ולהוריד את התוצאות.
3. מבנה משני ותחזיות נגישות ממס
- בדוק את השאילתה מעוצב FASTA רצף המוצג בראש הדף את התוצאה. אם כל איפוק תבנית נוספת / צוין במהלך הגשת הרצף, קישור אל דף האינטרנט המציג למשתמש מידע שצוין ניתן לראות (איור 1A).
- בדוק את התחזית המבנה המשני מוצג כ: סליל אלפא (H), גדיל בטא (S) או סליל (C) ציון אמון של חיזוי (0 = נמוך, 9 = גבוהה) עבור כל שאריות. חפש באזור עם ארוכים של המבנה המשני הרגיל (H או S) התחזיות, כדי להעריך את אזור הליבה של החלבון. בכיתה מבנית של חלבונים יכול גם להיות מנותח על בסיס ההפצה של אלמנטים משני מבנים. אלכך, אזורים רב של אלמנטים סליל בחלבון בדרך כלל מצביעים על אזורים בלתי מובנה / סדר.
- הצג את הנגישות ממס חזה (תרשים 1C) כדי לברר אזורים חשופים קבור ממס בשאילתה. ערכים של חזו טווח נגישות ממס בין 0 (שאריות קבור) עד 9 (שאריות חשוף). אזור המכיל שאריות קבור ברובו ניתן להשתמש כדי לשרטט את אזור הליבה של החלבון, בעוד אזורים עם שאריות חשוף הידרופילי ממס הם הידרציה פוטנציאל / אתרים פונקציונליים.
4. מבנה תחזיות השלישון
- גלול למטה כדי לראות את המבנים שלישוני חזה של חלבון השאילתה, מוצג יישומון Jmol אינטראקטיבי (איור 2). השמאל ללחוץ על היישומון כדי לשנות את המראה של המבנה מוצגת, זום לתוך אזור מסוים, בחר סוגי שאריות ספציפיים במודל חזה או לחשב שאריות בין מרחקים.
- ניתוח מודלים עבור נוכחות של אזורי זמן מובנה. אלה regions בדרך כלל מתאימות אזורים סדר בחלבון או להצביע על חוסר תיאום התבנית. אזורים אלה הם בדרך כלל בעלי דיוק נמוך דוגמנות והסרת אזורים אלה במהלך דוגמנות מ N & C-הסופית האזור יהיה לשפר את הדיוק דוגמנות.
- הורד את קבצי PDB מבנה מעוצב של המודל על ידי לחיצה על הקישורים "הורד מודל". אתה יכול לפתוח קבצים אלה כל תוכנה להדמיה מולקולרית (למשל Pymol, Rasmol וכו ') לצורך ניתוח נוסף של תכונות מבניות.
- ניתוח התפלגות ביטחון (C-ציון) של מודלים מבנה להעריך את איכות המבנים חזה. C-ציון (Eq. 2) הערכים הם בדרך כלל בטווח [-5, 2], שבו ניקוד גבוה יותר משקף מודל של איכות טובה יותר. כ-TM ציון ו RMSD של הדגם הראשון מוצג "דיוק משוער של דגם 1". עבור חלבונים ארוך, מומלץ להעריך את איכות המודל מבוסס על ניקוד-TM, כמו TM-ציון רגיש יותר לשינויים מאשר RMSD טופולוגי. < li> לחץ על "נוסף על C-ציון" הקישור לנתח C-ציון, גודל האשכול וצפיפות מקבץ של כל הדגמים. משוער TM-ציון ו RMSD מוצגים רק עבור דגם I-TASSER הראשון, כי C-ציון של מודלים מדורגות נמוך יותר אינו בקורלציה עם ציון-TM או RMSD. איכות נמוכה יותר בדירוג הדגמים ניתן להעריך חלקית המבוססת על צפיפות הצביר שלהם גודל האשכול ביחס לדגם הראשון, שבו מודלים מאשכול גדול יותר צפיפות גבוהה יותר בממוצע קרוב למבנה הילידים.
