

Summary
A combinação de cromatina imunoprecipitação e sequenciamento ultra-high-throughput (CHIP-seq) pode identificar e mapear as interações DNA-proteína em uma determinada linha de tecido ou célula. Delineado é como gerar um modelo de alta chip de qualidade para o seqüenciamento posterior, utilizando a experiência com o fator de transcrição TCF7L2 como um exemplo.
Abstract
Microplaqueta-seqüenciamento (Chip-seq) métodos diretamente oferecer cobertura de todo o genoma, onde a combinação de cromatina imunoprecipitação (CHIP) e sequenciamento massivamente paralelo pode ser utilizado para identificar o repertório de sequências de DNA de mamíferos vinculados por fatores de transcrição in vivo. "Próxima geração" tecnologias de seqüenciamento do genoma fornecer 1-2 ordens de magnitude aumento na quantidade de seqüência que pode ser rentável gerou mais de tecnologias mais antigas permitindo métodos ChIP-seq para fornecer diretamente a cobertura de todo o genoma para profiling eficaz de mamíferos interacções proteína-ADN.
Para o sucesso abordagens ChIP-seq, deve-se gerar modelo de DNA chip de alta qualidade para obter os melhores resultados de sequenciamento. A descrição é baseada em experiência com o produto de proteína do gene mais fortemente implicado na patogénese da diabetes de tipo 2, a saber, o factor de transcrição do factor de transcrição 7-like 2 (TCF7L2). Este factor também tem sido implicado em vários cancros.
Descritos é como ADN molde para gerar chip de alta qualidade derivada da linha celular de carcinoma colo-rectal, HCT116, a fim de construir um mapa de alta resolução através de sequenciação para determinar os genes ligados por TCF7L2, dando uma visão mais para o seu papel chave na patogénese de características complexas.
Introduction
Durante muitos anos, tem havido uma necessidade não satisfeita para identificar o grupo de genes ligados e regulada por um genoma proteína dada largura, em particular os da classe factor de transcrição.
Odom et al. 1 usado cromatina imunoprecipitação (CHIP), combinado com o promotor de microarrays para identificar sistematicamente os genes ocupados por órgãos reguladores de transcrição pré-especificados em ilhotas pancreáticas fígado humano e. Subsequentemente, Johnson et ai. Dois desenvolveram um ensaio de imunoprecipitação da cromatina em larga escala com base em ultra-sequenciação de DNA de alto rendimento directo (ChIP seguintes), a fim de mapear de maneira abrangente interacções proteína-ADN em toda genomas de mamíferos. Tal como um caso de teste, eles mapearam in vivo a ligação do factor de silenciador neurónio restritiva (NRSF) para 1946 locais no genoma humano. Os dados exibidos sharp resolução da posição de ligação (+ 50 pares de bases), o que facilitou tanto o isolation de motivos ea identificação dos motivos NRSF vinculativas. Estes dados do chip-seq também teve alta sensibilidade e especificidade e confiança estatística (P <10 -4), propriedades que são importantes para inferir novas interações candidatos.
Robertson et al. 3 também usado ChIP seq para mapear STAT1 alvos no interferão-γ (IFN-γ)-estimuladas e não estimuladas As células HeLa S3 humanas in vivo. Por Chip-seq, com 15,1 e 12,9 milhões seqüência mapeada unicamente lê, e uma taxa de falsa descoberta estimada em menos de 0.001, eles identificaram 41.582 e 11.004 STAT1 regiões de ligação putativos em células estimuladas e não estimuladas, respectivamente. Dos 34 loci conhecidos para conter STAT1 interferon-responsive ligação 4-8 locais, Chip-seq encontrados 24 (71%). Alvos ChIP-seq foram enriquecidos em sequências semelhantes aos conhecidos STAT1 motivos de ligação. Comparações com dois dados do chip-PCR existentes define sugeridoque a sensibilidade ChIP seq situou-se entre 70% e 92% e especificidade de, pelo menos, 95%. Além disso, ficou claro que o chip-seq oferece baixa complexidade analítica e sensibilidade que aumenta com a profundidade de seqüenciamento.
Como as tecnologias de seqüenciamento tal, genoma "next-generation" fornecer 1-2 ordens de magnitude aumento na quantidade de seqüência que pode ser custo-benefício gerado sobre as tecnologias mais velhos 9. Métodos ChIP-seq, portanto, fornecer diretamente a cobertura de todo o genoma para profiling eficaz de mamíferos interações DNA-proteína 3.
