

Summary
细胞总RNA提供了一种用于研究的短期变化,以及在RNA合成和衰变动力学的RNA加工模板差。这里,我们描述遵循与4 - thiouridine的新转录的RNA的代谢标记通过硫醇特异性的生物素化和纯化的新转录的RNA,来克服这些限制。
Abstract
全转录组的芯片和新一代测序的发展已经彻底改变了我们的细胞基因表达的复杂性的理解。随着更好地理解所涉及的分子机制,底层动力学的精确测量变得越来越重要。在这里,这些有力的方法面临的主要限制,因为他们学习的模板样本, 即细胞总RNA的内在属性。在许多情况下,细胞总RNA发生变化,要么过慢或过快,代表潜在的分子事件和动力学有足够的分辨率。此外,RNA的合成,加工,和衰减的贡献改变不容易区分。
我们最近开发高分辨率基因表达谱来克服这些限制。我们的做法是根据新转录RNA代谢标记4 thiouri用餐(因此也称为4SU标记),然后通过严格的纯化的新转录的RNA,使用硫醇特异性的生物素和链霉亲和素包被的磁珠。它是适用于范围广泛的生物体包括脊椎动物, 果蝇 ,酵母或细菌。我们成功地应用于4SU标记实时动力学研究转录因子的活动,提供精确的测量RNA半衰期,到RNA加工的动力学,并取得了新的见解。最后,计算模型可以被用来产生一个综合的,全面的分析,潜在的分子机制。
Introduction
基因表达谱是一个关键的工具,用来研究细胞过程和相关的复杂的相互作用网络。 mRNA丰度的研究,通常的首选方法,以获得基本的洞察潜在的分子机制。全转录组微阵列1和,最近,新一代的RNA测序(RNA-SEQ)2-4的发展助长了这种方法。虽然这些技术已经彻底改变了我们的细胞基因表达的复杂性的理解,他们面临着由于细胞总RNA模板样本, 即内在性能的主要限制。首先,短期变动的总RNA水平不相匹配的转录率的变化,但本质上是依赖于各自的转录的RNA的半衰期。虽然五倍一个短命的成绩单, 如编码转录因子的诱导,将随时检测总RNA中在一个小时之内,同样的一项长期的成绩单, 例如编码一种代谢酶的诱导,仍将几乎看不见。另外,即使是一个完整的关机的转录率的平均基因的RNA的5个小时的半衰期(> 1000倍的下调)将简单地需要5个小时,它的总的RNA水平降低仅由两方面。因此,总RNA的分析有利于短命的转录调节的检测,其中有许多编码的转录因子和基因监管的功能5。此外,调节掩盖真正的动能级联和主要信号事件的不能从二级分化的。 ,反过来,可能会导致大量的偏见在下游的生物信息学分析。其次,总RNA水平的改变不能被改变RNA的合成或腐烂。后者需要测量细胞侵袭的方法, 例如阻断transcripti使用6,放线菌素D和扩展监控正在进行的RNA随着时间的推移衰减。平均mRNA的半衰期在哺乳动物细胞中的5 - 10小时5,7,大多数基因的mRNA水平的减少小于双重转录停止在几个小时后。在这些相当小的差异,导致在大多数由于基本的数学方程的指数性质的细胞基因的mRNA的半衰期非常不精确的测量。最后,当细胞总RNA的RNA-seq中透露,大约有一半是我们的基因选择性剪接事件,底层动力学以及动力机制仍然知之甚少,指导组织和上下文特定的调控RNA加工。此外,贡献差异表达基因的RNA加工,特别是对于非编码RNA,仍有待确定。总而言之,这些限制主要障碍生物信息学动力学建模相关的分子机制。
我们最近开发出一种方法,称为高分辨率基因表达谱,以克服这些问题5,7,9。它是基于使用4 thiouridine(4SU标记),一种自然发生的尿嘧啶核苷衍生物的新转录RNA代谢标记,并提供直接访问新转录转录以最小的干扰细胞生长和基因表达( 见图1)5, 10-12。曝光的真核细胞快速吸收4SU磷酸,磷酸化,并纳入新转录RNA 4SU结果。细胞总RNA的分离,4SU标记的RNA部分是巯基的特异性生物素化的生物素和新转录的RNA之间产生一个二硫键。 “细胞总RNA'然后可以进行定量分离成标记(”新转录“)和未标记的('预existin的克)RNA具有高纯度,使用链霉亲和素包被的磁珠。最后,标记的RNA回收从有孔玻璃珠,通过简单地增加裂解二硫键的还原剂( 如二硫苏糖醇)和释放从有孔玻璃珠的新转录的RNA。
新转录的RNA描绘每一个基因的转录活性4SU曝光的时段内。 4SU 标记分钟的时间表,从而提供了一个快照图片真核生物基因表达和理想的模板,下游的生物信息学分析( 如启动子分析)。在稳态条件下可以假设的情况下,新转录/总的比例,新转录/未标记的未标记/,总RNA提供非侵入性的访问精确RNA半衰期7,13。此外,重要的是要注意4SU标记只需5分钟(5分钟4SU RNA)纯化后,新转录的RNA是年龄小于15分钟和60分钟4SU-RNA。当进行超短逐步不再4SU标记的RNA-seq的结合在一个单一的实验设置,RNA加工的动力学透露核苷酸分辨率9。最后,时间过程分析与计算模型相结合的新的转录总RNA允许RNA合成的综合分析和衰减14。
总之,该方法可以直接分析RNA的合成,处理,并在真核细胞中的降解动力学。这是适用于所有主要模式生物,包括哺乳动物,昆虫( 果蝇 ),两栖类( 爪蟾 )和酵母5,15,16。这是直接兼容微阵列分析5,17,9,13,14 RNA定序,适用于体内 12,15。在这里,我们详细的方法,在培养的哺乳动物细胞中的标签,隔离和净化新转录RNA。此外,电位人的问题和缺陷进行了讨论。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1。代谢标记4 thiouridine
实验装置的详细计划/调度, 例如 ,当添加4SU细胞培养和收获样品。计划在每个条件之间的至少5分钟。只有一个条件处理细胞的时间。处理最大。在一个给定的时间3 - 5个菜。尽可能快地处理细胞,以尽量减少在温度和CO 2水平的变化。避免明亮的光线后的细胞暴露4SU增加,因为这可能导致4SU标记的RNA的细胞蛋白的交联。
标签开始
- 解冻4 thiouridine(4SU)之前使用和吸管4SU每个条件进入无菌猎鹰管所需的金额。
- 以细胞培养液(5毫升每10厘米的培养皿)关闭菜肴所需金额,并添加含4SU猎鹰管调匀。取出并丢弃菜的剩余介质。 <LI> 4SU含介质倒流的菜肴。
结束标记
- 除去细胞的培养基从细胞中。加入5毫升Trizol法时,每盘。对于复杂的实验,其中包括多个时间点或条件,这一步最好由两个人,除去培养基,加入Trizol试剂和收获的溶胞产物。
- 完整的细胞裂解在室温下孵育5分钟。
- 使用10毫升吸管仔细冲洗板块,与增加的Trizol。这有助于完整的细胞裂解样品回收率。小心轻放的Trizol在与皮肤或眼睛接触时,是非常危险的!有苯酚烧伤手的解毒剂( 如聚乙二醇300 400在工业甲基化酒精(70:30))。将样品转移到聚丙烯管中。请注意标准猎鹰管不抵抗这些高克部队)。样品可以储存在-20℃下至少1个月,直至总RNA中,我准备投入战斗。
2。 RNA制备使用改良的Trizol协议
