

Summary
Руководящие принципы для компьютерных структурные и функциональные характеристики белка использованием I-TASSER трубопровода описывается. Начиная с запросом последовательности белка, 3D-модели создаются с использованием нескольких потоков выравнивания и итерационные структурного моделирования сборки. Функциональные выводы являются после этого сделать на основе матчей белков с известной структурой и функциями.
Abstract
Секвенирование генома проектов зашифрованные миллионы белковой последовательности, которая требует знания их структуры и функции для улучшения понимания их биологическая роль. Хотя экспериментальные методы могут дать подробную информацию для небольшой части этих белков, численное моделирование необходимо для большинства белковых молекул, которые экспериментально неохарактеризованных. I-TASSER сервер он-лайн верстак с высоким разрешением моделирования структуры белка и функции. Учитывая белковой последовательности, типичный выход из I-TASSER сервер включает вторичные предсказания структуры, предсказал растворителя доступности каждого из остатков, гомологичный шаблон белки обнаружены потоки и структуру трассы, до пяти полнометражных третичной структурной модели и структуры основе функциональной аннотации к классификации ферментов, Джин Онтология сроки и белок-лиганд сайтов. Все предсказания с меткой показатель достоверности которыхрассказывает, как точные предсказания, не зная экспериментальных данных. Чтобы облегчить специальные запросы конечных пользователей, сервер предоставляет каналы, чтобы принять заданные пользователем между остаток расстояния и контактные карты для интерактивного изменения I-TASSER моделирования; он также позволяет пользователям задавать любые белки, как шаблон, или исключить любой шаблон белков в процессе моделирования структуры сборки. Структурная информация может быть собрана пользователей на основе экспериментальных доказательств или биологического понимания с целью улучшения качества I-TASSER прогнозы. Сервер был оценен как лучших программ для белковую структуру и функции прогнозы в последнее всей общины экспериментах CASP. Есть в настоящее время> 20 000 зарегистрированных ученых из более чем 100 стран мира, которые используют онлайн-I-TASSER сервера.
Protocol
Метод обзор
После последовательность-к-структуры на функцию парадигмы, I-TASSER процедуру 1-4 для структуры и функции моделирования включает в себя четыре последовательных стадии: (а) шаблон идентификации LOMETS 5, (б) фрагмент структуры сборки на реплику- обмен Монте-Карло 6; (с) уточнение структуры атомного уровня, используя REMO 7 и FG-MD 8, и (г) структуры основе функции интерпретации использованием кофактора 9.
Шаблон идентификации: для запроса последовательность представленных пользователем, последовательность первых резьбовые через представителя PDB библиотеки структуру локально установленные LOMETS мета-Threading сервера. Threading представляет собой последовательность структуры выравнивание процедура, используемая для определения шаблонов белков, которые могут иметь подобные структуры или содержат аналогичные структурным мотивом, как запрос белка. Для увеличения охвата гомологичных Темплели обнаружений, LOMETS объединяет множество состоянии современных алгоритмов, охватывающих различные резьбы методологий. С другой потоковой программы имеют различные системы скоринга и выравнивания чувствительности, качество создаваемой потоками ряды с каждым резьбы программы оценивается нормированного Z-оценка, которая определяется как: 
где Z-оценка является оценка в единицах стандартного отклонения относительно среднестатистической цены всех выравнивания генерируются программой, и Z 0 является конкретным программам Z-счет отсечки определяется на основе крупномасштабных потоков тестах 5 отличить «хороший »и« плохими »шаблоны. Шаблона с высоким Z-счет означает, что топ шаблоны выравнивания оценка значительно выше, чем большинство других шаблонов, который обычно означает, что выравнивание соответствует хорошей моделью. Если большая часть верхней резьбы шаблоны приветGH нормированного Z-оценки, точность окончательного I-TASSER модели, как правило, высока. Однако, если белок крупных и охват резьбы выравнивания сводится к небольшой области запрос белка, высокое нормированного Z-оценка не обязательно означает высокую точность моделирования для полнометражного модели. Top две резьбы ряды с каждым резьбы программы собираются и используются для следующего шага структуры сборки.
Итерационные моделирования структуры сборки: После резьбы процедуры запроса последовательность разделяется на потоки выровнены и выровненным регионах. Непрерывная фрагментов в потоки выравнивания вырезали из шаблонов и использовать непосредственно для структуры сборки, в то время выровненным регионах цикла построены на основе моделирования первоначально. Процедура структуры Сборка осуществляется на решетке система руководствуется реплики обмена Монте-Карло 6. I-TASSER силовое поле включает в себя водород-бонахождении взаимодействий 10, основанной на знаниях статистической точки зрения энергии, получаемой из известных белковых структур в PDB-11, последовательный основе контактов с прогнозами SVMSEQ 12, и пространственных ограничений собраны из LOMETS 5 резьбы шаблонов. Конформационные приманки генерируется в низкотемпературной реплики во время моделирования сгруппированы по Spicker 13 для определения структуры низкой свободной энергией. Кластер центроиды верхней кластеров получены путем усреднения 3D-координаты всех кластерных структурных ложных целей и используются для окончательного поколения модели. Моделирования и кластеризации процедуру повторяют еще два раза для удаления стерических столкновения и дальнейшего совершенствования глобальной топологии.