- Low-C-ציון תחזיות בדרך כלל מצביעים על חיזוי דיוק נמוך. במקרים כאלה ביותר, חלבון השאילתה חסרה תבנית טוב בספריה יש גודל מעבר לטווח של הדוגמנות מלכתחילה (כלומר> 120 שאריות). במקרים אלה, משתמשים יכולים לחפש מעצורים מרחבי נוספים ולהשתמש בהם על מנת לשפר את הדוגמנות I-TASSER (ראה סעיף דיון). זה מעודד גם להגיש את הרצפים לשרת שלנו קווארק (קווארק / "> http://zhanglab.ccmb.med.umich.edu/QUARK/) דוגמנות ab טהור initio אם גודל החלבון היא מתחת 200 שאריות.
5. LOMETS תבנית היעד יישור
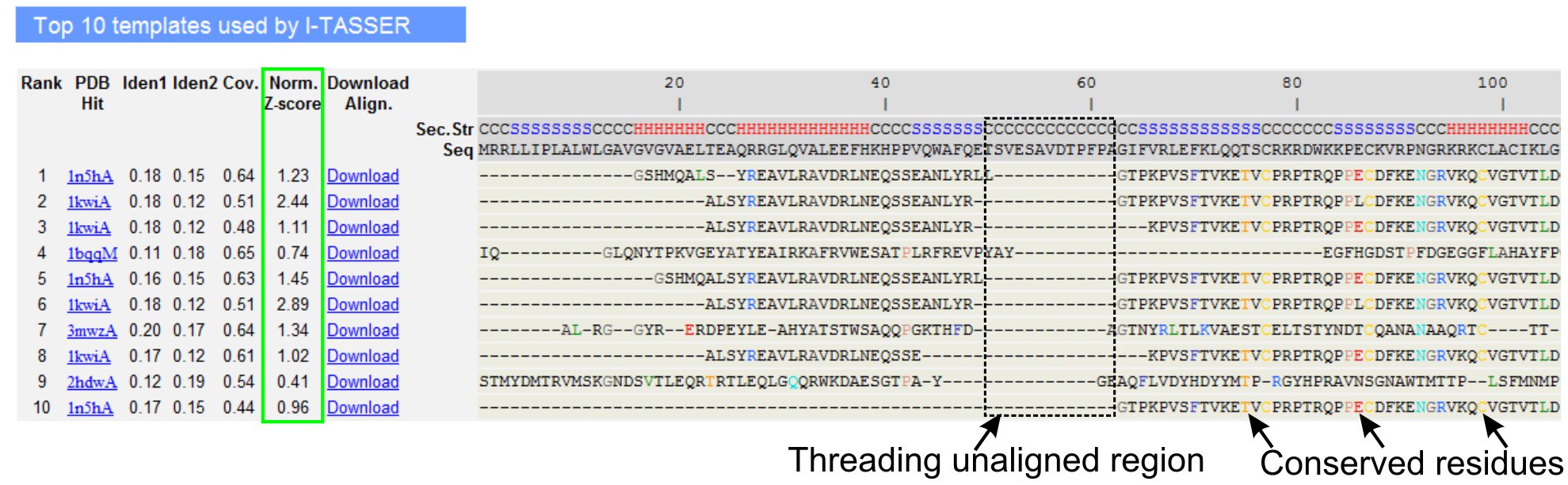
- גלול למטה כדי לנתח את עשרת תבניות השחלה של חלבון השאילתה, כפי שהוגדרו על ידי תוכניות LOMETS השחלה (איור 3). הצג את מנורמל Z-ציון (Eq. 1), שמוצג נורם ". Z-ציון 'טור, כדי לנתח את איכות מערכים השחלה. במערכים עם ציון-Z מנורמל> 1 משקף יישור ביטחון סביר להניח יש לקפל כמו חלבון השאילתה.
- ניתוח זהות רצף באזור השחלה מזדהות ("איידן. 1 'עמודה) ועבור כל שרשרת (" איידן. 2' עמודה) כדי להעריך את הומולוגיה בין השאילתה החלבונים התבנית. זהות רצף גבוה הוא אינדיקטור של קירבה אבולוציונית בין השאילתה וחלבונים התבנית.
- הצג את שאריות מיושר השחלה מוצג בצבע חזותית לזהות והחסרונותשאריות erved / מוטיבים בשאילתה ואת החלבונים התבנית. זהות רצף גבוה יותר באזור-threading מיושר, לעומת יישור כל שרשרת גם מצביע על נוכחות של מוטיב מבניים / תחומים נשמרת בשאילתה.
