

Summary
Directrices para el ordenador basado en la caracterización estructural y funcional de la proteína mediante la canalización I-TASSER se describe. A partir de secuencias de proteínas de la consulta, los modelos 3D se generan con múltiples alineaciones threading e iterativo simulaciones estructurales de montaje. Inferencias funcionales a partir de entonces son elaborados sobre la base de los partidos a las proteínas de estructura conocida y funciones.
Abstract
Los proyectos de secuenciación del genoma ha cifrado millones de secuencias de proteínas, que requieren el conocimiento de su estructura y función de mejorar la comprensión de su función biológica. Aunque los métodos experimentales pueden proporcionar información detallada para una pequeña fracción de estas proteínas, modelado computacional que se necesita para la mayoría de las moléculas de proteínas que son caracterizados experimentalmente. El servidor de I-TASSER es un banco de trabajo on-line de alta resolución de modelado de la estructura y función de proteínas. Dada una secuencia de la proteína, una salida típica del servidor I-TASSER incluye la predicción de estructura secundaria, prevé la accesibilidad solvente de cada residuo, las proteínas homólogas de plantilla detectado por roscado y alineaciones de la estructura, hasta cinco de larga duración en los modelos estructurales terciario, y basados en estructuras anotaciones funcionales para la clasificación de las enzimas, los términos de ontología de genes y la proteína-ligando sitios de unión. Todas las previsiones están marcadas con una puntuación de confianza quecuenta la precisión de la predicción son sin conocer los datos experimentales. Para facilitar las solicitudes especiales de los usuarios finales, el servidor proporciona canales para aceptar especificado por el usuario entre la distancia y el contacto con residuos de mapas para cambiar los modelos I-TASSER, sino que también permite al usuario especificar cualquier proteína como plantilla, o para excluir a cualquier plantilla proteínas durante las simulaciones de la estructura de montaje. La información estructural podría ser recogida por los usuarios sobre la base de evidencias experimentales o conocimientos biológicos con el fin de mejorar la calidad de la I-TASSER predicciones. El servidor fue evaluado como el mejor de los programas de estructura de las proteínas y las predicciones en función de los experimentos CASP reciente de toda la comunidad. Actualmente hay> 20.000 científicos registrados en más de 100 países que están utilizando el on-line I-TASSER servidor.
Protocol
Método de descripción
Siguiendo el paradigma de la secuencia a la estructura a la función, el procedimiento de I-TASSER 1-4 para la estructura y la función de modelado consta de cuatro pasos consecutivos de: (a) identificación de la plantilla por LOMETS 5, (b) la estructura de reensamblaje de fragmentos de replica- intercambio de simulaciones Monte Carlo 6; (c) el perfeccionamiento estructura atómica nivel con REMO 7 y FG-MD 8, y (d) estructura basada en la interpretación con la función cofactor 9.
Identificación de la plantilla: Para una secuencia de la consulta enviada por el usuario, la secuencia es la primera pasa a través de una biblioteca de la estructura por un representante de AP LOMETS instalado localmente meta-threading servidor. Threading es un procedimiento de alineación de secuencias-estructura que se utiliza para identificar proteínas de plantilla que puede tener una estructura similar o que contengan motivos estructurales similares, como la proteína de la consulta. Para aumentar la cobertura de los homólogos templdetecciones comió, LOMETS combina múltiples del estado de la técnica de algoritmos que cubren las diferentes metodologías de roscado. Desde diferentes programas de roscar tienen diferentes sistemas de puntuación y la sensibilidad de alineación, la calidad de los alineamientos generados hilos de rosca de cada programa es evaluado por normalizado Z-score, que se define como: 
donde Z es la puntuación en las unidades de desviación estándar con respecto a la media estadística de todas las alineaciones generado por el programa, y Z 0 es un programa específico de Z-score de corte determina con base en pruebas a gran escala de referencia para diferenciar los hilos 5 'buena "y" malos "de plantillas. Una plantilla con una alta puntuación Z significa que los modelos superiores tienen una puntuación significativamente más alta que la alineación la mayor parte de otras plantillas, que por lo general implica que la alineación se corresponde con un buen modelo. Si la mayoría de las plantillas de rosca superior tienen altanormalizado gh Z-score, la exactitud de la final I-TASSER modelo suele ser alto. Sin embargo, si la proteína es grande y la cobertura de las alineaciones de roscado se limita a una pequeña región de la proteína de la consulta, un alto normalizado Z-score no significa necesariamente un modelo de alta precisión para el modelo de cuerpo entero. Los dos primeros alineamientos hilos de cada programa de roscado se recogen y se utilizan para el siguiente paso del montaje de la estructura.
Iterativo de simulación de la estructura de montaje: Siguiendo el procedimiento de roscado, secuencia de la consulta se divide en regiones roscado alineados y no alineados. Fragmentos de continuo en la alineación hilos se cortan a partir de plantillas y utilizar directamente para el montaje de la estructura, mientras que las regiones de bucle no alineados se construyen mediante el modelado ab initio. El procedimiento de montaje de la estructura se realiza en un sistema de red guiado por la réplica de simulaciones de Monte Carlo de cambio 6. El campo I-TASSER fuerza incluye el hidrógeno-bonding interacciones 10, basado en el conocimiento de la energía términos estadísticos derivados de estructuras de proteínas conocidas en el AP 11, la secuencia basada en las predicciones de contacto de SVMSEQ 12, y las restricciones espaciales recogidos a partir de plantillas LOMETS 5 hilos. Los señuelos de conformación generados en las réplicas de baja temperatura durante las simulaciones se agrupan por Spicker 13 para identificar las estructuras de baja energía libre de los estados. Centroides de grupo de los grupos principales se obtiene promediando las coordenadas 3D de todos los señuelos agrupados estructurales y se utiliza para la generación del modelo final. La simulación y el procedimiento de agrupación se repiten dos veces para eliminar los enfrentamientos estéricos y refinar aún más la topología global.