Атомная уровне построения модели и уточнение: кластер центроиды, полученные после Spicker кластеризации сводятся белка модели (каждый остаток лице C α и боковой цепи центра масс) и чпр. ограниченной биологических приложений. Строительство полной атомной модели от моделей снижается осуществляется в два этапа. На первом этапе, REMO 7 используется для построения полной атомных моделей от C-альфа следы за счет оптимизации Н-связи сетей. На втором этапе, REMO полный атомных моделей дополнительно уточнены FG-MD 14, который улучшает позвоночник торсионных углов, длины связей и боковой цепи ориентации ротамер, по молекулярно-динамического моделирования, а руководствуются структурные фрагменты из искали PDB структур ТМ-выровнять. FG-MD изысканные модели используются в качестве окончательной модели для третичной структуры прогнозы по I-TASSER.
Качество создаваемой модели оцениваются на основе показатель достоверности (C-счет), который определяется на основе Z-счетом LOMETS резьбы выравнивания и сближения I-TASSER моделирования, математически сформулировать так: 
где
C-оценка имеет сильную корреляцию с качеством I-TASSER моделей. Объединив C-счет и белка длины, точность первых I-TASSER моделей можно оценить с Средняя ошибка 0,08 для ТМ-оценка и 2 Å для СКО 15. В общем, модели С-оценка> - 1.5 должны иметь правильную раза. Здесь, СКО и ТМ-оценка оба хорошо известные меры топологического сходства между моделью и нативной структуры. ТМ-оценка ценныеэс-диапазоне в интервале [0, 1], где более высокий балл указывает лучшей структуре соответствуют 16,17. Однако для более низким рейтингом модели (т.е. 2-й модели -5 е место), соотношение C-счет с ТМ-счет и СКО гораздо слабее (~ 0,5), и не могут быть использованы для надежной оценки абсолютного качества модели.
Это первая модель всегда лучшая модель в I-TASSER симуляции? Ответ на этот вопрос зависит от типа цели. Для легкой мишенью, первой моделью, как правило, лучшая модель и ее C-оценка, как правило, намного выше, чем остальные модели. Тем не менее, для твердых мишенях, где потоки не имеют значительные хиты шаблон, первая модель не обязательно лучшая модель, и я-TASSER на самом деле испытывает трудности в выборе лучших шаблонов и моделей. В этой связи рекомендуется, чтобы проанализировать все 5 моделей для жестких задач и выберите их на основе экспериментальных данных и биологических знаний.
Функция PREDictions: На последнем этапе, окончательный 3D-моделей, созданных с FG-MD используются для прогнозирования три аспекта функции белка, а именно: а) фермент комиссия (ЕК) числа 18 и (б) Гена Онтология (GO) 19 условий и ( в) сайты связывания для малых лигандов молекулы. Для всех трех аспектов, функциональные интерпретации генерируются с использованием кофактором, который является новым подходом, чтобы предсказать функцию белка, основанный на глобальном и локальном сходство с шаблоном белков в PDB с известной структурой и функциями. Во-первых, глобальной топологии предсказал модели сравнивается с функциональной библиотеки шаблонов с помощью программы структурных выравнивание ТМ-20 выровнять. Далее, набор белков наиболее близок к целевой модели выбираются из библиотеки на основе сходства их глобальные структуры, а также обширный локальный поиск производится для определения структуры и последовательности сходство рядом активных / сайт связывания региона. Результирующее глобальных и локальных оценки сходства используются для ранжированияШаблон белков (функциональных гомологов) и передача аннотации (ЕК номера и Джин Онтология 19 терминов) на основе хитов скоринга. Кроме того, связывание лигандов остатков сайте и связывание лигандов режиме выводятся на основе местных выравнивание запрос с известными лиганд остатков сайта в топ забил шаблоны функций 9.
Качество функции (ЕС и GO термин) предсказания в I-TASSER оценивается на основе функциональных оценка гомологии (Fh-счет), которая является мерой глобального и локального сходства между запросом и шаблон, и определяется как: 
где С-оценка является оценкой качества предсказал модель, как это определено в формуле. (2); ТМ-оценка мер глобальные структурные сходства модели и шаблоны белков; СКО Али СКО между моделью и шаблон структуры в соответствие структурно региона от ТМ-20 выровнять; Cov представляет охвата структурных выравнивания (т.е. отношение структурно соответствие остатков, деленная на запрос длины); ID Али идентичности последовательности в ТМ-выравнивание выравнивание. Оценкам оценка доверия к прогнозам ЕС количество также включает в себя термин для оценки соответствия активных сайтов (ACM) между запросом и шаблонов в пределах определенной локальной области, рассчитывается следующим образом: 
где N т представляет собой количество шаблонов присутствующих отложений в пределах района, N Али числа выровнены запросов шаблон пары остатков, D II является расстояние между C α я ю пару выровнены остатков, D 0 = 3,0 А Расстояние среза, M II является BLOSUM счетов между г-й паре выравниваются остатков. В общем, FH-оценка находится в диапазоне [0, 5] и ACM оценка составляет от [0, 2], Где высокие деления указывают на более уверенно функциональных обязанностей. ACM оценка также используется для оценки локальной структуры и последовательности сходство возле лиганд-связывающие сайты, которые называют BS-счет.