- הערכת כיסוי של יישור השחלה על ידי הצגת "Cov." עמודה לבדוק את היישור. אם הסיקור של מערכים העליון הוא נמוך מוגבל לאזור קטן בלבד של חלבון השאילתה או נעדר עבור קטע ארוך של רצף השאילתה, ולאחר מכן את החלבון שאילתה מכיל בדרך כלל אחד או יותר של תחום זה מומלץ לפצל את רצף מודל התחומים בנפרד (איור 3).
- הורד את PDB מעוצב רצף מבנה קבצים יישור על ידי לחיצה על "הורד יישור" קישורים. קובץ אלה יישור ניתן לפתוח כל תוכנית להדמיה מולקולרית המפורטים בסעיף החומרים, והוא יכול לשמש גם להוספת מגבלות נוספות במהלך דוגמנות המבנה (שלב 1.4).
6.אנלוגים מבניים PDB
- הצג את הטבלה הבאה (איור 4) של הדף התוצאה כדי לקבוע את עשרת אנלוגים מבניים של המודל החזוי הראשון, זוהה על ידי תוכנית יישור מבניים TM-ליישר 20. TM-ציון> 0.5 מציין אנלוגי לאתר מודל יש טופולוגיה דומה וניתן להשתמש בו כדי לקבוע את מעמד המשפחה מבניים / חלבון של חלבון השאילתה 16, בעוד אלו עם ניקוד-TM <0.3 אמצעי דמיון במבנה אקראי.
- ניתוח זהות רצף RMSD באזור מיושר מבנית שמוצג "IDEN 'ו' RMSD" עמודות כדי להעריך את שימור מוטיבים מרחבי במודל ואת אנלוגי מבניים. ראייה לבדוק את זוגות משקע בצבע מיושר יישור לזהות שאריות אלה נשמרת מבנית ומוטיבים.
- לחץ על קוד PDB המוצג בעמודה 'הכה PDB "כדי לבקר באתר האינטרנט RCSB וללמוד עוד על הסיווג המבני שלהם (SCOP, קת' ו PFAM) ומידע תפקודית (מספר EC, הקשורים GO התנאים ליגנד קשור).
7. פונקציה חיזוי באמצעות cofactor
- גלול מטה בדף התוצאות לנתח פרשנויות תפקודית של חלבון השאילתה. פונקציות חלבון הם המנויים שלושה שולחנות הקשר, מציג: אנזימים הנציבות (EC) מספרים, אונטולוגיה ג'ין (GO) במונחים, אתרי קשירה ליגנד.
- הצגת "TM-ציון", "RMSD", "IDEN 'ו' Cov." העמודות בטבלה כדי לנתח כל הפרמטרים של דמיון במבנה העולמי של שימור דפוסים מרחביים בין מודל homologues פונקציונלי מזוהה (תבניות).
8. אנזימים ועדת חיזוי מספר
- צפה חמשת homologues אנזים הפוטנציאל של חלבון השאילתה שמוצג "חזוי מספרים EC" שולחן (איור 5). רמת האמון של חיזוי מספר EC השימוש בתבניות אלה, מוצג בעמודה 'EC-ציון ". בהתבסס על benchmarking ניתוח 23, דמיון פונקציונלי (הראשונה 3 הספרות האחרונות של מספר EC) בין השאילתה חלבון התבנית ניתן לפרש באופן אמין באמצעות EC-ציון> 1.1.
- חפש קונצנזוס של הפונקציה (מספרים EC) בין תבניות, אשר יש לקפל דומה (כלומר TM-ציון> 0.5), כמו חלבון השאילתה. אם יש אותו מספר רב של תבניות מספר EC ו-EC-ציון> 1.1, רמת הביטחון של התחזית הוא גבוה מאוד. עם זאת, אם ציון-EC הוא גבוה, אבל יש חוסר הסכמה בין הלהיטים מזוהה, אז התחזית הופך אמין פחות המשתמשים מומלץ להתייעץ עם התחזיות לטווח GO.
- לחץ על הקישור המופיע המספרים EC לבקר באתר אנזימים ExPASy ולנתח את הפונקציה, כולל catalyzed התגובה, שיתוף גורם לדרישות מסלול מטבולי של החלבון תבנית בפירוט.