A nivel atómico modelo de construcción y perfeccionamiento: Los centroides cluster obtenidos después de la agrupación Spicker se reducen modelos de proteínas (cada residuo, representada por su α C y la cadena lateral del centro de masa) y have biológica limitada aplicación. La construcción de todo el modelo atómico de los modelos reducidos se hace en dos pasos. En el primer paso, REMO 7 se utiliza para construir modelos atómicos completo de C-alfa rastros mediante la optimización de las redes de enlace H-. En el segundo paso, REMO completo atómica modelos más refinados por FG-MD 14, que mejora los ángulos de torsión columna vertebral, longitudes de enlace, y la cadena lateral orientaciones rotámero, por simulaciones de dinámica molecular, guiado por los fragmentos estructurales de la búsqueda estructuras de AP en TM-align. Los modelos FG-MD refinado se utilizan como los modelos finales de las predicciones estructura terciaria de I-TASSER.
La calidad de los modelos generados se calcula sobre la base de una puntuación de confianza (C-score), que se define sobre la base de la Z-score de alineaciones LOMETS roscado y la convergencia de la I-TASSER simulaciones, formuladas matemáticamente como: 
donde
El C-score tiene una fuerte correlación con la calidad de los modelos I-TASSER. Mediante la combinación de C-score y la longitud de la proteína, la exactitud de los primeros I-TASSER modelos se puede estimar con un error promedio de 0,08 para el TM-score y un 2 para el RMSD 15. En general, los modelos de C-score> - 1.5 se espera que tengan un pliegue correcto. Aquí, RMSD y TM-score son medidas bien conocidas de la similitud topológica entre el modelo y la estructura nativa. TM-score valiosarango es de [0, 1], donde una mayor puntuación indica una mejor estructura coincide 16,17. Sin embargo, para los modelos de menor rango (es decir, 2 º -5 ª modelos), la correlación de C-score con el TM-score y RMSD es mucho más débil (~ 0,5), y no puede ser utilizado para la estimación fiable de la calidad del modelo absoluto.
Es el primer modelo siempre es el mejor modelo de I-TASSER simulaciones? La respuesta a esta pregunta depende del tipo de destino. Para un blanco fácil, el primer modelo suele ser la mejor modelo y su C-score es generalmente mucho mayor que el resto de los modelos. Sin embargo, para objetivos duros, donde rosca no tiene éxitos importantes de plantilla, el primer modelo no es necesariamente el mejor modelo y I-TASSER realmente tiene dificultades en la selección de las mejores plantillas y modelos. Por ello se recomienda analizar todas las 5 modelos para objetivos duros y seleccionarlos sobre la base de la información experimental y el conocimiento biológico.
Función predictions: En el último paso, último modelos 3D generados por FG-MD se utilizan para predecir tres aspectos de la función de las proteínas, a saber: a) Comisión de Enzimas (CE) número 18 y (b) Gene Ontology (GO) 19 términos y ( c) los sitios de unión de ligandos de moléculas pequeñas. Para todos los tres aspectos, interpretaciones funcionales se generan con cofactor, que es un nuevo enfoque para predecir la función de la proteína de la base de similitud global y local a las proteínas de la plantilla en el AP de estructura conocida y funciones. En primer lugar, la topología global de los modelos predijeron se compara con bibliotecas de plantillas funcionales mediante el programa de alineación estructural TM-align 20. A continuación, un conjunto de proteínas más similares a los modelos de destino son seleccionados de la biblioteca en base a su similitud estructura global, y una extensa búsqueda local se realiza para identificar la estructura y la similitud de secuencias, cerca de la región del sitio activo / vinculante. Las puntuaciones de similitud resultante global y lo local se utilizan para clasificar losproteínas plantilla (homólogos funcionales) y la transferencia de la anotación (números de la CE y Gene Ontology 19 términos) sobre la base de los éxitos con mayor puntuación. Del mismo modo, los residuos de unión al ligando del sitio y el modo de unión al ligando se infieren sobre la base de la alineación local de consulta con el conocido sitio de unión al ligando los residuos en las plantillas de función superior anotando 9.
La calidad de la función (CE y GO plazo) en la predicción de I-TASSER se evalúa según el puntaje de homología funcional (Fh-score) que es una medida de similitud global y local entre la consulta y la plantilla, y se define como: 
donde C-score es una estimación de la calidad del modelo de predecir como se define en la ecuación. (2), TM-score mide la similitud estructural global entre el modelo y las proteínas de la plantilla; RMSD Ali es el RMSD entre el modelo y la estructura de la plantilla en la región estructuralmente alineado de TM-align 20; Cov representa la cobertura de la alineación estructural (es decir, la proporción de los residuos alineados estructuralmente dividido por la longitud de la consulta); ID ali es la identidad de la secuencia en la alineación TM-align. Las medidas de confianza estimados para las predicciones de su número CE incluye también un plazo para la evaluación de coincidir con el sitio activo (AcM) entre la consulta y la plantilla dentro de una región local definida, calculada como: 
donde N representa el número t de residuos de la plantilla presentes en la zona local, Ali N es el número de los pares de residuos alineados plantilla de consulta, ii d es la distancia entre el par C α i-ésima de residuos alineados, d 0 = 3.0 Å es el punto de corte a distancia, M ii es la puntuación BLOSUM entre pares i de residuos alineados. En general, el FH-score se encuentra en el rango [0, 5] y la puntuación de AcM se encuentra entre [0, 2], Donde las puntuaciones más altas indican las asignaciones funcionales más confianza. Puntuación AcM también se utiliza para la evaluación de la estructura local y la similitud de secuencias cerca de los sitios de unión al ligando, lo que se conoce como BS-score.