1. Представление белковой последовательности
- Посетить I-TASSER веб-страницы на http://zhanglab.ccmb.med.umich.edu/I-TASSER начать со структурой и функцией моделирования эксперимента.
- Скопируйте и вставьте последовательность аминокислот в предоставленную форму или непосредственно загрузить его с вашего компьютера, нажав кнопку "Обзор". I-TASSER сервер в настоящее время принимает последовательностей с до 1500 остатков. Белки больше чем в 1500 остатков, как правило, несколькими доменами белков, и рекомендуются должен быть разбит на отдельные домены перед отправкой к I-TASSER.
- Укажите свой адрес электронной почты (обязательно) и название работы (по желанию).
- Пользователи могут дополнительно указать внешний среди разрешениемidue контакт / расстояние ограничения, надстройки дополнительных шаблонов или исключить какие-либо шаблон белков в процессе моделирования структуры. Подробнее об использовании этих опций в "Обсуждение" разделе.
- Чтобы представить последовательность, щелкните на "Run I-TASSER" кнопку. Браузер будет направлена на подтверждение страницы, где отображаются указанный пользователем информации, работа идентификации (Иов ID) номер и ссылку на веб-страницу, где результаты будут сданы на хранение после завершения работы. Пользователи могут отмечать эту ссылку или запишите номер задания идентификации для дальнейшего использования.
2. Наличие результатов
- Проверьте состояние вашего представленные работы, посетив I-TASSER очереди странице http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php . Нажмите на вкладку поиска и использовать номер ID Вакансии или последовательность запросов для поиска на представленные работы.
- После структуры и функции движенияdeling закончено, уведомление по электронной почте, содержащее образ предсказал структур и веб-ссылка будет отправлен к вам. Нажмите на ссылку или открыть ссылку в закладки Шаг 1,5 просматривать и загружать результаты.
3. Вторичная структура и растворителя прогнозы доступности
- Проверьте FASTA отформатирован последовательность запросов отображаются в верхней части страницы результатов. Если какие-либо дополнительные ограничения / шаблон был указан во время последовательности представления, ссылающиеся на веб-странице отображения указанных пользователем информацию также можно рассматривать (рис. 1А).
- Изучение вторичных предсказания структуры отображаются в виде: альфа-спирали (H), бета-нить (S) или катушки (C) и показатель достоверности прогнозирования (0 = низкий, 9 = высокий) для каждого остатка. Ищите области с длинными отрезками регулярной вторичной структуры (H или S) прогнозы, оценивать ядра региона в белке. Структурные класс белков также могут быть проанализированы на основе распределения элементов вторичной структуры. Alтаким образом, длинные регионов катушки элементы в белке обычно указывают неструктурированных / разупорядоченных областей.
- Посмотреть предсказал растворителя доступности (рис. 1С) для определения похоронен и растворителей подвергается регионов в запросе. Значения прогнозируемых растворителя диапазоне доступности от 0 (похоронен остаток) до 9 (контакт остатка). Регион содержит в основном похоронены остатки могут использоваться для обозначения основных региона в белке, а регионы с растворителем разоблачены и гидрофильные остатки являются потенциальными гидратации / функциональных сайтов.
4. Третичная структура прогнозы
- Прокрутите вниз, чтобы посмотреть предсказал третичной структуры белка запроса, отображается в интерактивном апплет Jmol (рис. 2). Щелкните левой кнопкой мыши на апплет для изменения внешнего вида отображаемых структуру, увеличить в конкретном регионе, выбрать конкретные типы остатков в предсказал модель или рассчитать между остаток расстояния.
- Анализ моделей на наличие длинных неструктурированных регионах. Эти тegions обычно соответствуют разупорядоченных областей в белок или указывают на отсутствие шаблона выравнивания. Эти регионы обычно имеют низкую точность моделирования и удаление этих регионах при моделировании из N и С-концом регионе улучшится моделирование точности.
- Скачать PDB файлы в формате структуры модели, нажав на кнопку "Загрузить модель" ссылки. Вы можете открыть эти файлы в любой молекулярной визуализации программного обеспечения (например, PyMOL, Rasmol и т.д.) для дальнейшего анализа структурных особенностей.
- Анализ показатель достоверности (C-оценка) структуры моделирования для оценки качества предсказал структур. C-счет (уравнение 2) значения, как правило, в диапазоне [-5, 2], в которой высокий балл отражает модель более высокого качества. Оценкам TM-счет и СКО первой модели показан как "Расчетная точность Модель 1". Долгие белков, рекомендуется для оценки качества модели на основе ТМ-счет, как ТМ-оценка является более чувствительной к топологическим изменениям, чем СКО. < LI> Нажмите на кнопку "Подробнее о C-оценка", чтобы проанализировать C-счет, размер кластера и кластера плотность всех моделей. Расчетное ТМ-счет и СКО представлены лишь за первый I-TASSER модели, так как С-счетом с более низким рейтингом моделей не сильно коррелирует с ТМ-счет или СКО. Качество более низким рейтингом модели могут быть частично оценивается по их плотности кластера и размер кластера по сравнению с первой моделью, в которой модели из больших кластеров и более высокой плотностью в среднем ближе к нативной структуры.