Em 2006, uma forte associação de variantes do fator de transcrição 7-like 2 (TCF7L2) gene com diabetes tipo 2 foi descoberto 10. Outros pesquisadores já replicado de forma independente este achado em diferentes etnias e, curiosamente, desde os primeiros estudos de associação ampla do genoma de diabetes tipo 2 publicados na revista Nature 11,12 Ciência 13-15 e em outros lugares 16,17, a associação mais forte foi de fato com TCF7L2, o que é agora considerado o mais importante descoberta genética na diabetes tipo 2 até à data 18-20. Além disso, TCF7L2 tem sido associada a risco de cancro 21,22; facto, esta ligação tornou-se mais evidente quando o locus 8q24 revelado pelos estudos genoma amplo associação de um certo número de cancros, incluindo os carcinomas colorrectais, foi demonstrado ser devido a um extremo a montante TCF7L2 elemento de ligação dirigir a transcrição de MYC 23,24. Como tal, há um grande interesse em determinar os genes a jusante regulados por este factor de transcrição chave.
Com base na experiência com TCF7L2 como um exemplo do método, este documento descreve como gerar o molde de ADN de chip de alta qualidade. Chip foi realizada na linha celular de carcinoma colo-rectal, HCT116, para sequenciação subsequente, a fim construir uma alta resolmapa buição dos genes vinculados por TCF7L2 25 em um esforço para produzir mais conhecimento para o seu papel fundamental na patogênese de características complexas.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Cruz-link Chromatin
- Cultivar células em placas de cultura de 100x20mm celulares. A quantidade de células pode variar de 1 a 10 milhões de células por placa, dependendo do tipo de célula. Aproximadamente 2 milhões de células é suficiente para uma imunoprecipitação.
- Células de ligação cruzada em formaldeído a 1% durante 10 min à temperatura ambiente com agitação ocasional.
- Quench de reticulação por adição de uma concentração final de glicina 125 mM e incubar durante 5 min à temperatura ambiente.
- Lavar as células com solução salina tamponada de fosfato 1X (PBS) duas vezes, decantar PBS e, em seguida, adicionar 0,2 ml de PBS.
- Células de colheita com um raspador de células de plástico dentro de um tubo de microcentrífuga.
- Girar as células a 2.000 rpm durante 5 min a 4 ° C.
- Aspirar o sobrenadante. Ressuspender as células em tampão de lise (SDS a 1% de SDS, 10 mM de EDTA, 50 mM Tris-HCl pH 8,1) para a célula lisado ou mantê-los como uma pastilha para a extracção nuclear.
- As células podem ser guardadas a -80 ° C, ou pode-se proceder immédiately com sonicação.
2. Prepare Núcleos (Avance para o Passo 3.5 para lisado celular inteiro)
- Suplemento de Lise Celular (5 mM de PIPES, pH 8,0, KCl 85 mM, 0,5% de NP-40) com 1X Inibidor de Protease de cada experimento.
- Ressuspender pellet celular descongelado em cerca de 10 vezes o volume de pelotas com Tampão de Lise Celular.
- DOUNCE-homogeneizar 10 vezes com pilão, em seguida, incubar no gelo por 10 min.
- Centrifugar amostra a 4000 rpm durante 5 min a 4 ° C, rejeitar o sobrenadante e guardar pelete nuclear.
3. Sonication *
- Aqueça SDS Tampão de Lise e quantidade de suplemento de tampão a ser utilizado com inibidor de proteinase.
- Ressuspender pellet nuclear em SDS Lysis Buffer (aproximadamente 0,5 ml de tampão por 1-10.000.000 células)
- Incubar no gelo por 10 min.
- Adicionar 0,5 mL de alíquotas de amostras para tubos de microcentrífuga.
- Sonicar em gelo húmido utilizando ultra-sons Misonix com 30 segundos ligado e 45 segundosfora com uma definição de amplitude de 2. O número de ciclos de tamanho ideal fragmento pode ser determinada pela primeira experimentar vários números de ciclo (ex. 2, 4, 8, 12, 16, e 20 ou mais ciclos). Um tipo diferente de ultrassons pode ser usada, no entanto, as condições podem variar. Experimentação com o número de ciclos e da quantidade de tempo ligado e desligado devem ser realizadas a fim de determinar as condições ideais.
