

Summary
Diretrizes para computador baseado caracterização estrutural e funcional da proteína usando o pipeline I-TASSER é descrito. A partir de seqüência de proteína de consulta, os modelos 3D são gerados usando vários alinhamentos threading e iterativo simulações de montagem estrutural. Inferências funcionais são posteriormente elaborado com base em correspondências às proteínas com estrutura conhecida e funções.
Abstract
Projetos de seqüenciamento do genoma tem ciphered milhões de seqüência de proteína, que exigem conhecimento de sua estrutura e função de melhorar a compreensão de seu papel biológico. Embora os métodos experimentais podem fornecer informações detalhadas para uma pequena fração dessas proteínas, modelagem computacional é necessário para a maioria das moléculas de proteínas que são experimentalmente descaracterizados. O servidor I-TASSER é uma bancada on-line de alta resolução de modelagem da estrutura da proteína ea função. Dada uma seqüência de proteína, uma saída típica do servidor I-TASSER inclui previsão de estrutura secundária, previu acessibilidade solvente de cada resíduo, proteínas homólogas modelo detectado por threading e alinhamentos estrutura, até cinco full-length terciário modelos estruturais, e baseado em estrutura anotações funcionais para a classificação das enzimas, termos Gene Ontology e proteína-ligante sítios de ligação. Todas as previsões são marcados com uma pontuação de confiança queconta como precisas as previsões são sem conhecer os dados experimentais. Para facilitar a pedidos especiais de usuários finais, o servidor fornece canais de aceitar especificado pelo usuário resíduos inter-distância e entre em contato mapas interativamente alterar a modelagem I-TASSER, mas também permite aos usuários especificar quaisquer proteínas como modelo ou excluir qualquer modelo proteínas durante as simulações de montagem da estrutura. As informações estruturais poderiam ser recolhidos pelos usuários com base em evidências experimentais ou insights biológicos com a finalidade de melhorar a qualidade de I-TASSER previsões. O servidor foi avaliado como o melhor programas para a estrutura da proteína e previsões função na recente experimentos CASP em toda a comunidade. Existem actualmente> 20.000 cientistas registrados em mais de 100 países que estão usando o servidor on-line I-TASSER.
Protocol
Resumo método
Seguindo o paradigma seqüência-a-estrutura-para-função, o procedimento I-TASSER 1-4 para a estrutura e função de modelagem envolve quatro etapas consecutivas de: (a) identificação do modelo, LOMETS 5; (b) estrutura de fragmento de remontagem por replica- troca de simulações Monte Carlo 6; (c) refinamento estrutura atômica nível usando REMO 7 e FG-MD 8, e (d) interpretações estrutura baseada em função usando cofator 9.
Identificação do modelo: Para uma seqüência de consulta apresentada pelo usuário, a seqüência é primeiro introduzido através de uma biblioteca de estrutura representativa APO num LOMETS instalado localmente meta-threading servidor. Threading é um procedimento de alinhamento da seqüência-estrutura utilizada para a identificação de proteínas que pode ter modelo de estrutura similar ou conter motivo estrutural semelhante como a proteína consulta. Para aumentar a cobertura de homólogos Templdetecções comeu, LOMETS combina várias state-of-the-art algoritmos cobrindo diferentes metodologias de threading. Uma vez que diferentes programas de threading têm sistemas de pontuação de diferentes sensibilidades e alinhamento, a qualidade dos alinhamentos gerados threading de cada programa é avaliado por threading normalizada Z-score, que é definida como: 
onde Z-score é a pontuação em unidades de desvio padrão em relação à média estatística de todos os alinhamentos gerados pelo programa, e Z 0 é um programa específico de corte Z-score determinado com base em grande escala testes de benchmark threading 5 para diferenciar "bom 'e' ruim 'modelos. Um modelo com uma alta Z-score significa que os modelos de topo têm uma pontuação de alinhamento significativamente maior do que a maioria dos outros modelos, o que geralmente implica que o alinhamento corresponde a um bom modelo. Se a maioria dos modelos top threading tem oigh normalizada Z-scores, a precisão do modelo I-TASSER final é geralmente alta. No entanto, se a proteína é grande ea cobertura dos alinhamentos threading está confinado a uma pequena região da proteína consulta, uma alta normalizada Z-score não significa necessariamente uma precisão de modelagem de alta para o modelo full-length. Top dois alinhamentos threading de cada programa de threading são coletados e usados para a próxima etapa da montagem da estrutura.
Iterativo de simulação de montagem da estrutura: Após o procedimento de threading, seqüência de consulta é dividida em regiões threading alinhados e desalinhados. Fragmentos contínuos em alinhamento threading são excisadas a partir de modelos e usados diretamente para a montagem da estrutura, enquanto as regiões de loop desalinhado são construídas por ab initio de modelagem. O procedimento de montagem da estrutura é executada em um sistema de rede guiada pela troca réplica simulações de Monte Carlo 6. O I-TASSER campo de força inclui hidrogênio-bonding interações 10, baseadas no conhecimento estatístico termos de energia derivada de estruturas de proteínas conhecidas no APO, 11 de previsões baseadas em seqüência de contato de SVMSEQ 12, e restrições espaciais coletados de LOMETS cinco modelos de threading. O chamariz conformacional gerada em réplicas de baixa temperatura durante as simulações são agrupados por SPICKER 13 a identificar estruturas de baixo de energia livre estados. Cluster centróides dos clusters de topo são obtidos pela média das coordenadas 3D de todos os chamarizes cluster estruturais e utilizado para a geração do modelo final. A simulação e procedimento de agrupamento são repetidas duas vezes para a remoção de confrontos estérico e um maior refinamento da topologia global.