9. אונטולוגיה ג'ין (GO) טווח התחזיות
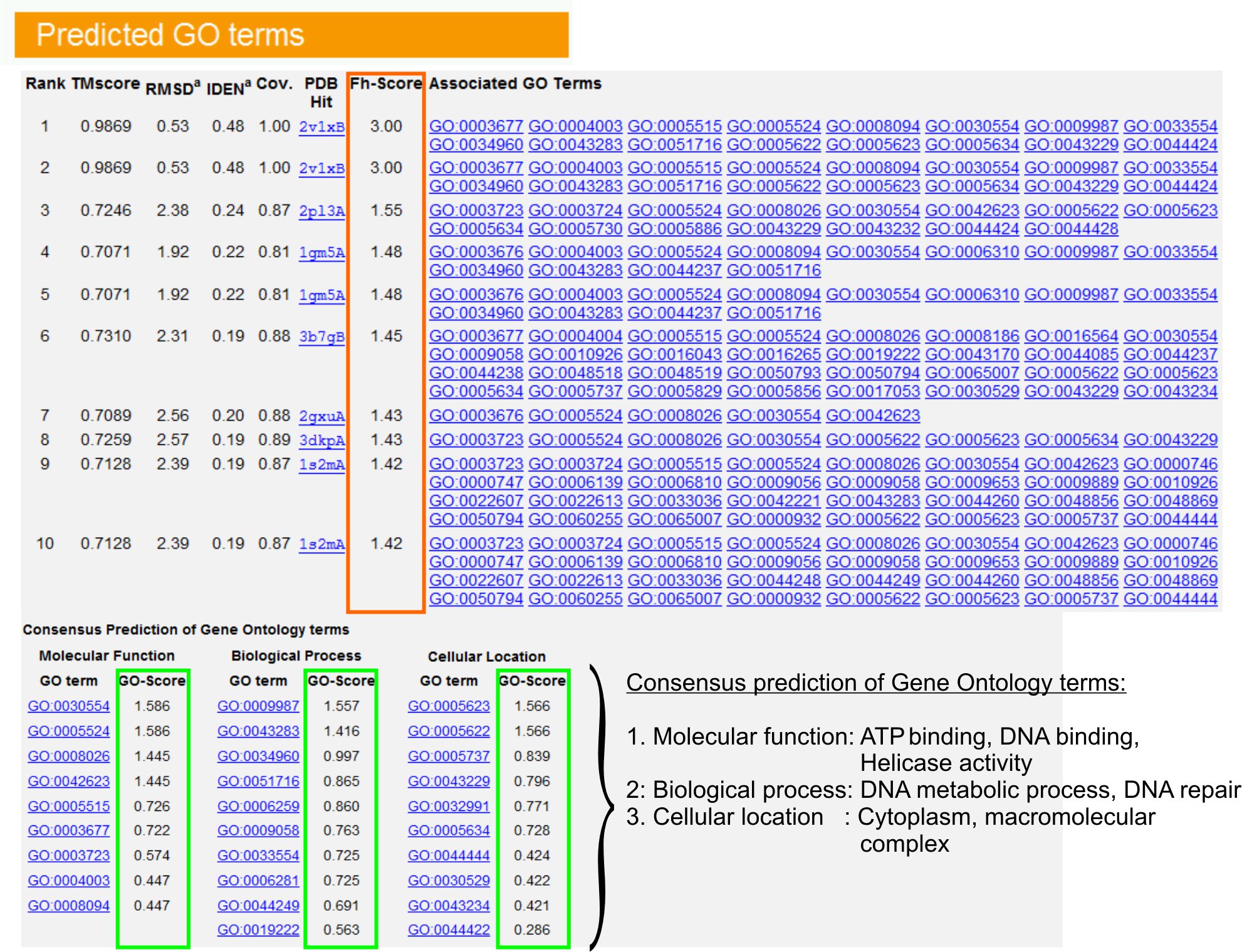
- הצגת "חזוי GO המונחים" שולחן (איוריור 6) כדי לזהות את עשרת homologues של חלבון השאילתה בספריה PDB, המבואר עם אונטולוגיה ג'ין (GO) תנאי. כל חלבון מזוהה בדרך כלל עם תנאי GO מרובים, המתארים פונקציות המולקולרי שלו (MF), תהליכים ביולוגיים (BP) ואת מרכיב הסלולר (CC). לחץ על כל מונח לבקר באתר אמיגו ולנתח הגדרתו השושלת.