1. Presentación de la secuencia de la proteína
- Visite la página web de I-TASSER en http://zhanglab.ccmb.med.umich.edu/I-TASSER para comenzar con la estructura y experimentar modelos de función.
- Copie y pegue la secuencia de aminoácidos en la forma prevista o directamente subir desde tu ordenador haciendo clic en el botón "Examinar". I-TASSER servidor acepta actualmente las secuencias con un máximo de 1.500 residuos. Las proteínas más de 1.500 residuos suelen ser multi-dominio de las proteínas, y se recomienda que se divide en dominios individuales antes de la presentación de I-TASSER.
- Proporcione su dirección de correo electrónico (obligatorio) y un nombre para el trabajo (opcional).
- Opcionalmente, puede especificar los usuarios externos inter-respóngase en contacto con idue / distancia restricciones, complemento de una plantilla adicional o excluir algunas de las proteínas de plantilla durante el proceso de modelado de la estructura. Más información sobre el uso de estas opciones en la "Discusión" sección.
- Para presentar la secuencia, haga clic en "Ejecutar I-TASSER" botón. El navegador se dirige a una página de confirmación que muestra la información especificada por el usuario, identificación del trabajo (Job ID) número y un enlace a una página web donde los resultados serán depositados después de terminar el trabajo. Los usuarios pueden marcar este enlace o anote el número de identificación del trabajo para futuras referencias.
2. Disponibilidad de los resultados
- Comprobar el estado de su trabajo presentado por visitar la página de la cola de I-TASSER en http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php . Haga clic en la pestaña de búsqueda y utilizar el número de identificación del trabajo o la secuencia de consulta para buscar en el trabajo presentado.
- Después de que la estructura y la función moDeling, se abrirá una notificación por correo electrónico que contiene la imagen de las estructuras predichas y un enlace en la web será enviado a usted. Haga clic en este enlace o abrir el enlace marcado en el paso 1.5 para ver y descargar los resultados.
3. Estructura secundaria y predicciones solventes accesibilidad
- Compruebe la secuencia de consulta FASTA formato que aparece en la parte superior de la página de resultados. Si ninguna restricción adicional / plantilla se especificó durante la presentación de secuencias, un enlace a la página web de visualización de la información especificada por el usuario también puede ser visto (Figura 1A).
- Examinar la predicción de estructura secundaria se muestra como: hélice alfa (H), beta hebra (S) o bobina (C) y la puntuación de confianza de la predicción (0 = bajo, 9 = alto) para cada residuo. Busque región con largos tramos de la estructura secundaria regular (H o S) las predicciones, para estimar el núcleo de la región de la proteína. Clase estructural de proteínas también pueden ser analizados sobre la base de la distribución de los elementos de estructura secundaria. AlPor lo tanto, largas regiones de elementos de la bobina de la proteína por lo general indican las regiones no estructurados / desordenada.
- Ver la accesibilidad prevista disolvente (fig. 1C) para determinar enterrado y solvente regiones expuestas en la consulta. Los valores de rango pronosticado disolvente accesibilidad de 0 (residuos enterrados) a 9 (residuos expuestos). Región que contiene los residuos enterrados en su mayoría pueden ser utilizados para delinear la región central de la proteína, mientras que las regiones con solvente residuos expuestos y son la hidratación hidrofílica potenciales / sitios funcionales.
4. Predicciones de estructura terciaria
- Desplácese hacia abajo para ver las estructuras predichas terciaria de la proteína de la consulta, aparece en una miniaplicación Jmol interactivo (Figura 2). Haz clic izquierdo en el applet para cambiar el aspecto de la estructura mostrada, hacer zoom en regiones específicas, seleccionar tipos específicos de residuos en el modelo de predecir o calcular distancias entre los residuos.
- Analizar los modelos de la presencia de largo regiones no estructurados. Estos rREGIONES suelen corresponder a las regiones desordenada en proteínas o indicar la falta de alineación de la plantilla. Estas regiones suelen tener una precisión de modelado de baja y la eliminación de estas regiones durante el modelado de N y C-terminal región mejorará la precisión de modelado.
- Descargar los archivos de formato PDB la estructura del modelo, haga clic en "Descargar" Modelo de enlaces. Puede abrir estos archivos en cualquier software de visualización molecular (por ejemplo, PyMOL, Rasmol etc) para su posterior análisis de las características estructurales.
- Analizar la puntuación de confianza (C-score) de modelar la estructura para estimar la calidad de las estructuras de predecir. Puntuación de C-(Ec. 2) Los valores están típicamente en el intervalo [-5, 2], en una puntuación más alta refleja un modelo de mejor calidad. La estimación de TM-score y RMSD del primer modelo se muestra como "la precisión estimada de Modelo 1". Para las proteínas de largo, se recomienda para evaluar la calidad del modelo basado en TM-score, el TM-score es más sensible a los cambios topológicos que RMSD. < li> Haga clic en "más información acerca de C-score" para analizar la C-score, el tamaño del clúster y la densidad del conjunto de todos los modelos. Estima TM-score y RMSD se presentan sólo para la primera I-TASSER modelo, ya que C-score de los modelos de menor puntuación no se correlaciona fuertemente con el TM-calificación o el RMSD. Calidad de los modelos de menor rango puede ser parcialmente evaluado en función de su densidad de racimo y el tamaño del clúster en relación con el primer modelo, en donde los modelos más grandes de racimo y una mayor densidad son, en promedio cerca de la estructura nativa.