Em nível atômico construção do modelo e refinamento: O aglomerado centróides obtidos após agrupamento SPICKER são reduzidos modelos de proteínas (cada resíduo representada pelo seu α C e cadeia lateral centro de massa) e hav aplicação limitada biológica. A construção do modelo full-atômica a partir dos modelos reduzidos é feito em duas etapas. Na primeira etapa, REMO 7 é usado para construir modelos full-atômicas de C-alfa traços através da otimização das redes de H-bond. Na segunda etapa, REMO full-atômica modelos são aperfeiçoados pela FG-MD 14, que melhora o ângulo de torção backbone, comprimentos de ligação, e orientações cadeia lateral rotamer, por simulações dinâmicas moleculares, conforme a orientação do fragmentos estruturais procurado a partir do estruturas PDB por TM-align. Os modelos FG-MD refinados são usados como modelos finais para as previsões estrutura terciária pelo I-TASSER.
A qualidade dos modelos gerados são estimados com base em uma pontuação de confiança (C-score), que é definido com base no escore Z de LOMETS alinhamentos threading ea convergência das I-TASSER simulações, matematicamente formulado como: 
onde
O C-score tem uma forte correlação com a qualidade dos modelos I-TASSER. Através da combinação C-score e comprimento de proteína, a precisão do primeiro I-TASSER modelos pode ser estimada com um erro médio de 0,08 para o TM-score e 2 Å para os 15 RMSD. Em geral, os modelos com C-score> - 1.5 deverão ter uma dobra correta. Aqui, RMSD e TM-score são medidas bem conhecidas de similaridade topológica entre o modelo e estrutura nativa. TM-pontuação values na faixa [0, 1], onde a maior pontuação indica uma estrutura melhor corresponder 16,17. No entanto, para os modelos de classificação mais baixa (ou seja, 2 ª -5 ª modelos), a correlação de C-score com TM-score e RMSD é muito mais fraco (~ 0,5), e não podem ser utilizados para a estimativa confiável da qualidade do modelo absoluto.
Primeiro modelo é sempre o melhor modelo em I-TASSER simulações? A resposta a esta pergunta depende do tipo de destino. Para alvos fáceis, o primeiro modelo é geralmente o melhor modelo e sua C-score é geralmente muito maior do que o resto dos modelos. No entanto, para alvos duros, onde threading não tem bate modelo significativo, o primeiro modelo não é necessariamente o melhor modelo e I-TASSER realmente tem dificuldade em selecionar o melhor modelo e modelos. É, portanto, recomenda-se analisar todos os 5 modelos para alvos difíceis e selecioná-los com base na informação experimental e do conhecimento biológico.
Função predictions: Na última etapa final, modelos 3D gerados a partir de FG-MD são usados para prever três aspectos da função da proteína, a saber: a) Enzima (CE) números 18 e (b) Gene Ontology (GO) 19 termos e ( c) sítios de ligação para pequenas moléculas ligantes. Para todos os três aspectos, as interpretações funcionais são gerados usando cofator, que é uma nova abordagem para predizer a função da proteína com base na similaridade global e local para proteínas modelo no PDB com estrutura conhecida e funções. Primeiro, a topologia global dos modelos previstos são comparados com bibliotecas de modelos funcionais usando o programa de alinhamento estrutural TM-align 20. Em seguida, um conjunto de proteínas mais semelhante aos modelos alvo são selecionados a partir da biblioteca com base em sua similaridade estrutura global, e uma extensa pesquisa local é realizada para identificar a estrutura e seqüência semelhança perto da região sítio ativo / binding. A resultante pontuações de similaridade global e local são utilizados para classificar osproteínas modelo (homólogos funcionais) e transferir a anotação (número CE e Gene Ontology 19 termos) com base nos sucessos de pontuação superior. Da mesma forma, resíduos de ligante de ligação local eo modo de ligação do ligando são inferidos com base no alinhamento local de consulta com ligante conhecido resíduos no local obrigatório em todos os modelos de scoring top função 9.
A qualidade da função (CE e GO termo) previsão em I-TASSER é avaliada com base na pontuação homologia funcional (Fh-score), que é uma medida de similaridade global e local entre a consulta eo modelo, e é definido como: 
onde C-score é uma estimativa da qualidade do modelo previu, tal como definido na equação. (2); TM-score medidas a semelhança estrutural global entre o modelo e proteínas modelo; RMSD ali é o RMSD entre o modelo ea estrutura do modelo na região estruturalmente alinhadas da TM-align 20; Cov representa a cobertura do alinhamento estrutural (isto é, a relação entre os resíduos alinhados estruturalmente dividido pelo tamanho da consulta); ID ali é a identidade de seqüência no alinhamento TM-align. A pontuação de confiança estimado para previsões número CE também inclui um termo para avaliar corresponder sítio ativo (AcM) entre consulta e modelo dentro de uma região definida local, calculado como: 
onde N t representa o número de resíduos de modelo de presentes dentro da área local, N ali é o número de pares alinhados consulta template-resíduos, ii d é a distância C α i entre pares º de resíduos alinhados, d 0 = 3,0 Å é o corte de distância, M ii é a pontuação BLOSUM entre om par de resíduos alinhados. Em geral, a Fh-score está na faixa [0, 5] e pontuação ACM é entre [0, 2], Onde as pontuações mais altas mais confiante atribuições funcionais. AcM pontuação também é utilizado para avaliar a estrutura local e similaridade de seqüência perto dos locais de fixação do ligando, o que é referido como BS-score.