- Низкая оценка C-прогнозы обычно указывают низкие точность прогноза. В большинстве подобных случаев, запрос белка не хватает хороших шаблонов в библиотеке и имеет размер за пределы неэмпирических моделирования (т.е.> 120 остатков). В этих случаях пользователи могут искать дополнительные пространственные ограничения и использовать их для улучшения I-TASSER моделирования (см. Обсуждение раздела). Кроме того, предлагается представить последовательности на наш сервер QUARK (QUARK / "> http://zhanglab.ccmb.med.umich.edu/QUARK/) для чистого моделирования неэмпирического если белок размером менее 200 остатков.
5. LOMETS целевой шаблон выравнивания
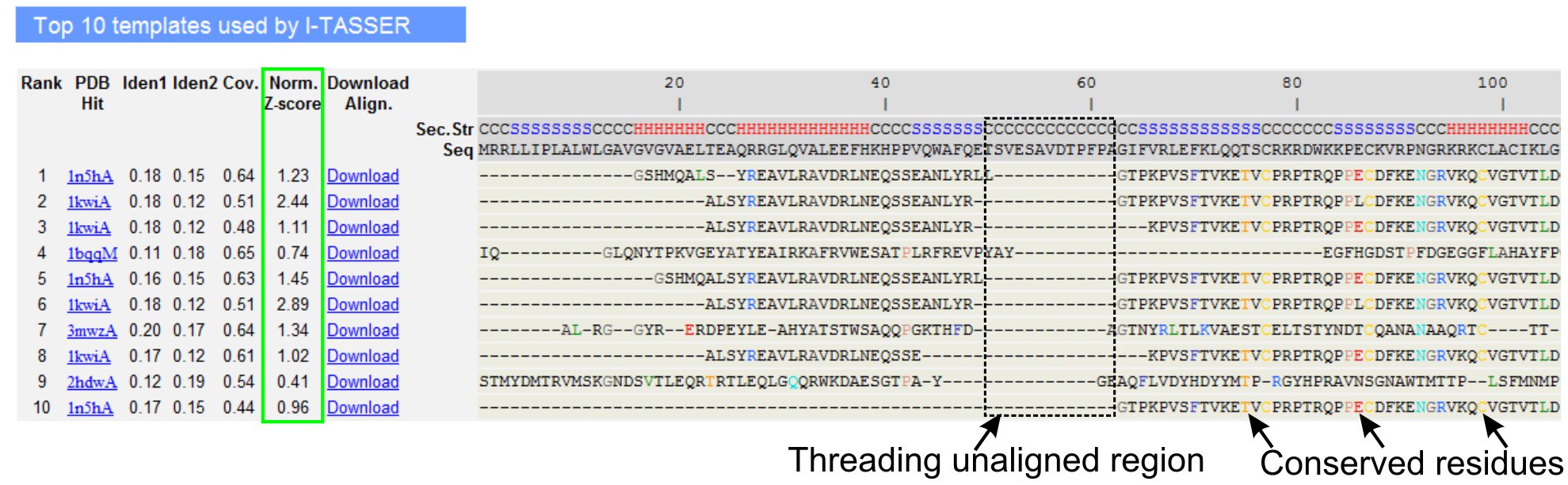
- Прокрутите вниз, чтобы проанализировать десятку резьбы шаблоны запросов белка, как это определено LOMETS программ резьбы (рис. 3). Посмотреть нормированного Z-счет (уравнение 1), показанном на 'Норма. Z-счет "столбец, анализировать качество резьбы выравнивания. Трассы с нормированной Z-оценка> 1 отражает уверенность выравнивание и, скорее всего, имеют те же раз по запросу белка.
- Анализ последовательности идентичность в резьбы краю области («Иден. 1 'столбец) и для всей цепочки (' Иден. 2 'колонка) оценить соответствие между запросом и шаблон белков. Высокая идентичность последовательности индикатора эволюционного родства между запросом и шаблон белков.
- Вид резьбы выровнены остатков показано на цветные визуально определить минусыerved остатков / мотивы в запрос и шаблон белков. Выше идентичности последовательности в резьбы краю области, по сравнению с цельной цепи выравнивание также указывает на присутствие сохраняется структурный мотив / доменов в запросе.
- Оценка охвата потоков выравнивания, просмотрев 'Соу. колонки и проверки выравнивания. Если освещение верхней выравнивания низка и ограничивается лишь небольшая область запроса белка или отсутствовал длинный сегмент запрос последовательность, то запрос белка обычно содержит более одного домена, поэтому рекомендуется разбить последовательность и модель областей в индивидуальном порядке (рис. 3).
- Скачать PDB отформатирован последовательность структуры файлов выравнивания, нажав на "Скачать Выравнивание" ссылки. Эти выравнивания файл можно открыть в любой программе молекулярной визуализации, перечисленные в разделе Материалы, а также может быть использован для добавления дополнительных ограничений в структуре моделирования (шаг 1.4).