- Baja puntuación de C-predicciones suelen indicar una predicción de baja precisión. En la mayoría de estos casos, la consulta proteína carece de una buena plantilla en la biblioteca y tiene un tamaño más allá del alcance de los modelos ab initio (es decir,> 120 residuos). En estos casos, los usuarios pueden buscar más restricciones espaciales y los utilizan para mejorar el modelo I-TASSER (véase la sección de discusión). También se anima a presentar las secuencias a nuestro servidor QUARK (QUARK / "> http://zhanglab.ccmb.med.umich.edu/QUARK/) para una modelización ab initio pura si el tamaño de la proteína está por debajo de 200 residuos.
5. LOMETS objetivo plantilla de alineación
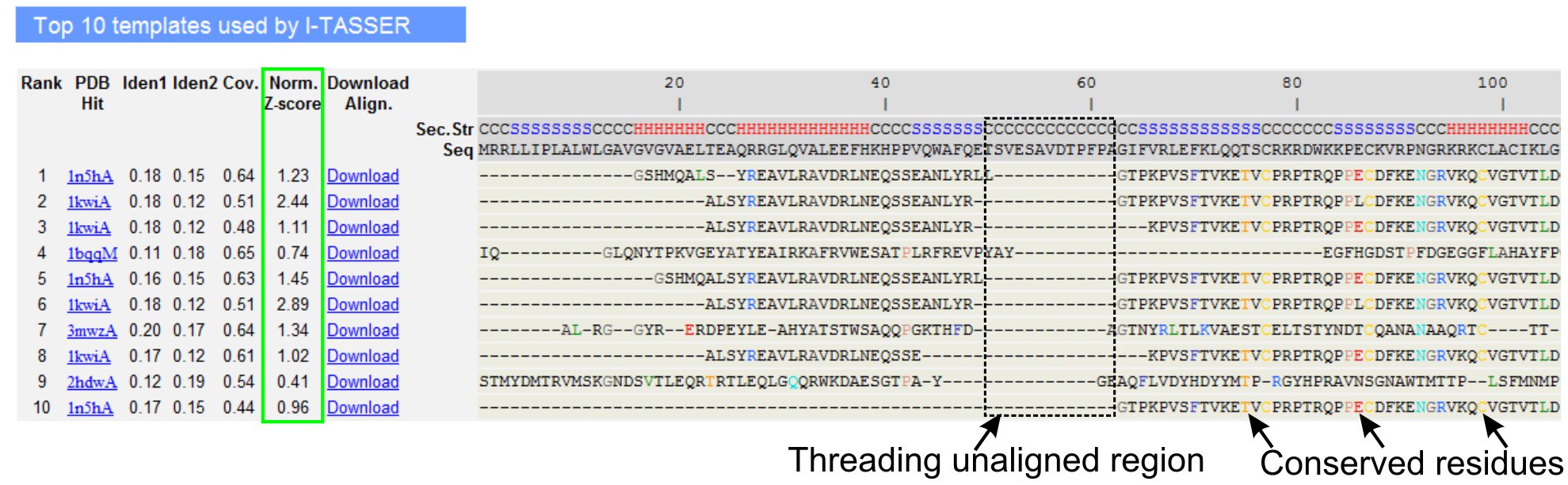
- Desplácese hacia abajo para analizar los diez mejores plantillas de rosca de la consulta proteína, identificada por LOMETS programas de threading (Figura 3). Ver el normalizado Z-score (Ec. 1), se muestra en la "norma. Columna de Z-score ", para analizar la calidad de las alineaciones de roscado. Alineaciones con una normalizado Z-score> 1 refleja una alineación segura y más probable es que el mismo redil como la proteína de la consulta.
- Analizar la identidad de secuencia en la región de rosca alineados ("Iden. 1 'columna) y para toda la cadena (la columna' Iden. 2 ') para evaluar la homología entre la consulta y las proteínas de la plantilla. Alta identidad de secuencia es un indicador de las relaciones evolutivas entre la consulta y las proteínas de la plantilla.
- Ver los restos de hilos alineados se muestra en colores para identificar visualmente los contrasresiduos erved / motivos de la consulta y las proteínas de la plantilla. Una identidad de secuencia más alto en el roscado alineados región, en comparación con toda la cadena de alineación también indica la presencia de motivos estructurales conservadas / dominios en la consulta.
- Evaluar la cobertura de la alineación de rosca por ver la "Cov. columna y la inspección de la alineación. Si la cobertura de las alineaciones superior es bajo y se limita a sólo una pequeña región de la proteína de consulta o ausente por un largo segmento de secuencia de la consulta, la consulta proteína por lo general contiene más de un dominio y se recomienda dividir la secuencia y el modelo los dominios de forma individual (Figura 3).
- Descargue el formato PDB estructura de la secuencia los archivos de la alineación, haga clic en "Descargar Align" enlaces. Estos archivos de alineación se puede abrir en cualquier programa de visualización molecular que figuran en el apartado de material, y también se puede utilizar para agregar restricciones adicionales en el modelado de la estructura (Paso 1.4).