- 加入1毫升氯仿(0.2毫升,每毫升Trizol法)和剧烈摇晃,持续15秒。在室温下孵育2 - 3分钟。
- 在13000×g离心15分钟,在4℃下
- 水的上层相(含有RNA)转移到一个新的15毫升的聚丙烯管中。
- 添加½两个RNA沉淀缓冲液和异丙醇的反应体积( 例如,3毫升上清液加1.5毫升的RNA沉淀缓冲液和1.5ml异丙醇)。
- 拌匀。在室温下孵育10分钟。
- 在13000×g离心10分钟,在4℃下弃上清。
- 后短暂离心(5000×g为30秒)和200微升移液器去除残留的异丙醇。
- 加入等体积的75%乙醇中,并摇动试管,直到颗粒分离。避免打破它成很多小块,因为这可能使残留物清除的L乙醇困难。
- 在13000×g离心10分钟,在4℃下弃上清。
- 简要降速RNA并取出剩余的乙醇200μl的吸管。重复步骤和除去剩余的乙醇与20微升吸管。这两个步骤之后,没有进一步的干燥沉淀应该被执行。
- 加入100μl每股H 2 O 100微克RNA产量预期,并拌匀上下吹打5 - 6次,以帮助溶解RNA。
- 溶解和变性的RNA通过加热到65℃下进行10分钟(振动筛),并立即在冰上放置。
- 测量RNA浓度使用NanoDrop分光光度计在260 nm处,按照制造商的说明。这种RNA可以储存在-80℃下至少1个月。
3。巯基新转录RNA的特定的生物素化
- 开始60 - 80微克的细胞总RNA。
- 组成贴标反应。以下吸管为了(每微克RNA):
- 1微升10X生物素缓冲区
- 7μlRNA提取(含1微克RNA稀释于无核酸酶的H 2 O)
- 2微升(1毫克/毫升DMF)生物素-HPDP
随时添加的生物素HPDP的最后,并立即混合吹打。的情况下,析出物的生物素,二甲基甲酰胺的含量可以增加至终浓度为40%。
- 在室温下孵育1.5小时,用旋转。
- 加入等体积的氯仿。大力混合。孵育2 - 3分钟,直到相开始分离,气泡开始消失。
- 在20000×g离心5分钟,在4℃下小心转移到一个新的管中的上层水相。
- 重复一次步骤3.4和3.5。您可能要执行此步骤2毫升锁相凝胶重型管,以减少损失的RNA。
- RNA沉淀:加入1/10体积的5 M氯化钠和等量的异丙醇到水相中。
- 在20000×g离心20分钟,在4℃下弃上清。
- 加入等体积的75%乙醇,离心,在20,000×g下在4℃下10分钟,弃去上清液。
- 短暂离心,用200微升吸管,去除残留的乙醇。
- 短暂离心,去除残留的乙醇与20微升吸管。
- 不要让RNA干。重新暂停在50 - 100微升H 2 O(〜1微升,每1微克输入RNA)。拌匀上下吹打5 - 6倍。
- 检查RNA质量通过electrophoretical分析的排除RNA降解。
4。斑点杂交分析4SU成立(可选)
4SU掺入可以很容易地由点生物素标记的RNA印迹分析。这是一个可选的步骤,允许4SU的掺入率相对生物素标记的DNA寡控制的故障排除和估计。这个实验我们recommend使用碘代乙酰基-生物素,而不是生物素-HPDP 4SU标记的RNA在步骤3.2中的生物素化。这将导致在一个不可逆的生物素化4SU-RNA。因此,基于列的方法( 例如 RNeasy试剂)可以用于恢复较少量的生物素化的RNA( 例如,5微克)。虽然RNA的生物素化使用生物素-HPDP也适于该测定的,所得到的信号弱,信噪比逊于( 图3)。
- 按照协议4SU细胞总RNA中所描述的第1和第2的标签和隔离。
- Biotinylate 4SU标记的RNA中所描述的更换生物素HPDP的第3与碘代素和执行两个氯仿抽提完全除去过量的碘代生物素残留。
- 异丙醇/乙醇沉淀恢复生物素化的RNA或使用基于列的方法( 例如 RNeasy试剂)的情况下,少量的RNA(<10微克)被使用。
- 孵育的泽塔膜在无核酸酶水10分钟摇动。
- 无核酸酶水出膜,将膜之间两个干净的纸巾用力按压,除去过量的液体。空气中干燥5分钟的膜,将导致更好的点。
- 对于每个样品,准备20μl的200纳克/μlRNA提取,用冰冷的斑点印迹结合缓冲液中(10mM氢氧化钠,1mM EDTA)中。应用5微升稀释( 即 1微克RNA),以及三个连续稀释10倍( 即 100,10,1毫微克RNA),通过移液的Zeta膜。通过一个空架移液器枪头可以提供均匀分布的间距。或者,使用斑点印迹装置,根据制造商的指示。
- 中应用5μl的生物素标记的DNA寡核苷酸的浓度范围从20毫微克/微升至20皮克/微升( 即 100将0.1 ng寡)作为阳性续ROL通过移液膜。使用生物素,4SU天真的样品作为阴性对照。
- 空气中干燥5分钟的膜。
- 孵育30分钟,在40ml封闭缓冲液中与摆动膜。
- 温育膜15分钟1:1,000链霉抗生物素蛋白 - 辣根过氧化物酶(5毫升PBS + 5毫升20%SDS +加入10μl链霉抗生物素蛋白 - 辣根过氧化物酶与10毫升)
- 在40毫升PBS + 10%SDS(20毫升PBS + 20毫升20%SDS)中洗膜两次,持续5分钟。
- 在40毫升PBS中洗膜两次+ 1%SDS(38毫升PBS和2毫升20%SDS),持续5分钟。
- 洗膜两次,在40毫升PBS + 0.1%SDS中(40毫升PBS +加入200μl20%SDS),持续5分钟。
- 除去过量的流体通过将膜之间两个干净的纸巾,并牢牢按住它们。
- 可视化膜结合HRP使用ECL每制造商的指示。
- 放置在塑料箔膜/袋,去除气泡和孵育2分钟,在黑暗。
- 公开膜影片1 - 5分钟。
5。标记和未标记的RNA链霉亲和素磁珠分离
- 热清洗缓冲液(3毫升每个样品)至65℃的水浴中。
- 准备新的100毫米二硫苏糖醇(DTT)在无核酸酶H 2 O做滗15 - 30毫克的DTT粉放入干净的50毫升猎鹰管上放置超精细的规模。称重,并添加所需金额无核酸H 2 O
- 热生物素化的RNA样品,以65℃下变性10分钟,并立即在冰上。
- 地点列μMacs成磁性立场。我们建议不要处理时间超过12个样品(6 - 8个样品是最优的)。
- 有预平衡美天旎列1毫升室温洗涤液。这将需要约15分钟。
- 同时,加100微升50 - 100μl的生物素标记的RNA链霉亲和素珠。在室温下孵育15分钟,用旋转。 < LI>如果还没有发起任何列排水到现在,这可以促进用戴手套的手指轻轻按下顶部的列。一旦流量已经开始列容易流失。
- 应用的RNA /有孔玻璃珠的列。丢弃的流量通过,除非你想收回未标记的RNA组分(见第7节)。
- 洗涤三次,用0.9毫升的65℃下洗涤缓冲液(1毫升移液吸头加样缓冲液,在65℃时,收缩)。
- 0.9毫升室温下洗涤缓冲液洗三次。
- 吸取700微升缓冲RLT(Qiagen公司的RNeasy MinElute清理套件,)进入新的2毫升管,并把它们列下方。
- 新转录的RNA进入RLT缓冲液洗脱,通过添加100μl的100mM DTT的列。