6.Структурные аналоги в PDB
- Просмотр следующей таблице (рис. 4) в результате странице, чтобы определить десятку структурных аналогов впервые предсказано моделью, как это определено программой структурных выравнивание ТМ-20 выровнять. ТМ-оценка> 0,5 означает, что обнаружена аналоговые и модели, аналогичной топологией и может быть использована для определения структурных классов / белок семейства запрос белка 16, а с ТМ-оценка <0,3 означает случайное сходство структуры.
- Анализ последовательности идентичность и СКО в структурно выровнены области, показанной на "IDEN» и «СКО» колонны для оценки сохранения пространственной мотивы в модели и структурным аналогом. Осмотрите цветных и приведены в соответствие пар остатков в выравнивание, чтобы определить эти структурно сохраняются остатки и мотивы.
- Нажмите на PDB код, указанный в колонке "PDB Hit 'посетить RCSB сайт и узнать больше об их структурной классификации (СКОП, CATH и PFAM) и функциональной информации (ЕС номер, связанный GO терминов и связанного лиганда).
7. Функция прогноз с помощью кофактора
- Прокрутите страницу результатов для анализа функциональных интерпретаций для запроса белка. Белки функции, перечисленные в три таблицы контексте отображения: Фермент комиссия (ЕК) номера, Джин Онтология (ГО) условиях, и связывание лигандов сайтов.
- Посмотреть "ТМ-оценка», «СКО», «IDEN» и «Соу. столбцов в каждой таблице для анализа параметров глобального сходства структуры и сохранения пространственных структур, между моделью и определены функциональные гомологов (шаблонов).
8. Фермент комиссии номер прогноз
- Посмотреть пятерку потенциальных гомологов фермента белка запроса показаны в "Прогнозируемая ЕС номеров" таблицы (рис. 5). Уровня доверия ЕС количество прогноз с помощью этих шаблонов показано в столбце "EC-Оценка». На основании benchmarking анализ 23, функциональное сходство (первые 3 цифры числа ЕК) между запросом и шаблон белка может быть надежно интерпретированы EC-оценка> 1.1.
- Ищите консенсус функции (ЕС номеров) среди шаблонов, которые имеют аналогичные раза (т.е. ТМ-оценка> 0,5) в качестве запроса белка. Если несколько шаблонов имеют одинаковое количество ЕС и EC-оценка> 1,1, уровень достоверности прогнозирования является очень высоким. Однако, если ЕС показатель высокий, но есть отсутствие консенсуса между определили хитов, то прогноз становится менее надежным и пользователям рекомендуется проконсультироваться GO долгосрочные прогнозы.
- Нажмите на ссылку, представленную на номера EC посетить ExPASy базы данных ферментов и анализировать функции, в том числе реакции, катализируемой, сопутствующим фактором требования и метаболический путь, по шаблону белка в деталях.
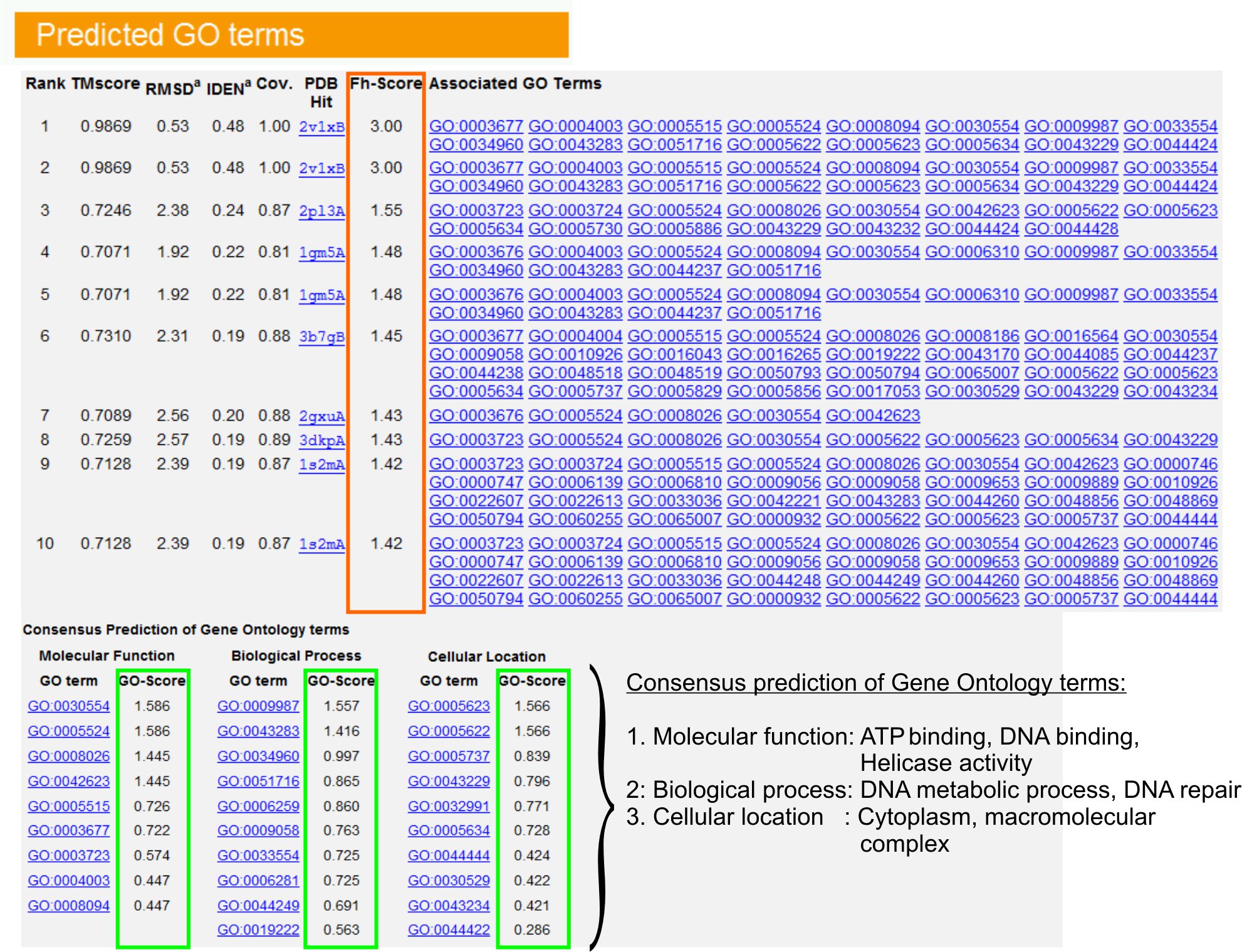
9. Онтология гена (ГО) прогнозы срок
- Посмотреть "Прогнозируемая GO термины" таблицы (рисЮр 6) определить десятку гомологов запрос белка в PDB библиотеке, аннотированный с Джином Онтология (ГО) условиях. Каждый белок, как правило, связан с несколькими условиями GO, описывающая его молекулярных функций (МФ), биологических процессов (БП) и клеточные компоненты (ГК). Нажмите на каждого члена, чтобы посетить веб-сайт Амиго и анализировать ее определение и происхождение.