- Coletar 20 ml de cada amostra para verificar os resultados sonicação e fazer a quantificação,. O restante da amostra pode ser armazenada a -80 ° C.
- Dilui-se a 20 uL da amostra por adição de 30 ul de 0,1 x tampão TE.
- Tratar amostra com 1 ml de RNase A a 37 ° C durante 1 hora, em seguida adicionar 1 ul de proteinase K, e incubar a 62 ° C durante 2 horas.
- Executar 20 ul da amostra num gel de agarose a 2%.
- Purificar o restante da amostra com QIAquick kit de purificação de PCR, em seguida, quantificar usando espectrofotômetro NanoDrop.
* Para ChI nativaP, microccocal nuclease de digestão pode ser usado como alternativa ao corte do DNA.
4. Bloco de agarose *
- Se contas já estão bloqueadas, vá para o passo 5.1.
- É uma proteína ou Proteína G agarose. Por 5 imunoprecipitações (IPs), use 600 ml de 50% chorume talão (300 ml talão pellet)
- Para lavar as contas, colocá-los para baixo a 800 rpm por 1 min a 4 ° C e descartar o sobrenadante. Adicionar um pouco mais do que 2 ml de Tampão de Diluição do chip (0,01% de SDS, EDTA 1,2 mM, NaCl 167 mM, 1,1% de Triton X-100, 16,7 mM Tris-HCl pH 8,1) e misturar lentamente invertendo o tubo de 10X. Spin-se novamente a 800 rpm durante 1 min a 4 ° C e desprezar o sobrenadante. Repetir a lavagem duas vezes mais.
- Bloquear a grânulos rodando a 4 ° C durante a noite em solução de bloqueio. Consulte a Tabela 1 para a receita da solução de bloqueio.
5. Cromatina pré-clear
- Thaw sonicado cromatina no gelo.
- Girar a 12.000 rpm fou 10 min a 4 ° C, em seguida, colocar no gelo imediatamente para remover o SDS (bolinha branca).
- Recolher o sobrenadante, descarte da pelota, e combinar as amostras, se necessário.
- Retire os montantes necessários para o experimento com base em cálculos (1-10 ug da cromatina por IP).
- Diluir cromatina 10X em Chip tampão de diluição suplementada com inibidor de protease.
- Adicionam-se 100 ul de contas bloqueadas por IP.
- Girar a 4 ° C durante 1 hora.
6. Immunoprecipitation
- Girar as amostras a 800 rpm durante 1 min, e transferir o sobrenadante para um tubo fresco.
- Girar o sobrenadante a 800 rpm durante 1 min e transferência para outro tubo limpo.
- Guardar 20 ul de sobrenadante, para servir como controlo de entrada, a -20 ° C.
- Alíquota da cromatina com o número de IPs a serem feitas no experimento.
- Adicionam-se 2 ug de anticorpo por 1-10 ug de cromatina para cada amostra.
- Incubar durante a noite de 4 ° C com rotação.
- Adicionam-se 100 ul de contas bloqueadas para cada amostra de IP.
- Incubar durante 1 hora a 4 ° C com rotação.
- Agregar as esferas para baixo, girando a 800 rpm durante 1 min e descartar tanto do sobrenadante quanto possível.
- Lavar contas uma vez com Low tampão de lavagem complexo imunológico sal. Adicionar 1 ml de tampão para cada tubo, girar à temperatura ambiente durante 5-8 minutos; girar para baixo a 800 rpm durante 1 min, em seguida, descartar o sobrenadante. Repita a lavagem uma vez com tampão de sal elevado complexo imune de lavagem e o tampão de lavagem complexo imunológico LiCl e duas vezes com tampão TE para um total de cinco lavagens (Tabela 2).
7. Eluição
- Descongelar amostras de entrada a partir do dia anterior a ser processadas com os eluentes.
- Faça eluição tampão fresco (Tabela 3).
- Faça uma mistura de mestre suficiente tampão de eluição necessário para IPs e amostras de controle de entrada mais 1-2 amostras extras.
- Adicionar 100 ul de tampão de eluição para cada uma das amostras de IP e incubar à temperatura ambiente durante 15 mcom rotação.