- 执行第二次洗脱轮3分钟后到同一管加入100微升100毫米的DTT。
6。新转录的RNA恢复
ontent“>继续的RNeasy MinElute清理协议(QIAGEN)按照制造商的说明25无核酸酶H 2 O的测量RNA浓度洗脱使用NanoDrop分光光度计,为了避免提交前需要解冻和重新冻结RNA高通量的检测,我们推荐的紧接之后的新转录的RNA制备的cDNA的纯化。2.5微升新转录的RNA的在20微升cDNA的合成混合用于cDNA合成按照制造商的说明使用。执行实时定量RT-PCR法用1控制:10稀释的cDNA混合。专区RNA在-80℃下7。恢复未标记的未绑定RNA(可选)
未结合的RNA的情况下,需要恢复的流量通过收集和组合(列后,加入RNA的链霉抗生物素蛋白微球溶液),进行后续沉淀的第一次洗涤。一般是足够的,只有50%的未结合的RNA沉淀氏s将含有> 80%的原料。
- 加入等体积的异丙醇(无盐需要添加洗涤缓冲液含有1M NaCl的)。
- 在20000×g离心20分钟,在4℃下弃上清。
- 加入等体积的75%乙醇,离心,在20,000×g下在4℃下10分钟,弃去上清液。
- 短暂离心,用200微升吸管,去除残留的乙醇。
- 短暂离心,去除残留的乙醇与20微升吸管。
- 不要让RNA干。重悬在100微升H 2 O的拌匀上下吹打5 - 6倍。孵育在65℃下振摇10分钟,并直接传送到冰。
- 检查RNA质量通过electrophoretical分析的排除RNA降解。
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
1。原料,预期产量
经过1小时(小时)4SU曝光新转录RNA约占1 - 4%的细胞总RNA。这将是较低的,因为他们生长被捕细胞不再合成RNA占细胞生长/复制。贴标签时1小时,我们建议60 - 80微克总RNA开始检测。小于30微克总RNA的小RNA颗粒生物素化步骤,是很难看后开始,因此可以很容易丢失。输入RNA水平可能会增加多达150微克的持续时间很短标签( 如 5 - 10分钟)。当RNA标记的持续时间缩短至1小时至5分钟的贡献短命的内含子序列在新转录的RNA增加〜60%〜80%9。由于内含子相比相当长的时间的量的编码序列,以及5'-和3'-非编码区,新转录RNA,它可以净化短期甚至是超短期4SU标记后,不会直线下降。因此,我们得到的> 0.5%的总RNA后,5分钟4SU标记在非贴壁的人B细胞株9。然而,它应该指出4SU稍长标签的持续时间,较高浓度可能会被要求实现类似4SU在贴壁细胞中的掺入率。虽然即使是低4SU掺入率将允许高效地捕捉和纯化大量,尿苷丰富的成绩单,很短的转录与尿嘧啶核苷含量低( 如 miRNA的)有可能逃脱净化即使使用4SU高浓度(> 1毫米)。在NIH-3T3小鼠成纤维细胞,1小时200微米4SU曝光新转录RNA标记4SU残留每50 - 100个核苷酸(NT)5。这应该让成绩单> 500 - 1000个核苷酸长度的高效回收。因此,我们只观察到轻微的成绩单大小偏置时1小时,使用200微米4SU在两个小鼠成纤维细胞和人类B细胞标记。虽然没有造成任何重大改变细胞转录水平在小鼠成纤维细胞,细胞长期暴露≥200微米4SU 1小时为200微米4SU导致可测量的增长在24小时内(未发表资料)的赤字。因此,这两个标签和经营4SU浓度的持续时间应尽量减少,以避免异位或毒性作用。一个简单的方法来确定最小4SU所需的高效回收新转录RNA浓度4SU浓度的增加( 例如:50 - 1600微米),是净化新转录RNA 4SU标签。如示于图2A和2B,标记为1小时,在原代人成纤维细胞中的新转录的RNA的回收大大提高,从50到200μM4SU,然后开始高原。
2。点吸干量化4SU团(可选)
在某些情况下可能会感兴趣4SU掺入量来衡量的总RNA中。最好的做法是使用生物素标记的RNA链霉亲共轭点印迹分析。由于其化学性质碘代乙酰基-生物素4SU残基几乎所有的新转录的RNA中的生物素化的生物素-HPDP硫醇组比更具有反应性。重要的是要注意,碘代乙酰基-生物素如生物素HDPD,不溶于水,从而有效地除去氯仿萃取法进行生物素-HPDP。因此,相同的反应条件和浓度可被用作在使用生物素-HPDP。然而,碘代乙酰基-生物素是不可逆的。因此,它可以用于在基于列的方法纯化的新转录的RNA。虽然使用碘代生物素可以量化注册4SU,基于生物素HPDP的测量同时考虑4SU注册成立及生物素化效率。采用两个生物素化试剂,以相同的采样允许4SU RNA注册的生物素化的效率的测量。 4SU 标记的RNA的生物素化的生物素-HPDP效率似乎是小于新转录的RNA,大约只有一个在三个4SU残基实际上是生物素化的生物素-HPDP( 图3)的碘乙酰基-生物素约三倍。通过比较DNA寡核苷酸的生物素化的对照样品的信号强度,生物素化的密度可以被测量。对于大多数的哺乳动物细胞系应该仍然是一个积极的信号在10毫微克生物素标记的RNA后1小时200微米4SU标签检测。疲软的背景信号通常是检测到的最高浓度(1微克)的未标记的RNA。
3。新转录RNA纯化
新转录RNA的恢复是非常全titative。如果具有相同的RNA浓度开始,对所有样品,你可以期望获得相同量的新转录的RNA。像许多基于列的检测,收集的新转录的RNA,用RNeasy MinElute试剂盒可能会导致额外的吸收在230 - 260纳米(洗涤剂的洗涤缓冲液是来自于存在),这可能会干扰OD 260测量。这被认为是在较小的程度时,每个离心步骤中使用新鲜的2 ml收集管。然而,任何不合理的高外径测量(> 2倍大于其它样品)应谨慎考虑,特别是如果OD 260/280比值<1.7。对于向下流的分析,因此,它是通常最好将所有的样品使用相同的模板RNA量的体积。在情况下,标记的RNA的产量低于预期支票RNA降解的迹象通过electrophoretical分析。新转录的RNA包含显着更大的金额大,拼接的成绩单典型rRNA的波段不太突出( 图4)。
4。新转录RNA定量
最后,在新转录RNA 4SU掺入率可以直接量化的分光光度分析的基础上的最大吸收峰在330 nm处的的OD 二百六十〇分之三百三十〇比5,18 4SU。这就要求在小体积(10 - 20微升)的异丙醇/乙醇沉淀浓缩的标记的RNA的3微克。为了避免丢失小RNA沉淀共沉淀用30微克无核酸糖原(Fermentas公司,#R0551)应该执行。在330 nm处反射到新转录的RNA( 图5)的掺入率4SU一个额外的峰是可见的。
/ files/ftp_upload/50195/50195fig1highres.jpg的“/>
图1。代谢标记与4 - thiouridine的4SU原理。4SU被添加到细胞所需的时间(5 - 120分钟),然后通过制备细胞总RNA。巯基的特异性生物素化后,被分离成细胞总RNA标记4SU,新转录的RNA,和未标记的,预先存在的RNA使用链霉亲和素包被的磁珠。新转录的RNA恢复使用还原剂,它劈开的二硫键连接新转录RNA的珠子珠。 点击这里查看大图 。