- Анализ FH-оценка (функциональная оценка гомологии) колонки для доступа к функциональным сходством между запросом и шаблон белков и оценить уровень достоверности передачи функциональной аннотации из этих белков. В нашем сравнительного исследования 23, 50% от родных условиях GO может быть определен неверно из впервые выявлен шаблонов с помощью РН-счет отсечки 0,8, с общей точностью 56%.
- Посмотреть "Консенсус прогноз ГО термины" таблица для анализа согласия функции между шаблонами. Эти общие функции используются для прогнозирования GO выражении (MF, ВР и КС) запросбелка и оценить уровень достоверности (GO-счет) ГО долгосрочные прогнозы. На основе тестирования тест 23, лучший ложноположительных и ложно отрицательные темпы получены для предсказания с GO-счет отсечки = 0,5, при уменьшении освещения прогноз на более глубоком уровне онтологии.
10. Белок-лиганд прогнозы сайт
- Прокрутите вниз до нижней части страницы, чтобы посмотреть в десятку лиганд прогнозы сайт для запроса белка. Прогнозируемая сайты связывания ранжируются в зависимости от количества прогнозируемых лиганд конформации, которые имеют общие связывающего кармана. Лучший сайт связывания определены уже отображается в Jmol апплет. Нажмите на переключатель, чтобы проанализировать и другие прогнозы и визуализировать остатков лиганд взаимодействует.
- Анализ BS-оценка колонки для оценки локального подобия между моделью и сайт связывания шаблона. На основании тестов 9, BS-оценка> 1,1 указывает на высокую последовательность и структура симilarity возле предсказал сайт связывания в модели и известный сайт связывания в шаблоне.
- Скачать файл в формате PDB структура комплекса, нажав на ссылку "Скачать". Пользователи могут открыть эти файлы в любую молекулярную программу визуализации и интерактивного просмотра предсказал сайт связывания и лиганд-белковых взаимодействий на своем локальном компьютере.
11. Представитель результаты

Рисунок 1 выдержка из I-TASSER результате страница, показывающая () FASTA запроса в формате последовательности;. (B) предсказал вторичной структуры и связанные с ними оценки уверенности, и (C) предсказал растворителя доступности остатков. Анализируемой области ядра и потенциальное место гидратации в запросе выделены голубым и красным прямоугольниками, соответственно.

Рисунок 2.

Рисунок 3. Пример I-TASSER результате страница, показывающая десятку определили резьбы шаблонов и выравнивания по LOMETS 5 резьбы программ. Качество резьбы выравнивания оценивается на основе нормированных Z-счет (выделено зеленым), где значение> 1 отражает уверенность выравнивания. Унифицированные остатков в шаблоне, идентичные соответствующие вычеты запроса выделяются цветом для обозначения присутствия сохраняется остаток / мотив, а отсутствие согласования в большинстве топ шаблонов указывает на наличие нескольких доменов в белке запрос и выровненным остатки соответствуют областям домена компоновщика. Нажмите здесь, чтобы посмотреть полноразмерные версии рисунке 3.