1. Apresentação de seqüência de proteína
- Visite a página web I-TASSER em http://zhanglab.ccmb.med.umich.edu/I-TASSER começar com estrutura e experiência de modelagem de função.
- Copie e cole a seqüência de aminoácidos na forma prevista ou diretamente faça o upload do seu computador clicando no botão "Procurar". I-TASSER servidor atualmente aceita seqüências com resíduos até 1500. Proteínas mais de 1500 os resíduos são geralmente multi-domínio proteínas, e são recomendados para ser dividida em domínios individuais antes de enviar para I-TASSER.
- Fornecer seu endereço de e-mail (obrigatório) e um nome para o trabalho (opcional).
- Os usuários podem, opcionalmente, especificar externo inter-residue contato / distância restrições, add-in um modelo adicional ou excluir algumas proteínas modelo durante o processo de modelagem da estrutura. Saiba mais sobre como usar essas opções na "Discussão" seção.
- Para enviar a seqüência, clique em "Run I-TASSER" botão. O navegador será direcionado para uma página de confirmação que exibe informações do usuário especificado, identificação do trabalho (Job ID) número e um link para uma página onde os resultados serão depositados após a conclusão do trabalho. Os usuários podem marcar esta link ou anotar o número de identificação do trabalho para referência futura.
2. Disponibilização dos resultados
- Verifique o status do trabalho apresentado, visitando a página fila I-TASSER em http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php . Clique na guia pesquisar e usar o número de identificação do trabalho ou a seqüência de consulta para procurar o seu trabalho apresentado.
- Após a estrutura e função moDeling é concluído, uma notificação de imagem e-mail contendo as estruturas previstas e uma web-link será enviado para você. Clique neste link ou abrir o link marcado na Etapa 1.5 para visualizar e fazer download dos resultados.
3. Estrutura secundária e previsões de acessibilidade solvente
- Verifique o FASTA seqüência de consulta formatado exibido no topo da página de resultados. Se qualquer restrição adicional / template foi especificada durante o envio seqüência, um link para a página exibindo especificado pelo usuário de informação também pode ser visto (Figura 1A).
- Examine a previsão estrutura secundária exibido como: alfa-hélice (H), beta fita (S) ou bobina (C) e pontuação de confiança da previsão (0 = baixa, 9 = alto) para cada resíduo. Olhe para a região com longos trechos de estrutura secundária regular (H ou S) previsões, para estimar o núcleo-região da proteína. Classe estrutural de proteínas também podem ser analisados com base na distribuição de elementos secundários estruturas. Alassim, as regiões longo dos elementos da bobina na proteína geralmente indicam regiões não estruturadas / desordenada.
- Ver a acessibilidade prevista solvente (Figura 1C) para verificar enterrado e solvente regiões expostas na consulta. Valores do previsto gama de acessibilidade solvente de 0 (resíduos enterrados) a 9 (resíduo expostos). Região que contém resíduos enterrados em sua maioria pode ser usada para delinear a região central da proteína, enquanto que as regiões com solvente resíduos expostos e hidrofílicos são hidratação potencial / sites funcionais.
4. Terciário previsões estrutura
- Scroll para baixo para ver as estruturas previstas terciária da proteína consulta, exibido em um applet Jmol interativa (Figura 2). À esquerda, clique no applet para alterar a aparência da estrutura apresentada, zoom em região específica, selecione os tipos de resíduos específicos no modelo previsto ou calcular distâncias inter-resíduos.
- Analisar os modelos para a presença de longas regiões não-estruturados. Estas rEGIÕES geralmente correspondem a regiões desordenadas na proteína ou indicar falta de alinhamento do modelo. Estas regiões têm geralmente precisão de modelagem baixo e remover essas regiões durante a modelagem da N & C-terminal região vai melhorar a precisão de modelagem.
- Baixe o PDB arquivos formatados estrutura do modelo clicando no "Download Model" links. Você pode abrir esses arquivos em qualquer software de visualização molecular (por exemplo PyMOL, Rasmol etc) para análise mais aprofundada das características estruturais.
- Analisar a pontuação de confiança (C-score) da modelagem de estrutura para estimar a qualidade das estruturas previstas. C-score (Eq. 2) Os valores são tipicamente no intervalo [-5, 2], em que uma pontuação mais elevada reflete um modelo de melhor qualidade. A estimativa TM-score e RMSD do primeiro modelo é mostrado como "precisão estimada de Modelo 1". Para proteínas prazo, recomenda-se avaliar a qualidade do modelo baseado em TM-score, como TM-score é mais sensível às mudanças topológicas que RMSD. < li> Clique em "mais sobre o C-score" link para analisar C-score, tamanho do cluster e densidade conjunto de todos os modelos. Estima-TM-score e RMSD são apresentados apenas para o modelo I-TASSER primeiro, porque C-score de menor modelos classificados não está fortemente correlacionada com a TM-score ou RMSD. Qualidade dos modelos de classificação mais baixa pode ser parcialmente avaliada com base em sua densidade de cluster e tamanho do cluster em relação ao primeiro modelo, em que modelos de maior cluster e de maior densidade são em média mais próxima da estrutura nativa.