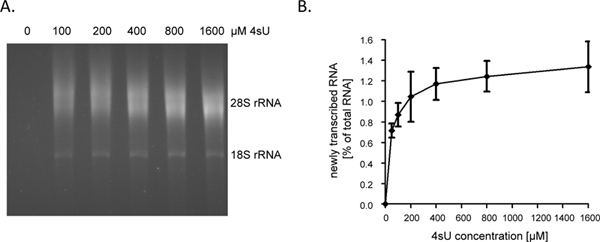
图2。恢复后新转录RNA 4SU 浓度的增加(A)主人包皮成纤维细胞(HFF)孵育100,200,400,800或1,600μM4SU。新转录的RNA纯化,从50微克总RNA,并进行electrophoretical分析。正如预期的那样,在恢复新转录的RNA浓度依赖性增加观察开始高原在较高浓度(B)应付新转录RNA纯化量化使用ImageJ的1.45s软件。新转录RNA的金额合并四个独立的实验数据恢复4SU标签不同浓度的范围为50 - 800微米4SU组(n = 2)或100 - 1,600μM4SU组(n = 2)所示。 查看大图 。
upload/50195/50195fig3.jpg“ALT =”图3“FO:内容宽度=”4.5英寸“FO:SRC =”/ files/ftp_upload/50195/50195fig3highres.jpg“/>
图3。 4SU 载入4SU标记的总RNA,用点印迹分析的估计中分离总RNA,从NIH-3T3小鼠成纤维细胞或培养的人包皮成纤维细胞(HFF)与200μM的4SU一小时。没有4SU被添加到一个菜作为阴性对照。对于HFF两个接触抑制组(n =非生长细胞)生长的细胞(Y)都包括在内。分离RNA使用Trizol试剂,随后结合到生物素-HPDP或碘代乙酰基-生物素。每个样品的浓度调节至200毫微克/微升和5微升在此稀释液中( 即 1微克RNA),以及三个连续稀释10倍( 即 100,10,1毫微克RNA),均点在一块泽塔膜。 5微升稀释的生物素标记的DNA寡核苷酸作为阳性对照concentrat膜被放置在离子为20 pg /μL的( 即 100至0.1纳克),分别从20纳克/微升。生物素密度探测用链霉亲和素 - 辣根过氧化物酶缀合物。
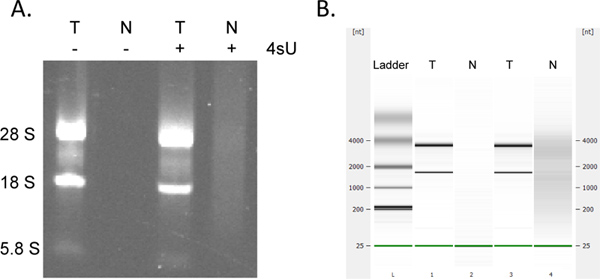
图4。 4SU 500μM的存在和不存在在培养1小时后,通过琼脂糖凝胶电泳分析小鼠NIH 3T3成纤维细胞(A)制备的总RNA(T)和新转录的RNA(N)的新的转录总RNA。electrophoretical分析 (以相同的顺序)使用Agilent Bioanalyser的(B)。没有回收RNA没有4SU细胞治疗。纯化新转录的RNA含有更大量的超高分子量的mRNA和明显不太成熟的核糖体rRNA比总RNA 28S,18S,5.8S rRNA的波段之间显着。 点击这里查看大图 。
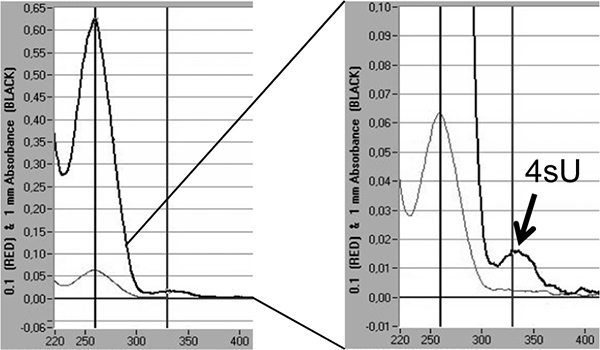
图5。 4SU 纳入新转录RNA定量分光光度分析新转录RNA纯化2×100μg总RNA后1小时为200微米4SU NIH-3T3小鼠成纤维细胞。新转录的RNA用异丙醇/乙醇沉淀后,加入30μg的无核酸酶的糖原。示出得到的新转录的RNA用的NanoDrop 1000分光光度计分光光度分析。浅灰色线代表测量为0.1毫米,而较厚,深灰色线代表在1毫米液柱测量。在右边,放大倍率为代表吨的高峰期灭绝他纳入4SU残留。灭绝基于高效4SU 18 4SU掺入率可估计。
| 时间标签[分钟] | 推荐4SU浓度[μM] |
| 120 | 100 - 200 |
| 60 | 200 - 500 |
| 15 - 30 | 500 - 1000 |
| <10 | 500 - 2000 |
表1中。推荐4SU浓度。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
新转录RNA代谢标记大幅提高电源的高通量技术,如芯片和RNA-SEQ提供更合适的模板来解决生物学问题的兴趣。本议定书进行了广泛的优化。它允许新转录RNA> 1000倍浓缩,并提供高度可重复性的结果。
4SU标记实验的实验设计是至关重要的新转录的RNA,将描绘仅在4SU细胞的曝光时间的实时的转录活性。如果刺激后转录率的实际变化已经消退,这些都将错过新转录RNA进行分析时,即使总RNA水平的变化仍可能检出。因此,良好的基础生物学的理解是非常重要的实验装置以及最佳时期Ø定义F时间4SU曝光。下面,我们提供建议,以及如何避免常见的陷阱,最关键的步骤。
股票的解决方案和塑料制品的制备
必须准备好所有股票的解决方案,使用无核酸酶水。在内部使用纯净去离子水,可能会造成问题,如果水中含有还原剂。在一个案例中,这导致完全丧失所有标记的RNA。因此,我们强烈建议购买预先无核酸的NaCl的Tris-CL,EDTA,柠檬酸钠和水。在任何时候都确保无核酸的条件。二甲基甲酰胺(DMF)溶解一些塑料材料。我们发现,用25毫升细胞培养塑料移液器转移DMF其股票玻璃瓶50毫升猎鹰管,到准备生物素HPDP的原液足以大幅降低新转录RNA的产量从整体上检测。有趣的是,这并不会受到负面影响的生物素通报BULLETIN效率(如测试通过斑点杂交法),但导致75到90%的损失可以从珠收回的新转录RNA。标签的持续时间从60个减少到30分钟或更少的亏损最为显着。最有可能的是,塑料移液管从由二甲基甲酰胺洗脱的物质部分被毁的链霉抗生物素蛋白的有孔玻璃珠的涂层。因此,通过各种手段,应避免使用塑料材料,不知道是用DMF兼容。出于同样的原因,不应该被用来刮细胞恢复的提升Trizol试剂样品,从细胞培养板。有趣的是,要注意的是推定的物质,从塑料洗脱DMF或Trizol试剂显然既不除去氯仿抽提,也不异丙醇/乙醇沉淀。
细胞培养
平板上的细胞密度是至关重要的。在一项实验中,细胞似乎是稍微太汇合(90 -100%),30分钟,100 U / ml的干扰素(IFN)治疗NIH-3T3小鼠成纤维细胞α或γ。在以下汇合的细胞已经导致干扰素治疗甚至15分钟的5 -至8倍的诱导基因IRF1或组socs3 5等。随着细胞略有太多融合的微阵列分析,并无发现任何诱导干扰素诱导基因,即使是最迅速的IRF1或SOCS3等诱导的基因。因此,细胞密度4SU标记实验和所有的细胞培养板标签开始前,应仔细检查是一个关键因素。
4SU是一个光活化的核苷4SU含RNA有效地交联蛋白质后暴露于365纳米光源。 4SU处理的细胞应培养在黑暗中暴露在强光下应尽量避免。搬迁后的细胞蛋白质的Trizol RNA分离这种风险大为降低。
<P类=“jove_content”> 4SU没有纳入到细胞DNA中。然而,它应该被注意的是,总RNA仍然会包含少量的细胞的DNA。当使用4SU标记和Q-RT-PCR分析研究病毒基因的表达,巨细胞病毒感染,我们发现有必要包括协议中的DNA酶I消化步骤以删除concatemeric的病毒基因组的19。这可能不是必要的时候使用下游DNA的存在是不敏感的协议。4SU掺入率和最佳浓度4SU