Рисунок 4. Пример результата страница, показывающая десятку определили структурные аналоги и структурных трасс, определенных ТМ-20 выровнять структурные программы выравнивания. Рейтинг аналогов показано на основано на ТМ-оценка (выделены синим) структурного выравнивания. ТМ-оценка> 0,5 указывает, что два сравниваемых структур аналогичную топологию, а ТМ-оценка <0,3 означает сходство между двумя случайными структурами. Структурно соответствие пар остатков выделяются цветом в зависимости от их аминокислотного собственности, в то время как выровненным регионах, обозначены «-».ove.com/files/ftp_upload/3259/3259fig4large.jpg "> Нажмите здесь, чтобы посмотреть полноразмерные версии рисунке 4.

Рисунок 5. Пример I-TASSER результате страница, показывающая определены фермента гомологов запрос белка в PDB библиотеки. Уровень достоверности прогнозирования числа ЕС, анализируется на основе ЕС-оценка (выделены зеленым цветом), где EC-оценка> 1,1 указывает функциональное сходство (те же первые 3 цифры числа ЕК) между запросом и шаблон белка.

Рисунок 6. Пример I-TASSER результате страница, показывающая GO долгосрочные прогнозы для запроса белка. Функциональные гомологов для запроса белка в библиотеку шаблонов Онтология гена оцениваются на основе их Fh-счет (в прямоугольник оранжевого цвета). Общие функциональные особенности этих топ-скоринга хитов выводятся на GENER ели окончательного GO долгосрочные прогнозы для запроса белка. Качество предсказал GO условий определяется на основе GO-оценка (показаны зеленым цветом), где GO-оценка> 0,5 указывает надежные предсказания. Нажмите здесь, чтобы посмотреть полноразмерные версии рисунке 6.

Рисунок 7. Пример I-TASSER результате страница, показывающая десятку белок лиганд прогнозы сайт с помощью кофактора 9 алгоритма. Рейтинг предсказал сайты связывания на основе числа предсказали лиганд конформации, которые имеют общие связывающего кармана в запросе. BS-оценка (выделено красным) является мерой местных последовательность и структура сходство между предсказанным и сайт связывания шаблона, а также полезен при анализе сохранения обязательных карманы сайта.
les/ftp_upload/3259/3259fig8.jpg "/>
Рисунок 8. Пример внешних файлов сдержанность используется для для определения остатков остатков контакт / расстояние ограничений.

Рисунок 9. Пример сдержанности файлов, используемых для определения шаблонов белка I-TASSER сервера. Пользователь может указать запрос-шаблон выравнивания либо в () FASTA формате, или (B) 3D-формате.

Рисунок 10. Пример файла, используемого для исключения шаблона во время I-TASSER процедура моделирования структуры. Первый столбец содержит PDB идентификатор шаблона белки должны быть исключены. Вторая колонка используется для указания отсечки идентичности последовательности, которая будет использоваться для других подобных шаблонов в библиотеку шаблонов.
Discussion
Протокол изложенная выше общего руководства для структуры и функции моделирования с использованием I-TASSER сервера. Хотя это автоматизированная процедура очень хорошо работает для большинства белков, вмешательство человека часто помогают значительно улучшить точность моделирования, особенно для белков, отсутствие близких шаблонов в PDB библиотеки. Пользователи могут выступать в ходе I-TASSER моделирования в следующих способов: (а) расщепление нескольких доменов белков, (б) предоставление внешних ограничений по совершенствованию структуры сборки и (в) удаление шаблонов во время моделирования.
Разделение нескольких доменов белка:
Многие длинные последовательности белка часто содержат несколько доменов, привязанных к гибким компоновщик регионах, что делает их структуру выяснение трудно с использованием как экспериментальных и расчетных методов. Тем не менее, как домены независимо складывания лицами и могут выполнять различные молекулярные функции, этоЖелательно разделить длинный нескольких доменов белков и модель каждого домена отдельно. Моделирование областей индивидуально не только ускорит процесс прогнозирования, но и повышает качество запросов шаблон выравнивания, что приводит к более надежной структурой и функцией предсказания.
Доменных границ в белковых последовательностей может быть предсказано использованием свободно доступных внешних онлайновых программ, таких как NCBI CDD 24, 25 или PFAM InterProScan 26. Кроме того, если LOMETS резьбы выравнивания доступны для запросов белка, доменные границы могут быть расположены по визуальной идентификации длинные отрезки выровненным остатков в верхней резьбы шаблонов (см. шаг 5.4). Эти выровненным регионах основном соответствуют областям домена компоновщика. Если несколько шаблонов доменов, которые уже доступны в шаблоне PDB библиотеку со всеми областями запроса выровнены, то запрос белка может быть смоделирована как всю длину.
Предоставление внешних ограничений
А. Укажите контактный / расстояние ограничений
Экспериментально характеризуется между остатком контакты / расстояния, например из ЯМР илисшивания экспериментов, может быть указано, загрузив файл сдержанность. Файл примера показано на рисунке 8, где Колонка 1 определяет тип ограничения, то есть "DIST" или "CONTACT". Для расстояния сдержанность (DIST), столбцы 2 и 4 содержат остатки позиции (I, J), графы 3 и 5 содержат атом-типов в остатке и столбце 6 указывается расстояние между двумя заданными атомов. Для контакта ограничения (КОНТАКТ), столбцах 2 и 3 содержат положения (I, J) вычетов, которые должны быть в контакте. Расстояние между центром боковые цепи этих контактов пар Остаток решили на основе наблюдаемых расстояний в известных структур PDB. I-TASSER постараемся привлечь этих атомных пар вблизи указанного расстояния во время моделирования структуры утонченности.