- Girar a 800 rpm durante 1 min e adicionar o sobrenadante para um tubo fresco.
- Adicionar mais 100 ul de tampão de eluição a cada tubo de esferas e incubar à temperatura ambiente durante 15 minutos com rotação.
- Vortex durante 15 segundos após a incubação; girar a 5000 rpm, durante 1 min, depois combinar o sobrenadante com o sobrenadante da primeira eluição. (Certifique-se que não há sobra contas no sobrenadante. Caso de dúvida, girar sobrenadante novamente a 5.000 rpm por 1 min e recolher o sobrenadante em um novo tubo.
- Adicionar 180 ul de tampão de eluição à 20 ul das amostras de controlo de entrada.
8. Cross-link reverso
- Para a 200 ul de eluentes e controles de entrada, adicionar 8 mL de 5 M de NaCl.
- Tubos de vedação com parafilme e incubar em banho-maria a 65 ° C durante a noite.
9. Purificação de DNA
- Tratar cada amostra com 1 ml de RNase A durante 1 hora a 37 ° C.
- Adicionar 4 μl de EDTA 0,5 M, 8 ul de 1M Tris-HCl, misturar, adicionar 1 ml de Proteinase K a cada amostra e incubar a 45 ° C durante 2 horas.
- Purifica-se as amostras usando o kit de purificação de PCR QIAquick. As amostras podem ser guardadas a -20 ° C e a PCR de verificação pode ser feito numa data posterior.
* Alternativamente, as esferas magnéticas ChIP grau pode ser usado em lugar de agarose para a porção de imunoprecipitação.
10. PCR cheque
- Para a verificação de PCR, use primers para regiões conhecidas em se comprometer com a proteína de interesse. Além disso, use primers para regiões não-vinculativos como controles negativos.
- Misturar os reagentes para a reacção. Diluir a amostra de entrada a 1:100 (Tabela 3).
- Executar reação. Programa de PCR:
Passo 1: 94 ° C 3 min
Passo 2: 94 ° C 20 seg
59 ° C 30 seg
72 ° C 30 seg
(Repita a Etapa 2, pelo menos, 30 cycles)
Passo 3: 72 ° C 2 min
- Executar amostras em gel de agarose a 1%.
- O enriquecimento pode também ser determinada quantitativamente, por PCR em tempo real.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Uma vez que a cromatina foi sonicada e foram tratados com RNase e Proteinase, as amostras são executados em gel de agarose a 2%, devem apresentar um esfregaço com a maior parte do ADN com o tamanho desejado. Se vários ciclos diferentes são testadas, uma redução gradual no tamanho deve ser visto como aumentar o número de ciclos (Figura 2).
Depois de completar a porção de imunoprecipitação do protocolo do enriquecimento pode ser verificada por PCR, ou PCR em tempo real. Para amostras de PCR são executados em um gel de agarose deve haver faixas na entrada e chip (com o anticorpo para a proteína de interesse, que é TCF7L2 neste caso) vias de amostras e nada ou, no máximo, um leve banda (ruído de fundo) no IgG (negativa) da pista de controlo para a região de ligação positivo. Para a região de ligação negativo não deve ser muito fraca ou nenhuma banda para o controle IgG e pistas chip. Deve haver uma banda na pista de entrada (Figura 3).
A Figura 4 mostra as mesmas amostras examinadas por PCR em tempo real. Tal como acontece com a figura anterior, deve haver um enriquecimento significativo de vezes a região de ligação positivo para a amostra em fichas sobre o controlo de IgG. Além disso, deve haver muito pouco de enriquecimento, eventualmente, visto na região de ligação negativa.

Figura 1. Diagrama de fluxo de processo de chip. Clique aqui para ver a figura maior .
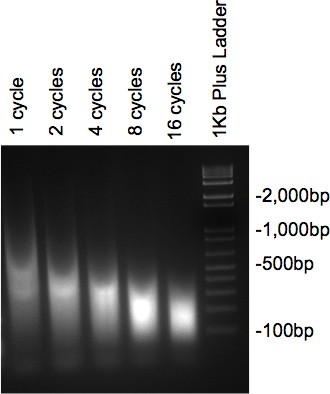
Figura 2. Gel verificação de sonicação DNA.

Figura 3. PCR Verificação de chip.