4SU 很容易被细胞内和细胞外的水平内平衡最有可能不到一分钟9,16。 4SU吸收和掺入率呈浓度依赖性。因此,可以方便地调节4SU浓度根据就业期限标签。 表1提供咨询4SU浓度相对通报BULLETIN标签我们最好的个人经验的基础上的持续时间。在哺乳动物细胞中的4SU标签1小时,200μM4SU足够对于大多数应用程序,导致约一4SU残余物,每50至100个核苷酸的新的成纤维细胞中的转录的RNA。
在过去几年中,我们已经4SU标记应用到广泛的人类和小鼠的起源,包括成纤维细胞,内皮细胞,上皮细胞,骨髓基质细胞,巨噬细胞和T细胞的细胞类型。此外,成功地用于从果蝇和爪蟾的细胞。 4SU掺入在所有这些实验中,被认为是高效4SU浓度为不同类型的细胞需要最低限度的调整。当设置了新的类型的细胞的方法,我们推荐在标记细胞的增加4SU浓度( 例如,从50到1600μM),分析纯化的新转录的关系ŗNA的应用4SU浓度(参见图2A / B)。 4SU浓度的纯化的新转录的RNA的量进入高原应选择。
高度融合,接触抑制细胞的情况下,我们会建议使用4SU浓度略高( 如:500,而不是200微米),以确保有效的4SU成立。此外,在很短的新转录的转录捕获(200个核苷酸)的情况下,是特别感兴趣的,4SU浓度也可能需要增加。这不应该被结合延长标记时间( 例如 1小时),以避免异位作用或毒性。最后,我们发现,使用量太少细胞培养基4SU掺入率可能会降低。因此,我们建议使用10厘米或15厘米的培养皿中每5毫升或10毫升,分别。
细胞总RNA的制备
如果在此协议的成功关键是要获得干净,无RNA酶的细胞总RNA。使用5毫升的Trizol每15厘米的菜干净RNA核酸。我们建议使用修改后的Trizol协议ChomczynskiP等等 。20。首先,它更适合于隔离坚实的粒料,在洗涤步骤,更容易处理增强的离心力导致的大的RNA量(> 100微克)。然而,这需要使用特殊的聚丙烯管和适配器定期15毫升实验室猎鹰管不生存超过6000×g下。其次,它提高了DNA和糖蛋白的去除。准备RNA器官或组织时,这一点变得尤其明显。第三,它不限制的最高金额可以分离出总RNA。虽然我们还发现基于列的RNA提取方法( 例如的RNeasy)提供RNA相适应的质量,标准列ARË只能够捕捉高达100微克总RNA,从而限制量的起始原料。最后,通过除去剩余的乙醇,用移液管的两倍,干燥的RNA,以去除残留的乙醇,不再需要。这消除了过度干燥的RNA,这可能是难以再溶解之后的风险。 4SU 标记的原则,适用于体内 , 例如,通过静脉注射小鼠。然而,我们注意到,RNA的纯度是一个重大的问题,需要净化波利亚转录纯化前新转录RNA(未发表资料)。
生物素和去除未结合的生物素
生物素-HPDP硫醇特异性为100%,生物素基和硫醇的新标记的RNA转录的分子之间形成二硫键。 4SU 标记的RNA的生物素化效率为30%左右,由墨点分析5。由于生物素-HPDP不溶于水它可以被有效地除去,用氯仿萃取。虽然一个单一的氯仿萃取步骤是我们足以除去未结合的生物素绝大多数定期重复此步骤,以确保彻底清除。 RNA的损失,以减少在氯仿提取步骤2毫升锁相凝胶重管(Eppendorf)中,按照制造商的说明,也可以使用。通常我们使用锁相管只为第二氯仿抽提步骤的第一步往往是模板量过高,这些管子直接兼容。除去未结合的生物素HPDP,RNA是由异丙醇/乙醇降水的恢复。重要的是要注意,商业的基于列的试剂盒回收生物素化的RNA( 例如从QIAGEN公司的RNeasy)不应该被使用,因为它们包含在所提供的缓冲液中,还原剂和去除生物素的新转录的RNA裂解二硫键。
纯化新LY转录RNA
不要添加超过100μL生物素标记的RNA到100μL链霉亲和素珠。添加量较少首选。然而,对所有样品,应添加相同体积的RNA。调节的RNA输入音量(样本)之间添加的链亲和素珠,通过简单地增加所需体积的1X TE珠。一个简单的方法,使新鲜无核酸100毫米DTT倒出成猎鹰管放置在一个超灵敏的规模有足够量的DTT粉,然后加入所需量无核酸H 2 O产生100毫米的DTT (64.8μl水每1毫克DTT)。
在4SU标记的发展过程中,我们测试了来自不同供应商的链霉亲和素珠。其中一些产生大量的背景。因此,我们强烈建议您使用美天旎链霉亲和素珠,到目前为止,我们从来没有经历过任何问题,结转未标记的RNA从组织c油菜栽培源性的RNA样品。以这种方式,只需在150 ng的标记的RNA可以具体从150微克生物素化的RNA(在100μl的水)纯化,用100μl的链霉亲和素磁珠。与有孔玻璃珠提供,用平衡缓冲液平衡的珠子可以执行可能略有提高捕获速率13。
质量控制
我们建议之前执行Q-RT-PCR检测控制新转录RNA受到高通量分析。这可能包括一些已知的参考基因差异调节在给定的实验设置的量化。 4SU 标记研究RNA衰变率的情况下,我们会建议一个短命的谈话内容( 如 myc基因,FOS)和长寿命( 如 GAPDH)总额和新转录RNA量化。新的转录/总RNA的比例应该是相当高的(约5 - 至10 - 倍)为短命的成绩单。基于RNA的半衰期为参照基因,RNA的半衰期可以被确定。如果所有三个RNA级分(总RNA,新转录的RNA和未标记的预先存在的RNA)分析为四个或更多的基因,归一化的不同的RNA的子集,可以通过线性回归分析和质量控制分数可以被确定为7描述,21。
Q-RT-PCR分析,我们建议使用2.5微升标记的RNA的20μLcDNA合成的混合。为了获得最佳的Q-RT-PCR结果的比较冻结的cDNA,在第一次使用前的5微升的等分试样。管使用前刚刚解冻,加入45微升的H 2 O和受5微升的稀释Q-RT-PCR分析。这大大提高了不同的PCR扩增运行之间的可比性。
新转录的RNA样品应RNA降解的使用安捷伦Bioanalyser的的迹象之前对他们进行检查高通量分析(微阵列或RNA-SEQ)。然而,它应该被注意的是,有时可观察到由安捷伦Bioanalyser的额外条带。的生物学意义尚不清楚。新转录的RNA含有核糖体RNA,这些样品偶尔失败的安捷伦Bioanalyser的质量控制。如果这不是由于可见RNA降解样品可接受的质量通常是细要进行高通量分析。
新转录RNA的兼容性与下游分析
新转录的RNA含有比总RNA,mRNA的大幅度增加。这主要是由于大量的内含子序列在新增加4SU标记的持续时间被缩短时,转录的RNA。因此,我们不定期进行新转录的RNA样品的核糖体rRNA耗尽,因为这需要大量的起始材料,同时提供而少(双重)增益非rRNA的读取。最后,我们仍然未拼接的,高分子量的成绩单新转录的RNA中存在的较大百分比的要注意的是,可能需要额外的碎片在新一代测序制备cDNA文库时。因此应仔细质量控制结果的大小碎片步骤。
RNA半现场测量的数据正常化