Б. Укажите шаблона белковую структуру
LOMETS резьбы программы используют представитель PDB библиотеку, чтобы найти правдоподобное дворы для запроса протЭйн. Хотя использование библиотеки представитель структура помогает сократить время, необходимое для вычисления последовательности структуры трасс, вполне возможно, что хороший белок шаблон пропустили в библиотеке или шаблон, возможно, не были определены LOMETS резьбы программы, даже если это присутствует в библиотеке. В этих случаях пользователь должен указать желаемую структуру белка в качестве шаблона.
Чтобы определить структуру белка в качестве дополнительного шаблона, пользователи могут загрузить файл в формате PDB структуры или указать идентификатор PDB хранение структуры белка в PDB библиотеки. I-TASSER будет генерировать запрос-шаблон выравнивания использованием MUSTER программы 23 и будет собирать пространственные ограничения с обеих пользователя, указанного шаблона и LOMETS шаблоны для руководства моделирования структуры сборки. Поскольку точность LOMETS ограничений различна для разных целей, вес LOMETS ограничения сильнее в легкой (гомологичны) таrgets, чем в твердых (не гомологичны) цели, которые всесторонне настроены в нашем тесте обучения.
Пользователь может также указать свой запрос-шаблон выравнивания. Сервер принимает выравнивания в двух форматах: формат FASTA (рис. 9А) и формате 3D (рис. 9Б). Формат FASTA является стандартным и описан в http://zhanglab. ccmb.med.umich.edu / FASTA / . 3D формат похож на стандартный формат PDB ( http://www.wwpdb.org/documentation/format32/sect9.html ), но две дополнительные столбцы, производные от шаблонов добавляются ATOM записей (см. рис 9В):
Столбцы 1-30: Atom (С-альфа только) и остаток имен для запроса последовательность.
Столбцы 31-54: Координаты С-альфа атомов запрос скопированы из соответствующих атомов в шаблоне.
Столбцы 55-59: Соответствующий номер остатка в шаблон, основанный на выравнивание
Столбцы 60-64: Соответствующий имя остатков в шаблоне
Исключить шаблоны белков
Белки являются гибкие молекулы и может принять несколько конформационных состояний изменить свою биологическую активность. Например, структуры многих протеинкиназ и мембранных белков, были решены в активной и неактивной конформации. Кроме того, наличие или отсутствие связанного лиганда может привести к большим структурным движений. Хотя все конформационных состояний шаблон одинаковых для резьбы программ, желательно модель запроса с использованием шаблонов только в одном определенном состоянии. Новая опция на сервере позволяет пользователю исключить шаблон структуры белков в процессе моделирования. Эта функция также позволит пользователю выбрать гомологии уровне шаблонов, которые будут использоваться для моделирования. Пользователи могут исключить шаблон белков птОМ I-TASSER библиотеке:
А. Указание отсечки идентичности последовательности
Пользователи могут использовать эту опцию, чтобы исключить гомологичных белков из I-TASSER библиотеки шаблонов. Гомологии уровня устанавливается на основе отсечки идентичности последовательности, т.е. количество идентичных остатков между запросом и шаблон белок делится на последовательность длины запроса последовательности. Например, если пользователь вводит "70%" в соответствующем виде, все шаблоны белков, которые имеют идентичности последовательности> 70% до запроса белка I-будет исключен из I-TASSER библиотеки шаблонов.
Б. Исключение специфических белков шаблон
Специальные белки шаблон может быть исключен из I-TASSER библиотеки шаблонов, загружая список, содержащий идентификаторы PDB структур должны быть исключены. Пример файла показан на рисунке 10. Как же белка может существовать в виде нескольких записей в PDB библиотека, I-TASSER себеrver будет по умолчанию исключить указанные шаблоны (в Столбец1), а также все другие шаблоны из библиотеки, которые идентичности> 90% на указанный шаблонов. Пользователи также могут указать другой отсечки идентичности, например, 70%, где все шаблоны с единицей> 70% к указанным белкам шаблон будет исключена.
Disclosures
Нет конфликта интересов объявлены.
Acknowledgments
Проект осуществляется при поддержке, в частности Альфреда П. Слоуна, NSF Карьера премии (DBI 1027394), и Национальный Институт общей медицинских наук (GM083107, GM084222).
Materials
| Name | Company | Catalog Number | Comments |
| Material Name | Type | Company | Catalogue Number |
| FASTA formatted amino acid sequence of the protein to be modeled (see, http://www.ncbi.nlm.nih.gov/BLAST/fasta.shtml). | |||
| A personal computer with access to the internet and a web browser. | |||
| Molecular visualizing software, e.g. RASMOL or PYMOL, for analyzing the predicted tertiary structure and functional sites. |
References
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H. CD-Search: protein domain annotations on the fly. Nucleic. Acids. Res. 32, W327-W331 (2004).
- Finn, R. D. The Pfam protein families database. Nucleic. Acids. Res. 38, D211-D222 (2010).
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).

{kind=link}
{kind=link}