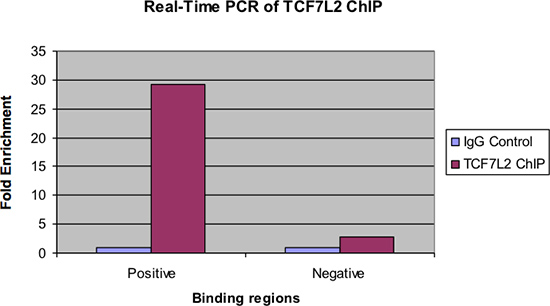
Figura 4. PCR em tempo real de TCF7L2 chip.
| Reagente | Volume |
| Bead pellet | 300 ul |
| BSA (50 mg / ml) | 30 ul |
| 100X Inibidor de Proteinase | 10 ul |
| Tampão de diluição ChIP | 660 mL |
| Total | 1000 mL |
Tabela 1. Receita para o bloqueio de agarose.
| Tampão | Componentes |
| Low sal tampão de lavagem Complexo Imune | SDS a 0,1% 1% de Triton X-100 2 mM de EDTA 20 mM Tris-HCl pH 8,1 150 mM de NaCl |
| Sal tampão de lavagem complexo imunológico alta | SDS a 0,1% 1% de Triton X-100 2 mM de EDTA 20 mM Tris-HCl pH 8,1 500 mM de NaCl |
| Tampão de Lavagem complexo imunológico LiCl | 0,25 M LiCl 1% de NP-40 1%, Desoxicolato 1 mM de EDTA 10 mM Tris-HCl pH 8,1 |
| Tampão TE | 10 mM Tris-HCl pH 8,1 EDTA 1 mM, pH 8,0 |
Tabela 2. Buffers de lavagem chip.
| Reagente | Volume |
| 10 ul | |
| NaHCO3 1M | 20 ul |
| H2O | 170 mL |
Tabela 3. Tampão de eluição para um IP.
| Reagente | 50 Reação mL | 20 Reação mL |
| Água | 27 mL | 10,8 mL |
| De tampão de reacção de PCR 5X | 10 ul | 4 ul |
| MgCl2 | 4 ul | 1,6 mL |
| de dNTP (10 mM) | 1 ul | 0,4 mL |
| Mix Primer (5 uM cada) | 2 ul | 0,8 mL |
| Taq (Promega Hotstart) | 1 ul </ Td> | 0,4 mL |
| DNA ChIP | 5 ul | 2 ul |
Tabela 4. Volumes de reacção de PCR.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
É agora possível realizar um perfil de todo o genoma de ADN-proteína utilizando as interacções associação ChIP seq, como foi recentemente demonstrado com outros factores de transcrição de 2,3. A chave para um resultado bem sucedido de sequenciação é a geração de um modelo de alta qualidade de ADN de cromatina imunoprecipitação.
Uma vez que o molde de ADN foi gerado e verificado ser adequadamente enriquecida, pode-se, em seguida, levá-lo na preparação de biblioteca para a sequenciação subsequente. Por exemplo, pode-se usar o protocolo de sequenciação biblioteca fornecida pelo vendedor, Ilumina. A selecção do tamanho dessa biblioteca pode ser realizada por electroforese em gel e subsequente excisão e purificação de ADN na ~ 200 - à escala de 700 pb. Reduzindo o tamanho e diminuir o intervalo de tamanho de ADN obtidos a partir de purificação em gel destina-se a melhorar a resolução de posicionamento do chip de seq. Por enriquecedora para pequenos pedaços de DNA de entrada ligados ao fator de interest, seria de esperar que a localização do site ganhará resolução. Selecção do tamanho mais apertado também melhora a uniformidade de tamanho das colónias moleculares produzidos na plataforma Illumina. Tal tamanho da colónia uniformidade também aumenta o número de leitura efectiva obtida. Menor tamanho de entrada de ADN também produz colónias mais robustos na plataforma Illumina, e isto pode significar que mais curtas peças de ADN dentro de uma determinada distribuição de amostras de entrada será representada de forma mais eficiente na saída final de sequência do que são os mais pedaços de entrada a partir da mesma distribuição.