正常化RNA半衰期测量的实验数据的标准方法是正常化的所有数据的RNA以及其特征在于看家基因的半寿期或中位数的RNA的半衰期在以前的实验确定在给定的信元类型。在哺乳动物细胞中,后者位于在6,7 5分至10小时的范围内。虽然这种方法也能使很好地4SU为基础的测量,以及其他装置,用于归一化是必需的,如果中值RNA的半衰期不知道,或者如果它毫安Ÿ甚至会受到改变下的蜂窝系统研究, 例如击倒RNA降解途径。 4SU 标记提供了一个独特的方式推定的中位数的RNA根据所有三个RNA成分, 即总细胞RNA,新转录的RNA,和未标记的预先存在的RNA的分析半衰期。作为细胞总RNA被分离成一个简单的线性回归模型,可以采用彼此三个RNA成分正常化,并确定中位数RNA半衰期7,16后两者的RNA级分。软件包可在网上进行这些分析22。
尿苷含量低成绩单捕获效率低下可能会影响RNA半衰期造成人为的低新转录/总RNA比率和长期的RNA半衰期的测量。这个问题的严重程度进行评估绘制RNA半衰期或日志(新建转录/总RNA比)对uridi所有成绩单7,15 NE含量。这也提供了一个良好的质量控制,以评估4SU掺入率之间的差异不同样品或条件。尿苷内容观察到大量相关的情况下,这可能是生物信息学手段15纠正。然而,应该指出的新转录的RNA的成熟转录本的贡献不能很容易地从更大,因而更富含尿苷前体分化。除非处理一个给定的成绩单动力学(他们通常不会),只需校正尿苷含量低(低效捕捉)可能严重扭曲RNA半衰期。因此,我们最近发现处理人类最核仁,是非常低效的9。如果我们已经纠正了新的转录/总RNA比尿苷含量低而小核仁(70 - 300个核苷酸),这将导致在极短的snoRNA的半LIVES(<5分钟),与众多新转录/总RNA比超过100%。所以,我们一般不建议尿苷含量低时,校正测量RNA半衰期。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
作者宣称,他们有没有竞争经济利益。
Acknowledgments
我们想感谢阿米里根仔细阅读的手稿。支持这项工作是由NGFN加授#01GS0801,MRC奖学金授予G1002523 NHSBT授予WP11-05,LD和DFG授予FR2938/1-1 CCF
Materials
| Name | Company | Catalog Number | Comments |
| 4-thiouridine | Carbosynth | T4509 | Prepare 50 mM stock in sterile H2O, store at -20 °C in aliquots of 50-500 μl, discard unused reagent, do not refreeze. |
| Trizol | Invitrogen | 15596026 (100 ml), 15596018 (200 ml) | WARNING - CORROSIVE and HAZARDOUS TO HEALTH! Ensure immediate access to Phenol antidote (PEG-Methanol); Store at 4 °C. |
| Chloroform | Sigma | 372978 | WARNING - HAZARDOUS TO HEALTH |
| Isopropanol | Sigma | 650447 | |
| Sodium citrate, nuclease-free | Sigma | C8532 | Prepare 1.6 M stock solution using nuclease-free water. |
| 5M nuclease-free NaCl | Sigma | 71386 | Stock solution |
| Nuclease-free H2O | Sigma | W4502 | Make 1 ml aliquots in nuclease-free tubes. |
| RNA precipitation buffer | 1.2 M NaCl, 0.8 M sodium citrate in nuclease free water. Prepare in advance under strictly nuclease-free conditions. Store at room temperature in 50 ml falcon tubes. | ||
| Ethanol | Sigma | 459844 | Use with nuclease-free water to prepare 80% ethanol, store at -20 °C. |
| 1 M nuclease-free Tris Cl, pH 7.5 | Lonza | 51237 | Stock solution |
| 500 mM nuclease-free EDTA, pH 8.0 | Invitrogen | 15575-020 | Stock solution |
| 10x Biotinylation Buffer (BB) | 100 mM Tris pH 7.4, 10 mM EDTA in nuclease-free water, make aliquots of 1 ml. | ||
| Dimethylformamide (DMF) | Sigma | D4551 | |
| EZ-Link biotin-HPDP | Pierce | 21341 | Prepare 1 mg/ml stock solution by dissolving 50 mg biotin-HPDP in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. |
| Phase Lock Gel Heavy tubes 2.0 ml | Eppendorf | 0032 005.152 | Optional for the chloroform extraction step. |
| Zeta membrane | BIORAD | 162-0153 | |
| 10x Dot blot binding buffer | 100 mM NaOH, 10 mM EDTA | ||
| Biotin-oligo | 5'-biotin, 25 nucleotides, any sequence | ||
| Sodium dodecyl sulphate | Fisher | BPE9738 | For 100 ml 20% stock solution, add 20 g SDS to 80 ml PBS pH 7-8 and adjust volume to 100 ml. Keep all high-percentage SDS solutions above 20 °C. Warm the solutions slightly should SDS precipitate. |