6.Análogos estructurales en el AP
- Ver la siguiente tabla (Figura 4) de la página de resultados para determinar los diez primeros análogos estructurales del modelo predijo por primera vez, identificados por el programa de alineación estructural TM-align 20. A TM-score> 0,5 indica que el analógico y el modelo han detectado una topología similar y se puede utilizar para determinar la estructura de clase / familia de proteínas de la proteína de la consulta 16, mientras que aquellos con TM-score <0.3 significa una similitud estructura al azar.
- Analizar la secuencia de identidad y RMSD en la región estructuralmente alineado muestra en 'IDEN a' y 'RMSD un "columnas para evaluar la conservación de los motivos espaciales en el modelo y la estructura análoga. Inspeccione visualmente los pares de residuos de color y se alinea en la alineación para identificar estos restos estructurales conservados y motivos.
- Haga clic en el código de AP se muestra en la columna de "Hit AP para visitar la página web RCSB y aprender más acerca de su clasificación estructural (SCOP, Cath y Pfam) y la información funcional (número CE, asociados GO términos y ligando).
7. Función de predicción utilizando cofactor
- Desplácese hacia abajo en la página de resultados para analizar las interpretaciones funcionales de la proteína de la consulta. Funciones de las proteínas se enumeran en tres mesas de contexto, que muestra: Comisión de Enzimas (CE) número, Gene Ontology (GO) los términos, y ligando sitios de unión.
- Ver el "TM-score", "RMSD a ',' IDEN a" y "Cov. columnas de cada tabla para analizar los parámetros de similitud estructura global y la conservación de los patrones espaciales entre el modelo y se identificaron homólogos funcionales (templates).
8. Comisión de Enzimas de predicción número
- Ver las cinco homólogos de la proteína enzimática potencial de consulta que se muestra en la "Predicción de números de la CE" mesa (Figura 5). El nivel de confianza de la predicción del número CE uso de estas plantillas se muestra en la "CE-Score" de la columna. Con base en benchmarking análisis de 23 de similitud funcional (3 primeros dígitos del número CE) entre la consulta y la proteína de la plantilla puede ser interpretado de forma fiable con CE-score> 1.1.
- Buscar el consenso de la función (números de la CE), entre las plantillas, que tienen el pliegue similar (es decir, TM-score> 0,5) como la proteína de la consulta. Si varias plantillas tienen el mismo número de CE y la puntuación> 1,1, el nivel de confianza de la predicción es muy alta. Sin embargo, si la CE-Score es alto, pero hay una falta de consenso entre los golpes identificados, entonces la predicción se vuelve menos confiable y los usuarios se les recomienda consultar las predicciones GO plazo.
- Haga clic en el enlace que aparece en los números de la CE para visitar la base de datos de la enzima ExPASy y analizar la función, incluyendo la reacción catalizada, los requisitos de co-factor y la vía metabólica de la proteína modelo en detalle.
9. Gene Ontology (GO) las predicciones de largo plazo
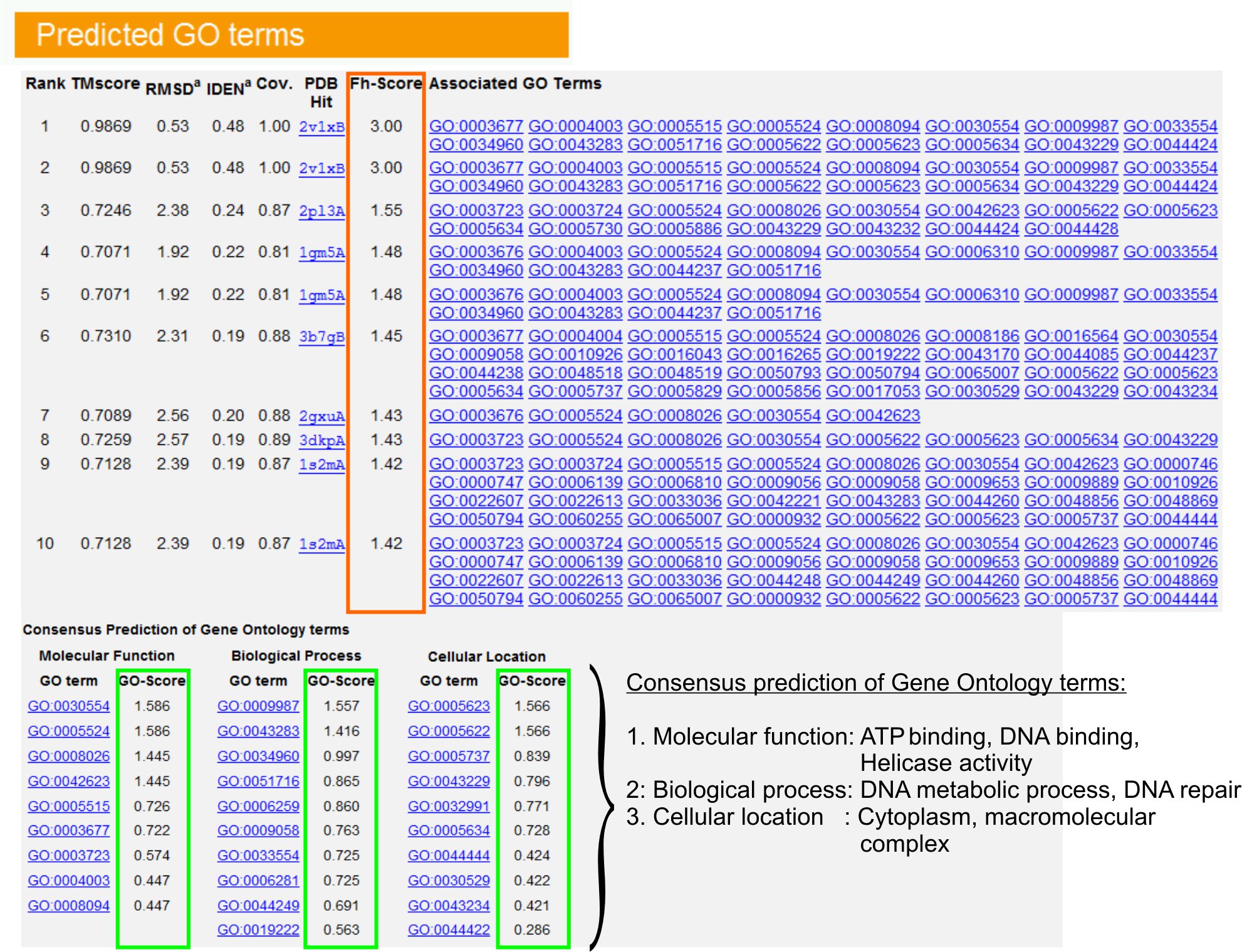
- Vea el tema "Predicción de los términos de GO" mesa (Fig.Ure 6) para identificar a los diez homólogos de la proteína de la consulta en la biblioteca de AP, anotado con Gene Ontology (GO) términos. Cada proteína se asocia generalmente con los términos de GO múltiples, describiendo sus funciones moleculares (MF), biológicos (BP) y los componentes celulares (CC). Haga clic en cada término para visitar la página web de Amigo y analizar su definición y su linaje.
- Analizar la columna Fh-score (puntuación de homología funcional) para acceder a la similitud funcional entre la consulta y las proteínas de la plantilla y estimar el nivel de confianza de la transferencia de anotación funcional de estas proteínas. En nuestro estudio comparativo, 23, 50% de los términos GO nativo podría ser correctamente identificados a partir de la primera plantilla identificados con un punto de corte Fh puntuación de 0,8, con una precisión del 56%.