- נתח את הטור FH-ציון (ציון הומולוגיה פונקציונלית) כדי לגשת דמיון פונקציונאלי בין השאילתה וחלבונים התבנית להעריך את רמת הביטחון של העברת ביאור תפקודית של חלבונים אלו. במחקר שלנו בהשוואות 23, 50% במונחים GO יליד ניתן היה לזהות נכונה מהתבנית זוהה לראשונה באמצעות הפסקת FH-ציון של 0.8, עם דיוק הכולל של 56%.
- הצג את "תחזית הקונצנזוס של מונחים GO" שולחן לנתח את ההסכמה של תפקוד בין התבניות. פונקציות אלה הנפוצים המשמשים לחיזוי תנאי GO (MF, BP ו CC) של השאילתהחלבון להעריך את רמת הביטחון (GO-ציון) של GO טווח התחזיות. בהתבסס על הבדיקה בהשוואות 23, שיעורי מיטב שווא שלילי חיובי כוזב מתקבלים על התחזיות עם הפסקת GO-ציון = 0.5, עם הפחתת הכיסוי של תחזית ברמות האונטולוגיה עמוק.
10. חלבון ליגנד אתר תחזיות מחייב
- גלול מטה לתחתית העמוד כדי להציג עשרת מחייב ליגנד אתר תחזיות של חלבון השאילתה. אתרי הקישור חזוי מדורגות על פי מספר תצורות של ליגנד חזו כי נתח בכיס מחייב משותף. האתר מחייב הכי מזוהה מוצג כבר יישומון Jmol. לחץ על לחצני הבחירה לנתח את תחזיות אחרות להמחיש את שאריות אינטראקציה ליגנד.
- נתח את הטור BS-ציון להעריך דמיון מקומי בין המודל אתר הקישור של התבנית. בהתבסס על 9 benchmark, BS-ציון> 1.1 מציין רצף גבוה ומבנה ה-SIMilarity סמוך לאתר מחייב חזה במודל והאתר מחייב ידוע בתבנית.
- הורד את הקובץ PDB מבנה מעוצב של המתחם על ידי לחיצה על הקישור "הורד". משתמשים יכולים לפתוח את הקבצים הללו בכל תוכנית להדמיה מולקולרית אינטראקטיבי להציג את האתר מחייב חזה ליגנד אינטראקציות חלבון במחשב המקומי שלהם.
11. נציג תוצאות

איור 1 קטע של הדף I-TASSER התוצאה מראה (א) FASTA מעוצב רצף השאילתה;. (ב) ניבא מבנה משני ועשרות אמון הקשורים; ו (ג) ניבא נגישות ממס של שאריות. אזור הליבה נותחו באתר הידרציה פוטנציאל בשאילתה מודגשים ציאן מלבנים אדומות, בהתאמה.

באיור 2.

באיור 3. דוגמה לדף I-TASSER התוצאה מראה עשרת תבניות השחלה מזוהה על ידי יישור LOMETS 5 תוכניות השחלה. איכות של מערכים השחלה מוערך על בסיס מנורמל Z-ציון (מסומן בירוק), שם ערך> 1 משקף יישור בטוח. שאריות מסודרים בתבנית זהים שאריות שאילתה המקביל מודגשים בצבע כדי לציין נוכחות של שאריות מוטיב משומר /, בעוד חוסר יישור ברוב תבניות העליון מציין נוכחות של מספר תחומים בחלבון את השאילתה שאריות המעוות מתאימות האזורים מקשר מושלם. לחץ כאן כדי לצפות בגרסה בגודל מלא של הדמות 3.

איור 4. דוגמה של דף התוצאות מראה עשרת אנלוגים מבניים זוהו יישור מבניים, שזוהו על ידי TM-20 ליישר תוכנית יישור מבניים. הדירוג של אנלוגים המוצג מבוסס על ניקוד-TM (מודגשות בכחול) של יישור המבני. TM-ציון> 0.5 מציין כי שני מבנים בהשוואה יש טופולוגיה דומה, בעוד TM-ציון <0.3 אמצעי דמיון בין שני מבנים אקראיים. זוגות שאריות מיושר מבנית מודגשים בצבע על בסיס רכושם חומצה אמינית, בעוד אזורים המעוות מסומנים על ידי "-".ove.com/files/ftp_upload/3259/3259fig4large.jpg "> לחץ כאן כדי לצפות בגרסה בגודל מלא של הדמות 4.