- C-score baixo previsões geralmente indicam uma previsão baixa precisão. Na maioria dos casos, a proteína consulta carece de um bom modelo na biblioteca e tem um tamanho fora do alcance da ab initio de modelagem (ie> 120 resíduos). Nestes casos, os usuários podem buscar adicionais restrições espaciais e usá-los para melhorar a modelagem I-TASSER (ver secção Discussão). É também encorajados a submeter as seqüências para o nosso servidor QUARK (QUARK / "> http://zhanglab.ccmb.med.umich.edu/QUARK/) para uma modelagem pura ab initio se o tamanho da proteína é inferior a 200 resíduos.
5. LOMETS alinhamento template-alvo
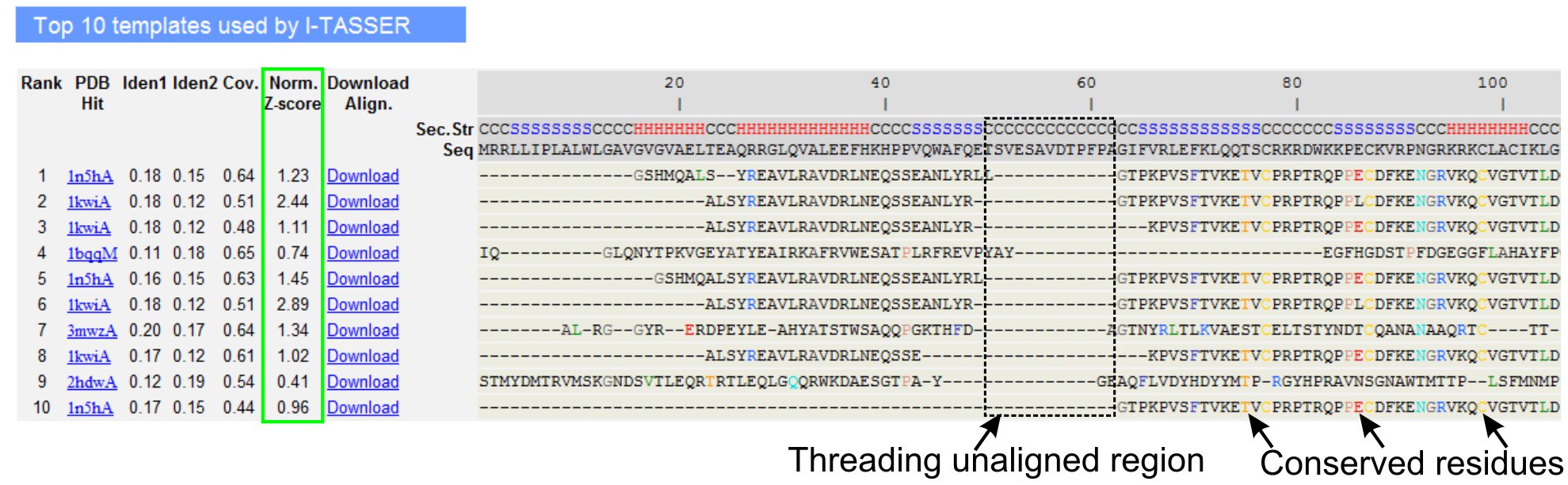
- Desloque-se para analisar os dez modelos de threading da proteína de consulta, conforme identificado por programas LOMETS threading (Figura 3). Ver o normalizado Z-score (Eq. 1), mostrado em 'Norm. Coluna Z-score ", para analisar a qualidade de alinhamentos threading. Alinhamentos com um normalizada Z-score> 1 reflete um alinhamento confiante e provavelmente terá a mesma vezes como a proteína consulta.
- Analisar a seqüência de identidade na região threading-alinhados ("Iden. 1 'coluna) e para toda a cadeia (coluna" Iden. 2') para avaliar a homologia entre a consulta e as proteínas modelo. Identidade de seqüência de alta é um indicador de parentesco evolutivo entre a consulta e proteínas modelo.
- Ver os resíduos threading alinhada mostrada na cor para identificar visualmente os contrasresíduos erved / motivos da consulta e as proteínas modelo. A identidade maior seqüência em threading alinhado região, em comparação com todo o alinhamento da cadeia também indica presença de motivo estrutural conservada / domínios na consulta.
- Avaliar a cobertura do alinhamento threading visualizando o 'Cov. coluna e inspecionar o alinhamento. Se a cobertura dos alinhamentos superior é baixo e confinado a apenas uma pequena região da proteína consulta ou ausente por um longo segmento de seqüência de consulta, em seguida, a proteína consulta geralmente contém mais de um domínio e recomenda-se dividir a seqüência e modelo os domínios individualmente (Figura 3).
- Baixe o PDB formatado arquivos de seqüência-estrutura de alinhamento, clicando no "Download Align" links. Esses arquivos de alinhamento pode ser aberto em qualquer programa de visualização molecular listados na seção Materiais, e também pode ser usada para adicionar restrições adicionais durante a modelagem de estrutura (Passo 1.4).