- Ver el "consenso de predicción de términos GO" mesa para analizar la concurrencia de funciones entre las plantillas. Estas funciones comunes se utilizan para la predicción de los términos GO (MF, BP y CC) de la consultaproteína y evaluar el nivel de confianza (GO-score) de GO predicciones a largo plazo. Con base en la prueba de referencia 23, las mejores tasas de falsos positivos y falsos negativos obtenidos para las predicciones de corte IR-score = 0,5, con la disminución de la cobertura de la predicción de los niveles de la ontología más profunda.
10. Ligando-proteína predicciones sitio de unión
- Desplácese hasta la parte inferior de la página para ver diez principales predicciones del sitio de unión al ligando de la proteína de la consulta. Predijo sitios de unión se clasifican en función del número de conformaciones ligando predijo que comparten bolsillo de unión común. El mejor sitio de unión identificado ya aparece en la miniaplicación Jmol. Haga clic en los botones de radio para analizar otras predicciones y visualizar los residuos de la interacción ligando.
- Analizar la columna BS-puntuación para evaluar la similitud local entre el modelo y el sitio de unión de plantilla. Con base en el punto de referencia 9, BS-score> 1,1 indica la secuencia de alta y la estructura similarity cerca del sitio de unión previsto en el modelo y el sitio conocido obligatorio en la plantilla.
- Descargue el archivo PDB la estructura del complejo formato haciendo clic en el vínculo "Descargar". Los usuarios pueden abrir estos archivos en cualquier programa de visualización molecular y de forma interactiva ver el sitio predicho vinculante y las interacciones ligando-proteína en su computadora local.
11. Los resultados representativos

Figura 1 Un extracto de la página de resultados de I-TASSER muestra (A) FASTA formato secuencia de la consulta;. (B) predijo la estructura secundaria y los resultados asociados a la confianza, y (C) predijo disolvente accesibilidad de los residuos. Región analizada principal y el sitio de hidratación potencial en la consulta están resaltados en azul verdoso y rectángulos rojos, respectivamente.

Figura 2.

Figura 3. Un ejemplo de página de resultados de I-TASSER muestra los diez mejores plantillas de identificar hilos y las alineaciones por LOMETS 5 programas de threading. La calidad de las alineaciones de roscado se evalúa en función normalizada Z-score (resaltado en verde), donde un valor> 1 refleja una alineación segura. Residuos alineados en la plantilla que son idénticos a los residuos de consulta correspondiente se resaltan en color para indicar la presencia de residuos conservados / motivos, mientras que la falta de alineación en la mayoría de los plantillas de arriba indica la presencia de varios dominios en la proteína de la consulta y los residuos no alineados corresponden a regiones enlazador de dominio. Haga clic aquí para ver la versión en tamaño completo de la figura 3.

Figura 4. Un ejemplo de página de resultados muestra los diez primeros identificado análogos estructurales y las alineaciones estructurales, identificados por TM-align 20 programa de alineación estructural. El ranking de los análogos se muestra en la se basa en el TM-score (en azul) de la alineación estructural. A TM-score> 0,5 indica que las dos estructuras comparadas tienen una topología similar, mientras que un TM-score <0.3 significa una similitud entre dos estructuras al azar. Estructuralmente alineado de pares de residuos se resaltan en color sobre la base de sus aminoácidos de propiedad, mientras que las regiones no alineados se indican con "-".ove.com/files/ftp_upload/3259/3259fig4large.jpg "> Haga clic aquí para ver la versión en tamaño completo de la figura 4.