איור 5. דוגמה לדף I-TASSER התוצאה מראה homologues אנזים זיהו חלבון השאילתה בספריה PDB. רמת האמון של חיזוי מספר EC מנותח על בסיס EC-ציון (מסומן בירוק), שם EC-ציון> 1.1 מציין דמיון פונקציונלי (זה תחילה 3 ספרות של מספר EC) בין שאילתה חלבון התבנית.

איור 6. דוגמה לדף I-GO TASSER התוצאה מראה תחזיות לטווח של חלבון השאילתה. Homologues פונקציונלית של חלבון השאילתה הספרייה אונטולוגיה ג'ין תבנית מדורגות על בסיס ציון FH-שלהן (מלבן כתום). תכונות תפקודית נפוצים אלה העליונה הבקיע להיטים נגזרות כדי gener אכלו הסופי GO תחזיות לטווח של חלבון השאילתה. איכות של GO מבחינת חזה מוערכת על בסיס ניקוד-GO (מוצג ירוק), שם הציון GO-> 0.5 מציין חיזוי אמין. לחץ כאן כדי לצפות בגרסה בגודל מלא של הדמות 6.

איור 7. דוגמה לדף I-TASSER התוצאה מראה עשרת ליגנד חלבון מחייב אתר תחזיות באמצעות אלגוריתם 9 cofactor. הדירוג של אתרי הקישור חזה מבוססת על מספר תצורות ליגנד חזו כי נתח בכיס מחייב משותף בשאילתה. BS-ציון (מסומן באדום) הוא מדד של רצף המקומית הדמיון בין מבנה חזה אתר הקישור של התבנית, והיא שימושית לניתוח שימור כיסים האתר מחייב.
les/ftp_upload/3259/3259fig8.jpg "/>
איור 8. דוגמה קבצים איפוק חיצוני המשמש לציון שאריות, שאריות קשר / מגבלות מרחק.

איור 9. דוגמה של קבצים איפוק משמש ציון חלבון תבנית לשרת I-TASSER. משתמש יכול לציין את היישור שאילתה, בתבנית או בתבנית FASTA (א) או (ב) בפורמט 3D.

איור 10. קובץ דוגמה המשמש למעט תבנית במהלך הליך I-TASSER דוגמנות המבנה. העמודה הראשונה מכילה את מזהה PDB של חלבונים תבנית כדי להיות שליליים. הטור השני משמש כדי לציין את זהות רצף הפסקת אשר ישמשו עבור תבניות דומות אחרות בספריה את התבנית.
Discussion
פרוטוקול שהוצגו לעיל הוא קו מנחה כללי מבנה דוגמנות פונקציה באמצעות שרת I-TASSER. אמנם, הליך זה אוטומטית עובד טוב מאוד עבור רוב החלבונים, התערבות האדם לעתים קרובות לעזור לשפר באופן משמעותי את הדיוק דוגמנות, במיוחד עבור חלבונים אשר חסרים תבניות קרוב בספריה PDB. משתמשים יכולים להתערב במהלך דוגמנות I-TASSER בדרכים הבאות: (א) פיצול רב תחום חלבונים (ב) מתן מגבלות חיצוניות על מנת לשפר את מבנה הרכבה: (ג) הסרת תבניות במהלך הדוגמנות.
חלבון פיצול רב תחום:
רבים רצפי חלבון ארוך לעתים קרובות מכילים מספר תחומים קשורים לפי אזורים מקשר גמיש, מה שהופך הבהרה המבנה שלהם קשה תוך שימוש בטכניקות ניסיוניות הן חישובית. עם זאת, כפי תחומים הן באופן עצמאי מתקפלים הגופים והוא יכול לבצע פונקציה מולקולרית ברורים, היא רצוי לפצל זמן רב תחום חלבונים מודל כל תחום בנפרד. תחומים דוגמנות בנפרד לא רק להאיץ את תהליך החיזוי, אך גם מעלה את האיכות של יישור שאילתה בתבנית, וכתוצאה מכך מבנה אמין יותר תחזיות לתפקד.