6.Análogos estruturais no AO
- Visualizar a tabela seguinte (Figura 4) da página de resultados para determinar os dez melhores análogos estruturais do modelo previsto pela primeira vez, conforme identificado pelo programa de alinhamento estrutural TM-align 20. A TM-score> 0,5 indica que o analógico detectados e modelo têm uma topologia semelhantes e podem ser usados para determinar a família de classe / proteína estrutural da proteína consulta 16, enquanto aqueles com TM-score <0,3 significa uma semelhança estrutura aleatória.
- Analisar a identidade de seqüência e RMSD na região estruturalmente alinhados mostrado em 'IDEN a' e 'RMSD um "colunas para avaliar a conservação de motivos espaciais no modelo analógico eo estrutural. Inspecionar visualmente os pares de resíduos coloridos e alinhados no alinhamento para identificar estes resíduos estruturalmente conservadas e motivos.
- Clique no código PDB mostrado na coluna 'Hit PDB "para visitar o site RCSB e aprender mais sobre a sua classificação estrutural (SCOP, E Pfam CATH) e informação funcional (número CE, associado GO termos e ligante ligado).
7. Previsão função usando cofator
- Desloque-se na página de resultado para analisar interpretações funcional para a proteína consulta. Funções da proteína são enumerados em três mesas de contexto, mostrando: Enzima Comissão (CE) números, Gene Ontology (GO) termos, e ligando sítios de ligação.
- Ver o "TM-score ',' RMSD a ',' IDEN a 'e' Cov. colunas em cada tabela para analisar parâmetros de similaridade estrutura global ea conservação de padrões espaciais entre modelo e identificados homólogos funcionais (templates).
8. Enzima previsão número Comissão
- Ver o top cinco homólogos enzima potencial de proteína consulta mostrada na "Previsão de números CE" tabela (Figura 5). O nível de confiança de CE previsão número usando esses modelos é mostrado na coluna 'CE-Score'. Baseado em benchmarking análise 23, a similaridade funcional (3 primeiros dígitos do número CE) entre a consulta ea proteína modelo pode ser interpretado de forma confiável usando CE-score> 1.1.
- Procure por consenso de função (número CE) entre os modelos, que têm a dobra similar (ie TM-score> 0,5) como a proteína consulta. Se vários modelos têm número CE mesma e CE-score> 1,1, o nível de confiança de previsão é muito alto. No entanto, se a CE-Score é alto, mas há uma falta de consenso entre os hits identificados, em seguida, a previsão torna-se menos confiável e os usuários são aconselhados a consultar as previsões GO prazo.
- Clique no link fornecido sobre os números da CE para visitar o banco de dados de enzimas ExPASy e analisar a função, incluindo a reação catalisada, co-fator requisitos e a via metabólica, da proteína modelo em detalhes.
9. Gene Ontology (GO) previsões prazo
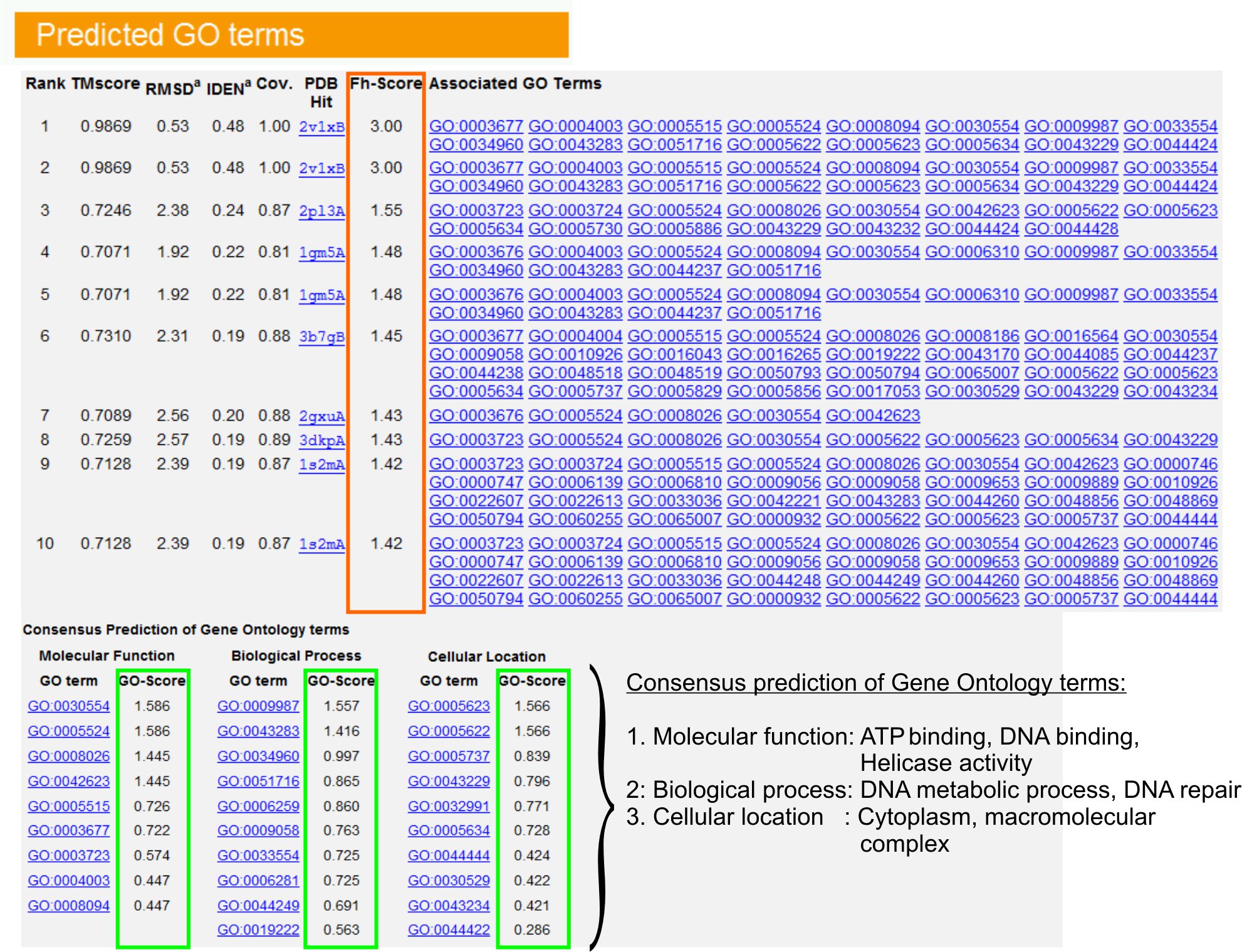
- Ver o "Previsão GO termos" tabela (Fig.ure 6) para identificar dez principais homólogos da proteína de consulta na biblioteca da APO, anotado com Gene Ontology (GO) termos. Cada proteína é geralmente associada a termos GO múltiplas, descrevendo suas funções molecular (MF), processos biológicos (BP) e componente celular (CC). Clique em cada termo para visitar o site Amigo e analisar a sua definição e da linhagem.
- Analisar a coluna Fh-score (pontuação homologia funcional) para acessar a semelhança funcional entre a consulta e proteínas modelo e estimar o nível de confiança de transferência de anotação funcional destas proteínas. Em nosso estudo de benchmarking 23, 50% dos termos GO nativa poderia ser corretamente identificadas a partir do modelo identificado pela primeira vez usando um corte Fh-score de 0,8, com uma precisão geral de 56%.