| EZ-Link Iodoacetyl-LC-Biotin | Pierce | 21333 | Prepare 1 mg/ml stock solution by dissolving 50 mg iodoacetyl-biotin in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. Generates irreversible, thiol-specific biotinylation. |
| Phosphate buffer saline | Gibco | 10010-015 | |
| Dot blot blocking buffer | Mix 20 ml 20% SDS with 20 ml 1 x PBS pH 7-8 and add EDTA to the final concentration of 1 mM. | ||
| Streptavidin-horseradish peroxidase | Vector Laboratories | SA5004 | Store at -20 °C. Mix 10 ml 20% SDS with 10 ml 1 x PBS. Add 20 μl Streptavidin-HRP before use. |
| ECL reagent | GE Healthcare | RNP2109 | Use following the manufacturer's instructions. |
| Super RX, X-RA Film, 18x24 cm | Fujifilm | 47410 19236 | |
| μMacs Streptavidin Kit | Miltenyi | 130-074-101 | Store the beads at 4 °C. |
| Tween 20 | Sigma | P1379 | |
| Washing buffer | 100 mM Tris pH 7.4, 10 mM EDTA, 1 M NaCl, 0.1% Tween 20 in nuclease-free H2O. | ||
| Dithiothreitol (DTT) | Sigma | 43817 | Prepare as 100 mM DTT in nuclease-free H2O, always prepare fresh before use. |
| RNeasy MinElute Kit | Qiagen | 74204 | Store columns at 4 °C, remaining components of the kit at room temperature. |
| 1.5 ml screw-top polypropylene tubes | Sarstedt | 72.692.005 | Compatible with Dimethylformamide |
| 2.0 ml screw-top polypropylene tubes | Sarstedt | 72.694.005 | Compatible with Dimethylformamide |
| 15 ml tubes | BD Falcon | 352096 | Compatible with Dimethylformamide |
| 50 ml tubes | BD Falcon | 352070 | Compatible with Dimethylformamide |
| All solutions/reagents should be stored at room temperature unless otherwise specified. | |||
| Equipment | |||
| UV/VIS spectrophotometer | Thermo Scientific | NanoDrop 1000 | Or equivalent. Use low volume (1-2 μl) for measurements of low RNA concentrations to avoid excessive sample loss. |
| Polypropylene 15 ml centrifuge tubes | VWR International | 525-0153 | In contrast to standard 15 ml tubes, these tolerate up to 15,000 × g |
| High-speed centrifuge | Beckman Coulter | Avanti J-25 | Or equivalent equipment capable of reaching 13,000×g |
| High-speed rotor | Beckman Coulter | JLA-16250 | Or equivalent equipment capable of reaching 13,000×g |
| Adaptors for 15 ml tubes | Laborgeräte Beranek | 356964 | Or equivalent equipment capable of reaching 13,000×g |
| Refrigerated table-top centrifuge | Eppendorf | 5430 R | Or equivalent. |
| Thermomixer | Eppendorf | Thermomixer compact | Or equivalent. |
| Magnetic stand | Miltenyi Biotec | 130-042-109 | One stand holds 8 μMacs columns. |
| Waterbath | Grant | SUB Aqua 5 | Or equivalent. |
| Ultra-fine scale | A&D | GR-202 | Or equivalent. |
| E-Gel iBase Power System | Invitrogen | G6400UK | For RNA gels; or equivalent. |
| E-Gel EX 1% agarose precast gels | Invitrogen | G4020-01 | For RNA gels; or equivalent. |
References
- Brown, P. O., Botstein, D. Exploring the new world of the genome with DNA microarrays. Nature Genetics. 21, 33-37 (1999).
- Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 5, 621-628 (2008).
- Nagalakshmi, U., Wang, Z., et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 320, 1344-1349 (2008).
- Wilhelm, B. T., Marguerat, S., et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 453, 1239-U1239 (2008).
- Dölken, L., Ruzsics, Z., et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 14, 1959-1972 (2008).
- Yang, E., van Nimwegen, E., et al. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 13, 1863-1872 (2003).
- Friedel, C. C., Dölken, L., Ruzsics, Z., Koszinowski, U. H., Zimmer, R. Conserved principles of mammalian transcriptional regulation revealed by RNA half-life. Nucleic Acids Res. 37, e115 (2009).
- Nilsen, T. W., Graveley, B. R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 463, 457-463 (2010).
- Windhager, L., Bonfert, T., et al. Ultra short and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res. , (2012).
- Melvin, W. T., Milne, H. B., Slater, A. A., Allen, H. J., Keir, H. M. Incorporation of 6-thioguanosine and 4-thiouridine into RNA. Application to isolation of newly synthesised RNA by affinity chromatography. Eur. J Biochem. 92, 373-379 (1978).
- Cleary, M. D., Meiering, C. D., Jan, E., Guymon, R., Boothroyd, J. C. Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nature Biotechnology. 23, 232-237 (2005).
- Kenzelmann, M., Maertens, S., et al. Microarray analysis of newly synthesized RNA in cells and animals. Proc. Natl. Acad. Sci. U.S.A. 104, 6164-6169 (2007).
- Schwanhäusser, B., Busse, D., et al. Global quantification of mammalian gene expression control. Nature. 473, 337-342 (2011).
- Rabani, M., Levin, J. Z., et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. , (2011).
- Miller, M. R., Robinson, K. J., Cleary, M. D., Doe, C. Q. TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nat. Methods. 6, 439-441 (2009).
- Miller, C., Schwalb, B., et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 7, 458 (2011).
- Weintz, G., Olsen, J. V., et al. The phosphoproteome of toll-like receptor-activated macrophages. Mol. Syst. Biol. 6, 371 (2010).
- Lipsett, M. N. The isolation of 4-thiouridylic acid from the soluble ribonucleic acid of Escherichia coli. Journal of Biological Chemistry. 240, 3975-3978 (1965).
- Marcinowski, L., Liedschreiber, M., et al. Real-time Transcriptional Profiling of Cellular and Viral Gene Expression during Lytic Cytomegalovirus Infection. PLoS Pathog. 8, e1002908 (2012).
- Chomczynski, P., Mackey, K. Short technical reports. Modification of the TRI reagent procedure for isolation of RNA from polysaccharide- and proteoglycan-rich sources. Biotechniques. 19, 942-945 (1995).
- Friedel, C. C., Dölken, L. Metabolic tagging and purification of nascent RNA: implications for transcriptomics. Mol. Biosyst. 5, 1271-1278 (2009).
- Friedel, C. C., Kaufmann, S., Dölken, L., Zimmer, R. HALO - A Java framework for precise transcript half-life determination. Bioinformatics. , (2010).