Figura 5. Un ejemplo de página de resultados de I-TASSER mostrando identificado homólogos de la enzima de la proteína de la consulta en la biblioteca de AP. El nivel de confianza de la predicción del número CE se analiza sobre la base de la CE-score (resaltado en verde), en el CE-score> 1,1 indica similitud funcional (lo mismo los 3 primeros dígitos del número CE) entre la consulta y la proteína de la plantilla.

Figura 6. Un ejemplo de página de resultados de I-TASSER mostrando GO predicciones plazo para la proteína de la consulta. Homólogos funcionales de la proteína de la consulta en la biblioteca de Gene Ontology plantilla se clasifican en función de su Fh-score (en el rectángulo naranja). Común las características funcionales de estos éxitos puntaje más alto-se derivan para generar se comió la última GO predicciones plazo para la proteína de la consulta. La calidad de la predicción de los términos de GO se calcula sobre la base de GO-score (en verde), donde un GO-score> 0,5 indica una predicción fiable. Haga clic aquí para ver la versión en tamaño completo de la figura 6.

Figura 7. Un ejemplo de página de resultados de I-TASSER mostrando diez principales predicciones ligando de unión a proteínas sitio utilizando el algoritmo de cofactor 9. El ranking de los sitios de unión prevista se basa en el número de conformaciones ligando predijo que comparten bolsillo de unión común en la consulta. BS-score (en rojo) es una medida de la secuencia local y la similitud entre la estructura prevista y sitio de unión de la plantilla, y es útil para analizar la conservación de los bolsillos de sitio de unión.
les/ftp_upload/3259/3259fig8.jpg "/>
Figura 8. Un ejemplo de moderación archivos externos utilizados para la especificación de residuos de residuos de contacto / restricciones a distancia.

Figura 9. Ejemplo de archivos de inmovilización utilizados para la especificación de una proteína de la plantilla en el servidor I-TASSER. El usuario puede especificar la alineación de consulta de plantilla, ya sea en (A) en formato FASTA, o (B) en formato 3D.

Figura 10. Un archivo de ejemplo que se usa para excluir a la plantilla durante el procedimiento de la estructura de I-TASSER modelado. La primera columna contiene la identificación del anteproyecto de presupuesto de las proteínas de la plantilla para ser excluidos. La segunda columna se utiliza para especificar el punto de corte de identidad de secuencia que se utiliza para otros modelos similares en la biblioteca de plantillas.
Discussion
El protocolo presentado es una guía general para la estructura y la función de modelado usando el servidor de I-TASSER. A pesar de que este procedimiento automatizado funciona muy bien para la mayoría de las proteínas, las intervenciones humanas a menudo ayudan a mejorar significativamente la precisión de modelos, especialmente para las proteínas que carecen de las plantillas de cerca en la biblioteca de AP. Los usuarios pueden intervenir en los modelos I-TASSER de las siguientes maneras: (a) separación de multi-dominio de las proteínas, (b) la prestación de restricciones externas para mejorar el montaje de la estructura, y (c) la eliminación de las plantillas durante el modelado.
Dividir multi-dominio de la proteína:
Muchas secuencias de proteínas de largo con frecuencia contienen múltiples dominios atados por la flexibilidad enlazador regiones, lo que hace su difícil elucidación de la estructura mediante técnicas experimentales y computacionales. Sin embargo, como los dominios son independientes plegables entidades y puede realizar la función molecular distinta, esconveniente dividir a largo multi-dominio de las proteínas y el modelo de cada dominio por separado. Dominios de modelado de forma individual, no sólo acelerará el proceso de predicción, sino que también aumenta la calidad de la alineación de consulta de plantilla, lo que resulta en una estructura más fiable y predicciones de la función.
Los límites del dominio de las secuencias de proteínas se puede predecir el uso de programas externos disponibles gratuitamente en línea, tales como NCBI CDD 24, 25 o PFAM InterProScan 26. Además, si las alineaciones LOMETS threading están disponibles para la proteína de la consulta, los límites del dominio puede ser localizado por la identificación visual de los residuos de largos tramos no alineados en las plantillas de rosca superior (ver Paso 5.4). Estas regiones no alineados en su mayoría corresponden a las zonas de dominio enlazador. Si varios dominios las plantillas ya están disponibles en la biblioteca de plantillas AP con todos los dominios de consulta alineados, entonces la consulta proteína puede ser modelado como de larga duración.
Proporcionar restricciones externas
A. Especificar contacto / distancia restricciones
Experimentalmente caracteriza residuos entre contactos / distancias, por ejemplo, a partir de RMN oentrecruzamiento experimentos, se puede especificar mediante la subida de un archivo de la moderación. Un archivo de ejemplo se muestra en la Figura 8, donde la columna 1, se especifica el tipo de restricción, es decir, "DIST" o "Contacto". A la moderación a distancia (DIST), las columnas 2 y 4 contienen posiciones de residuos (i, j), las columnas 3 y 5 contienen el átomo-tipos en el residuo y la columna 6 se especifica la distancia entre los dos átomos especificado. Para sistemas de retención (contacto), las columnas 2 y 3 contienen las posiciones (i, j) de los residuos que deben estar en contacto. La distancia entre el centro de cadenas laterales de estos pares de residuos de contacto se decidió sobre la base de las distancias observadas en estructuras conocidas en el AP. I-TASSER a tratar de sacar estos pares de átomos cerca de la distancia especificada en las simulaciones de la estructura de refinamiento.