- Ver o "Consenso de previsão de termos GO" mesa para analisar a concordância de função entre os modelos. Essas funções comuns são usados para prever os termos GO (MF, BP e CC) da consultaproteína e avaliar o nível de confiança (GO-score) das previsões GO prazo. Com base no teste de benchmarking 23, o melhor falsos positivos e falsos negativos são as taxas obtidas para previsões com GO-score de corte = 0,5, com a diminuição da cobertura de previsão em níveis mais profundos ontologia.
10. Proteína ligante de ligação previsões do site
- Desça até a parte inferior da página para ver top ten ligante previsões sítio de ligação para a proteína consulta. Previu sítios de ligação são classificados com base no número de conformações ligante previu que partes do bolso de ligação comum. O melhor site de ligação é identificada já exibido no applet Jmol. Clique nos botões de rádio para analisar outras previsões e visualizar os resíduos interagindo ligante.
- Analisar a coluna BS-score para avaliar similaridade local entre o modelo eo sítio de ligação modelo. Com base na referência 9, BS-score> 1,1 indica alta seqüência e estrutura similarity próximo ao local previsto obrigatório em todos os modelos e conhecido sítio de ligação no modelo.
- Baixe o arquivo de APO estrutura formatada do complexo, clicando no link "Download". Os usuários podem abrir esses arquivos em qualquer programa de visualização molecular e interativamente visualizar o site previu vinculativo e ligante-proteína interações em seu computador local.
11. Resultados representativos

Figura 1 Um trecho de I-TASSER página de resultado mostrando (A) FASTA formatado seqüência de consulta;. (B) previu estrutura secundária e dezenas de confiança associados, e (C) prevê a acessibilidade dos resíduos de solventes. Região do núcleo local analisado e hidratação potencial na consulta são destacadas em ciano e retângulos vermelhos, respectivamente.

Figura 2.

Figura 3. Um exemplo de I-TASSER página de resultados mostra dez identificados modelos de segmentação e alinhamentos por LOMETS 5 programas de threading. A qualidade dos alinhamentos threading é avaliado com base normalizada Z-score (destacado em verde), onde um valor> 1 reflete um alinhamento confiante. Resíduos alinhados no modelo que são idênticos aos resíduos de consulta correspondentes estão em destaque na cor para indicar presença de resíduo conservado / motivo, enquanto a falta de alinhamento na maioria dos modelos top indica a presença de vários domínios na proteína de consulta e os resíduos não alinhado correspondem a regiões linker domínio. Clique aqui para ver a versão completa do tamanho da figura 3.

Figura 4. Um exemplo de página de resultados mostra dez identificados análogos estruturais e alinhamentos estruturais, identificadas por TM-align 20 programa de alinhamento estrutural. O ranking dos análogos mostrado na baseia-se no TM-score (destaque em azul) do alinhamento estrutural. A TM-score> 0,5 indica que as duas estruturas comparados têm uma topologia similar, enquanto a TM-score <0,3 significa uma semelhança entre duas estruturas aleatórias. Pares de resíduos alinhados são estruturalmente em destaque na cor com base em sua propriedade amino-ácidos, enquanto as regiões desalinhado são indicados por "-".ove.com/files/ftp_upload/3259/3259fig4large.jpg "> Clique aqui para ver a versão completa do tamanho da figura 4.