גבולות המתחם רצפי חלבונים ניתן לחזות שימוש זמין באופן חופשי ברשת תוכניות חיצוניים, כגון צמח השדה CDD 24, 25 או PFAM InterProScan 26. כמו כן, אם LOMETS יישור השחלה זמינים עבור חלבון השאילתה, גבולות תחום יכול להיות ממוקם על ידי זיהוי חזותי ארוכים של שאריות המעוות של תבניות השחלה העליון (ראה שלב 5.4). אלו אזורים המעוות בעיקר מתאימות האזורים מקשר מושלם. אם רב תחום התבניות כבר זמין בספריית תבנית PDB עם כל התחומים שאילתה מיושר, אז חלבון השאילתה יכול להיות המודל כפי באורך מלא.
ספק מעצורים חיצוניים
class = "jove_content"> הרכבה מבנה סימולציות של I-TASSER מונחים בעיקר על ידי מגבלות מרחביות שנאסף LOMETS השחלה תבניות. עבור חלבונים שאילתה פגעו השחלה טוב (Z-Norm. ציון> 1) בספריית תבנית, מגבלות מרחביות נגזרת בעיקר של דיוק גבוהה ואני-TASSER יפיק מודלים ברזולוציה גבוהה מבני החלבונים האלה. לעומת זאת, עבור חלבונים שאילתה יש חלש או לא פגע השחלה (Norm. Z-ציון <1), מגבלות מרחביות נאסף לעיתים קרובות מכילים טעויות בגלל חוסר הוודאות של התבנית ואת יישור. עבור המטרות האלה חלבון, המשתמש שצוין מידע מרחבי יכול להיות מאוד מועיל לשפר את איכות של המודל החזוי. משתמשים יכולים לספק מעצורים חיצוניים לשרת I-TASSER בשתי דרכים:
א 'ציין קשר / מרחק מעצורים
מאופיין ניסיוני בין שאריות צור קשר / מרחקים, למשל מן התמ"ג אוcross-linking ניסויים, ניתן לציין זאת על ידי טעינת קובץ איפוק. קובץ למשל מוצג באיור 8, שם טור 1 מציין את סוג של איפוק, כלומר "Dist" או "צור קשר". עבור ריסון מרחק (Dist), עמודות 2 ו -4 עמדות מכילים שאריות (i, j), טורים 3 ו 5 להכיל את אטום סוגי למשקע ועמודה 6 מציין את המרחק בין שני האטומים שצוינו. עבור מעצורים מגע (קשר), עמודים 2 ו - 3 מכילים את העמדות (i, j) של שאריות שאמור להיות במגע. המרחק בין המרכז לצד רשתות אלה זוגות שאריות קשר הוא החליט מבוסס על מרחקים שנצפתה מבנים ידועים PDB. I-TASSER ינסה למשוך זוגות אלה אטום קרוב מרחק שצוין במהלך סימולציות עידון המבנה.
ב ציין תבנית מבנה החלבון
LOMETS תוכניות השחלה להשתמש בספריית PDB נציג למצוא קפלי מתקבל על הדעת prot את השאילתהעין. למרות שהשימוש ספריה מבנה נציג עוזר להפחית את הזמן הנדרש כדי לחשב את רצף מבנה מערכים, זה אפשרי כי חלבון תבנית טוב הוא החמיץ בספריה או התבנית לא זוהו על ידי תוכניות LOMETS השחלה, גם אם היא כיום בספריה. במקרים כאלה, המשתמש צריך לציין את מבנה החלבון הרצוי כתבנית.
כדי לציין את מבנה החלבון כתבנית נוספת, משתמשים יכולים גם להעלות קובץ PDB מבנה מעוצב או לציין את מזהה PDB של מבנה החלבון שהופקדו בספריית PDB. I-TASSER יפיק את יישור שאילתה-תבנית באמצעות תוכנית לגייס 23 ו תאסוף מגבלות מרחביות מן המשתמש הן שצוין התבנית LOMETS תבניות להנחות את הסימולציה הרכבה מבנה. בגלל הדיוק של מעצורים LOMETS שונה עבור מטרות שונות, משקל את המגבלות LOMETS הוא חזק קל (הומולוגיים) targets מזה קשה (לא הומולוגיים) מטרות, אשר היה מכוון באופן שיטתי הכשרה benchmark שלנו.