B. Especificar una plantilla de estructura de proteínas
LOMETS programas de threading utilizar una biblioteca de AP representante para encontrar pliegues plausible para la prot consultaein. Aunque el uso de una estructura representativa de la biblioteca ayuda a reducir el tiempo necesario para calcular las alineaciones estructura de la secuencia, es posible que una proteína de buena plantilla se pierde en la biblioteca o la plantilla no han sido identificados por los programas de LOMETS threading, a pesar de que es presentes en la biblioteca. En estos casos, el usuario debe especificar la estructura de la proteína deseada en la plantilla.
Para especificar la estructura de la proteína como una plantilla adicional, los usuarios pueden subir un archivo de formato PDB estructura o especificar el ID de AP de una estructura de la proteína depositada en la biblioteca de AP. El I-TASSER va a generar la alineación de consulta de plantilla mediante el programa de Muster 23 y recogerá las limitaciones espaciales tanto de la plantilla especificada por el usuario y las plantillas LOMETS para guiar a la simulación de montaje de la estructura. Debido a la precisión de las restricciones LOMETS es diferente para diferentes objetivos, el peso de las restricciones LOMETS es más fuerte en fácil (homóloga) targets que en el disco duro (no homólogos) los objetivos, que han sido sistemáticamente sintonizado en nuestra formación de referencia.
Los usuarios también pueden especificar su propia plantilla de consulta alineaciones. El servidor acepta la alineación en dos formatos: el formato FASTA (Figura 9) y el formato 3D (Figura 9B). El formato de FASTA es estándar y se describe en http://zhanglab. ccmb.med.umich.edu / FASTA / . El formato 3D es similar al formato estándar PDB ( http://www.wwpdb.org/documentation/format32/sect9.html ), pero dos columnas adicionales derivados de las plantillas se agregan a los registros ATOM (véase la Figura 9B):
Columnas 1-30: Atom (C-alfa solamente) y los residuos de los nombres de la secuencia de consulta.
Columnas 31-54: Coordenadas de los átomos de C-alfa de la consulta copiada de los átomos correspondientes en la plantilla.
Columnas 55-59: número de residuo correspondiente en la plantilla sobre la base de la alineación
Columnas 60-64: nombre de residuo correspondiente en la plantilla
Excluir a las proteínas de plantillas
Las proteínas son moléculas flexibles y pueden adoptar varios estados conformacionales de cambiar su actividad biológica. Por ejemplo, las estructuras de las proteínas quinasas y muchas proteínas de membrana se han resuelto en la conformación de activos e inactivos. También la presencia o ausencia de ligando puede causar grandes movimientos estructurales. Si bien todos los estados de conformación de la plantilla son iguales para los programas de threading, es deseable que el modelo de la consulta el uso de plantillas en un solo estado en particular. Una nueva opción en el servidor permite al usuario excluir las proteínas de la plantilla durante el modelado estructura. Esta característica también permite al usuario elegir el nivel de homología de los modelos que se utilizarán para el modelado. Los usuarios pueden excluir plantilla de proteínas from la biblioteca I-TASSER por:
A. Especificación de un corte de identidad de secuencia
Los usuarios pueden utilizar esta opción para excluir las proteínas homólogas de la biblioteca de plantillas I-TASSER. El nivel de homología se establece en función del punto de corte de identidad de secuencia, es decir, el número de residuos idénticos entre la consulta y la proteína de la plantilla dividida por la longitud de la secuencia de la secuencia de la consulta. Por ejemplo, si el usuario escribe en el "70%" en la forma prevista, todas las proteínas de las plantillas que tienen una identidad de secuencia> 70% a la proteína de consulta I-serán excluidos de la biblioteca de plantillas I-TASSER.
B. Excluir las proteínas específicas de plantilla
Proteínas específicas de la plantilla puede ser excluido de la biblioteca de plantillas I-TASSER al cargar una lista que contiene los identificadores de AP de las estructuras a ser excluidos. Un archivo de ejemplo se muestra en la Figura 10. A medida que la misma proteína puede existir en varias entradas en la biblioteca de AP, I-TASSER síRVer de forma predeterminada excluye las plantillas especificadas (en la columna 1), así como todas las otras plantillas de la biblioteca que tienen una identidad> 90% de las plantillas especificadas. Los usuarios también pueden especificar una identidad de corte diferentes, por ejemplo, 70%, en todas las plantillas con la identidad de> 70% a las proteínas de la plantilla será excluido.
Disclosures
No hay conflictos de interés declarado.
Acknowledgments
El proyecto es apoyado en parte por la Fundación Alfred P. Sloan, Premio NSF carrera (DBI 1027394), y el Instituto Nacional de Ciencias Médicas Generales (GM083107, GM084222).
Materials
| Name | Company | Catalog Number | Comments |
| Material Name | Type | Company | Catalogue Number |
| FASTA formatted amino acid sequence of the protein to be modeled (see, http://www.ncbi.nlm.nih.gov/BLAST/fasta.shtml). | |||
| A personal computer with access to the internet and a web browser. | |||
| Molecular visualizing software, e.g. RASMOL or PYMOL, for analyzing the predicted tertiary structure and functional sites. |
References
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H. CD-Search: protein domain annotations on the fly. Nucleic. Acids. Res. 32, W327-W331 (2004).
- Finn, R. D. The Pfam protein families database. Nucleic. Acids. Res. 38, D211-D222 (2010).
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).

{kind=link}
{kind=link}