Figura 5. Um exemplo de I-TASSER página de resultado mostrando homólogos enzima identificada da proteína consulta na biblioteca do PDB. O nível de confiança de CE previsão número é analisada com base na EC-score (destacado em verde), onde CE-score> 1,1 indica similaridade funcional (mesmo 3 primeiros dígitos do número CE), entre consulta e proteína modelo.

Figura 6. Um exemplo de I-TASSER página de resultado mostrando GO previsões prazo para a proteína consulta. Homólogos funcional para a proteína de consulta na biblioteca de modelos Gene Ontology são classificados com base em suas Fh-score (no retângulo laranja). Comum características funcionais a partir desses top hits pontuação são derivados de Gener comeu a final previsões GO prazo para a proteína consulta. A qualidade do previsto GO termos é estimado com base na GO-score, onde um GO-score> 0,5 indica uma previsão confiável. (Mostrado em verde) Clique aqui para ver a versão completa do tamanho da figura 6.

Figura 7. Um exemplo de I-TASSER página de resultados mostra dez proteína ligante previsões sítio de ligação com o cofator 9 algoritmo. O ranking dos sites de ligação é previsto com base no número de conformações ligante previu que partes do bolso de ligação comum na consulta. BS-score (destaque em vermelho) é uma medida da seqüência de local e estrutura de similaridade entre o previsto eo sítio de ligação do modelo, e é útil para analisar a conservação dos bolsos sítio de ligação.
les/ftp_upload/3259/3259fig8.jpg "/>
Figura 8. Um exemplo de arquivos de contenção externa usada para especificar a resíduo de bagaço de contato / restrições à distância.

Figura 9. Exemplo de arquivos de contenção utilizada para especificar um modelo de proteína para o servidor I-TASSER. O usuário pode especificar o alinhamento template-consulta ou em (A) de formato FASTA, ou (B) formato 3D.

Figura 10. Um arquivo de exemplo usado para a exclusão de modelo durante o procedimento de modelagem I-TASSER estrutura. A primeira coluna contém o ID PDB das proteínas modelo a ser excluídos. A segunda coluna é usada para especificar o corte seqüência identidade que será usada para outros modelos similares na biblioteca de modelo.
Discussion
O protocolo apresentado acima é uma diretriz geral para a estrutura e função de modelagem usando o servidor I-TASSER. Embora, este procedimento automatizado funciona muito bem para a maioria das proteínas, as intervenções humanas muitas vezes ajudam a melhorar significativamente a precisão de modelagem, especialmente para as proteínas que faltam modelos de perto na biblioteca PDB. Os usuários podem intervir durante a modelagem I-TASSER das seguintes formas: (a) divisão de multi-domínio proteínas; (b) fornecimento de restrições externas para melhorar a estrutura de montagem, e (c) remover modelos durante a modelagem.
Proteína multi-domínio divisão:
Muitas seqüências de proteínas longo freqüentemente contêm vários domínios amarrados por regiões linker flexível, o que torna difícil a sua elucidação da estrutura através de técnicas experimentais e computacionais. No entanto, como os domínios são dobráveis independentemente entidades e pode executar a função molecular distinta, édesejável para dividir longas multi-domínio proteínas e modelo de cada domínio separadamente. Domínios de modelagem, individualmente, não só acelerar o processo de previsão, mas também aumenta a qualidade da consulta de modelo de alinhamento, resultando em uma estrutura mais confiável e previsões função.
Limites de domínio em seqüências de proteínas pode ser predita usando livremente disponível externo programas online como o NCBI CDD 24, 25 ou Pfam InterProScan 26. Além disso, se LOMETS alinhamentos threading estão disponíveis para consulta a proteína, os limites de domínio podem ser localizados por identificar visualmente longos trechos de resíduos não alinhado nos modelos top threading (veja o Passo 5.4). Estas regiões não alinhado em sua maioria correspondem a regiões linker domínio. Se multi-domínio modelos já estão disponíveis na biblioteca de modelos PDB com todos os domínios de consulta alinhados, então a proteína consulta pode ser modelado como comprimento total.
Fornecer restrições externas
A. Especifique contato / distância restrições
Experimentalmente caracterizadas inter-resíduos contatos / distâncias, por exemplo, de NMR oucross-linking experimentos, pode ser especificado pelo upload de um arquivo moderação. Um arquivo de exemplo é mostrado na Figura 8, onde Coluna 1 especifica o tipo de restrição, ou seja, "DIST" ou "CONTATO". De contenção à distância (DIST), colunas 2 e 4 contêm resíduos de posições (i, j), colunas 3 e 5 contêm o átomo tipos de resíduos e na coluna 6 especifica a distância entre os dois átomos especificado. No caso dos apoios de contato (CONTACT), colunas 2 e 3 contêm as posições (i, j) de resíduos que deve estar em contato. A distância entre o centro cadeias laterais desses pares resíduos entrar em contato é decidido com base em distâncias observadas em estruturas conhecidas no PDB. I-TASSER vai tentar tirar esses pares de átomos perto da distância especificada durante as simulações refinamento estrutura.