משתמשים יכולים גם לציין שאילתה לתבנית שלהם מערכים. השרת מקבל יישור בשני פורמטים: הפורמט FASTA (איור 9 א) לבין בפורמט 3D (איור 9 ב). הפורמט FASTA הוא תקן תיאר http://zhanglab. ccmb.med.umich.edu / FASTA / . הפורמט 3D דומה לפורמט PDB סטנדרטי ( http://www.wwpdb.org/documentation/format32/sect9.html ), אבל שתי עמודות נוספות הנגזרות התבניות מתווספים הרשומות ATOM (ראה איור 9 ב):
עמודים 10-30: Atom (C-alpha בלבד) שאריות שמות רצף השאילתה.
עמודים 31-54: הקואורדינטות של אטומי C-alpha של השאילתה שהועתקו אטומי המתאים בתבנית.
עמודים 55-59: מספר שאריות מקבילים בתבנית בהתבסס על יישור
עמודים 60-64: שם שאריות מקבילים בתבנית
תכלול חלבונים תבניות
החלבונים הם מולקולות גמישה יכול לאמץ מדינות קונפורמציה מרובים לשנות את פעילותם הביולוגית. לדוגמה, מבנים של קינאזות חלבון חלבונים בממברנה רבים נפתרו בתוך קונפורמציה שני פעילים פעיל. כמו כן, קיומו או אי קיומו של ליגנד כבול עלולה לגרום לתנועות מבניים גדולים. בעוד כל המדינות קונפורמציה של תבנית דומים עבור התוכניות השחלה, רצוי מודל השאילתה באמצעות תבניות במצב היחיד בפרט. אפשרות חדשה על השרת מאפשר למשתמש להוציא חלבונים תבנית במהלך דוגמנות המבנה. תכונה זו גם תאפשר למשתמש לבחור את רמת הומולוגיה של תבניות לשמש הדוגמנות. משתמשים יכולים לכלול חלבונים תבנית frאום I-TASSER הספרייה על ידי:
א ציון הפסקת זהות ברצף
משתמשים יכולים להשתמש באפשרות זו כדי לכלול חלבונים הומולוגיים מספריית I-TASSER את התבנית. רמת הומולוגיה נקבע בהתבסס על הפסקת זהות ברצף, כלומר את מספר שאריות זהה בין השאילתה לבין חלבון תבנית מחולק לפי אורך רצף של רצף השאילתה. לדוגמה, אם משתמש מקליד את המילה "70%" בטופס המיועד לכך, כל התבניות חלבונים אשר יש זהות ברצף> 70% חלבון השאילתה I-ייכללו מספריית I-TASSER את התבנית.
ב תכלול חלבונים תבנית ספציפית
חלבונים תבנית ספציפית ניתן לשלול מספריית I-TASSER את התבנית על ידי העלאת רשימה המכילה מזהי PDB של המבנים להיות מורחק. קובץ דוגמה מוצגת באיור 10. כאשר החלבון אותו יכול להתקיים כישות ערכים מרובים בספריה PDB, I-TASSER server יהיה כברירת מחדל להוציא את תבניות שצוין (ב Column1), כמו גם את כל התבניות האחרות מהספריה כי יש זהות> 90% על תבניות שצוין. משתמשים יכולים גם לציין הפסקת זהות שונה, למשל 70%, כאשר כל התבניות עם זהות> 70% חלבונים תבנית שצוין ייכללו.
Disclosures
אין ניגודי אינטרסים הכריז.
Acknowledgments
הפרויקט נתמך בחלקו על ידי אלפרד פ 'סלואן קרן, פרס NSF קריירה (DBI 1,027,394), לבין המכון הלאומי למדעי הרפואה הכללית (GM083107, GM084222).
Materials
| Name | Company | Catalog Number | Comments |
| Material Name | Type | Company | Catalogue Number |
| FASTA formatted amino acid sequence of the protein to be modeled (see, http://www.ncbi.nlm.nih.gov/BLAST/fasta.shtml). | |||
| A personal computer with access to the internet and a web browser. | |||
| Molecular visualizing software, e.g. RASMOL or PYMOL, for analyzing the predicted tertiary structure and functional sites. |
References
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H. CD-Search: protein domain annotations on the fly. Nucleic. Acids. Res. 32, W327-W331 (2004).
- Finn, R. D. The Pfam protein families database. Nucleic. Acids. Res. 38, D211-D222 (2010).
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).

{kind=link}
{kind=link}