B. Especifique um modelo de estrutura de proteínas
Programas LOMETS threading usar uma biblioteca representativa PDB para encontrar dobras plausível para a prot consultaein. Embora o uso de uma biblioteca de estrutura representativa ajuda a reduzir o tempo necessário para calcular a alinhamentos de seqüência-estrutura, é possível que uma proteína bom modelo é perdida na biblioteca ou o modelo não pode ter sido identificado por LOMETS programas threading, embora seja presentes na biblioteca. Nestes casos, o usuário deve especificar a estrutura da proteína desejada como modelo.
Para especificar a estrutura da proteína como um modelo adicional, os usuários podem fazer upload de um arquivo de estrutura PDB formatado ou especificar o ID PDB de uma estrutura de proteína depositada na biblioteca PDB. O I-TASSER irá gerar o alinhamento template-consulta usando o programa Muster 23 e irá recolher as restrições espaciais de tanto o usuário especificado modelo e modelos LOMETS para orientar a simulação de montagem da estrutura. Porque a precisão das restrições LOMETS é diferente para diferentes alvos, o peso das restrições LOMETS é mais forte em fácil (homóloga) targets do que em disco rígido (não-homólogos) metas, que têm sido sistematicamente ligado em nosso treinamento benchmark.
Os usuários também podem especificar suas próprias consulta template-alinhamentos. O servidor aceita o alinhamento em dois formatos: o formato FASTA (Figura 9A) eo formato 3D (Figura 9B). O formato FASTA é padrão e descrito em http://zhanglab. ccmb.med.umich.edu / FASTA / . O formato 3D é semelhante ao formato padrão PDB ( http://www.wwpdb.org/documentation/format32/sect9.html ), mas duas colunas adicionais derivados dos modelos são adicionados aos registros ATOM (ver Figura 9B):
Colunas 1-30: Atom (C-alfa apenas) e resíduo de nomes para a seqüência de consulta.
Colunas 31-54: Coordenadas do C-alfa átomos da consulta copiado dos átomos correspondentes no modelo.
Colunas 55-59: Número de resíduos correspondente no modelo baseado em alinhamento
Colunas 60-64: Nome do resíduo correspondente no modelo
Excluir proteínas modelos
Proteínas são moléculas flexíveis e podem adotar vários estados conformacionais de mudar sua atividade biológica. Por exemplo, as estruturas de proteínas quinases e proteínas da membrana muitos foram resolvidos na conformação ativos e inativos. Também presença ou ausência de ligante ligado pode causar grandes movimentos estruturais. Enquanto todos os estados conformacionais do modelo são iguais para os programas de threading, é desejável que o modelo de consulta utilizando templates em apenas um estado particular. Uma nova opção no servidor permite ao usuário excluir proteínas modelo durante a modelagem da estrutura. Este recurso também permite ao usuário escolher o nível de homologia de modelos a serem utilizados para a modelagem. Os usuários podem excluir template proteínas from a biblioteca I-TASSER por:
A. A especificação de um corte de identidade de seqüência
Os usuários podem usar esta opção para excluir proteínas homólogas da biblioteca de modelos I-TASSER. O nível de homologia é definida com base no corte de identidade de seqüência, ou seja, o número de resíduos idênticos entre a consulta ea proteína modelo, dividido pelo comprimento da seqüência da seqüência de consulta. Por exemplo, se o usuário digita em "70%" na forma prevista, todas as proteínas modelos que têm uma identidade de seqüência> 70% para a proteína I-consulta será excluído da biblioteca de modelos I-TASSER.
B. Exclude proteínas modelo específico
Proteínas modelo específico pode ser excluído da biblioteca de modelos I-TASSER fazendo o upload de uma lista contendo IDs PDB das estruturas a serem excluídos. Um arquivo de exemplo é mostrado na Figura 10. Como a mesma proteína pode existir como várias entradas na biblioteca PDB, I-TASSER siRVer, por padrão, excluir os modelos especificados (em coluna1), bem como todos os outros modelos da biblioteca que tem uma identidade> 90% para os modelos especificados. Os usuários também podem especificar uma identidade diferente de corte, por exemplo, 70%, onde todos os modelos com a identidade> 70% às proteínas modelo especificado serão excluídos.
Disclosures
Não há conflitos de interesse declarados.
Acknowledgments
O projeto é apoiado em parte pela Alfred P. Sloan Foundation, NSF Prémio Carreira (DBI 1027394), eo Instituto Nacional de Ciências Médicas Gerais (GM083107, GM084222).
Materials
| Name | Company | Catalog Number | Comments |
| Material Name | Type | Company | Catalogue Number |
| FASTA formatted amino acid sequence of the protein to be modeled (see, http://www.ncbi.nlm.nih.gov/BLAST/fasta.shtml). | |||
| A personal computer with access to the internet and a web browser. | |||
| Molecular visualizing software, e.g. RASMOL or PYMOL, for analyzing the predicted tertiary structure and functional sites. |
References
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H. CD-Search: protein domain annotations on the fly. Nucleic. Acids. Res. 32, W327-W331 (2004).
- Finn, R. D. The Pfam protein families database. Nucleic. Acids. Res. 38, D211-D222 (2010).
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).

{kind=link}
{kind=link}