As abordagens de bioinformática para análise de seqüência "next-generation" continuam a evoluir, com muitos vendedores fazendo seu software open-source para posterior refinamento. Pode-se transformar a lê que o mapa de locais genómicos únicos em um fragmento de ADN do perfil de sobreposição. Picos significativos podem ser identificados pelos perfis threshholding a uma altura equivalente a uma taxa de detecção de falsas estimativa. A posição spmatrizes de frequências ecific derivados deste trabalho pode ser utilizado para identificar e localizar os sítios de ligação de ADN através do genoma humano de um determinado factor de.
Mas é preciso ser cauteloso com relação a fatores que se deseja estudar com Chip-seq. Antes de iniciar um tal estudo deve-se avaliar se um anticorpo disponível no mercado, que é utilizável no contexto de chip, como um anticorpo pobre pode ter efeitos bastante nocivos sobre um de resultados experimentais. Além disso, deve-se considerar, se houver isoformas de splicing da proteína em estudo, na verdade, TCF7L2 é conhecido por ter muitas isoformas portanto, foram particularmente cauteloso na selecção de um anticorpo que se liga a aminoácidos consistentemente presente em todos os principais isoformas deste factor de transcrição 25.
Em resumo, a combinação de imunoprecipitação da cromatina e sequenciação de ultra-high-throughput (ChIP seq) pode identificar e mapear as interacções proteína-ADN em um determinado tecido ou clinha ell. Delineamos como gerar um modelo de chip de alta qualidade para o seqüenciamento subseqüente.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Os autores declaram que não têm interesses financeiros concorrentes.
Acknowledgments
O trabalho é apoiado por uma concessão do Instituto de Desenvolvimento do Hospital Infantil da Filadélfia.
Materials
| Name | Company | Catalog Number | Comments |
| QIAquick PCR Purification Kit | Qiagen | 28104 | |
| EZ-ChIP Kit | Millipore | 17-371 | |
| GoTaq Hot Start Polymerase | Promega | M5001 | |
| Misonix Sonicator | Qsonica | XL-2000 | |
| NanoDrop 1000 Spectrophotometer | Thermo-Scientific | ||
| Positive control primer sequences (TCF7L2-1) Forward- 5'-TCGCCCTGTCAATAATCTCC-3' Reverse- 5'-GCTCACCTCCTGTATCTTCG-3' Negative control primer sequences (CTRL-1) Forward-5'-ATGTGGTGTGGCTGTGATGGGAAC-3' Reverse- 5'-CGAGCAATCGGTAAATAGGTCTGG-3' |
|||
References
- Odom, D. T., et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 303, 1378-1381 (2004).
- Johnson, D. S., Mortazavi, A., Myers, R. M., Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 316, 1497-1502 (2007).
- Robertson, G., et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nature Methods. 4, 651-657 (2007).
- Reich, N. C., Liu, L. Tracking STAT nuclear traffic. Nat. Rev. Immunol. 6, 602-612 (2006).
- Lodige, I., et al. Nuclear export determines the cytokine sensitivity of STAT transcription factors. The Journal of Biological Chemistry. 280, 43087-43099 (2005).
- Schroder, K., Sweet, M. J., Hume, D. A. Signal integration between IFNgamma and TLR signalling pathways in macrophages. Immunobiology. 211, 511-524 (2006).
- Vinkemeier, U. Getting the message across, STAT! Design principles of a molecular signaling circuit. The Journal of Cell Biology. 167, 197-201 (2004).
- Brierley, M. M., Fish, E. N. Stats: multifaceted regulators of transcription. J. Interferon Cytokine Res. 25, 733-744 (2005).
- Bentley, D. R. Whole-genome re-sequencing. Current Opinion in Genetics & Development. 16, 545-552 (2006).
- Grant, S. F., et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nature Genetics. 38, 320-323 (2006).
- Sladek, R., et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 445, 881-885 (2007).
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 447, 661-678 (2007).
- Saxena, R., et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 316, 1331-1336 (2007).
- Zeggini, E., et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 316, 1336-1341 (2007).
- Scott, L. J., et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 316, 1341-1345 (2007).
- Steinthorsdottir, V., et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nature Genetics. 39, 770-775 (2007).
- Salonen, J. T., et al. Type 2 Diabetes Whole-Genome Association Study in Four Populations: The DiaGen Consortium. American Journal of Human Genetics. 81, 338-345 (2007).
- Zeggini, E., McCarthy, M. I. TCF7L2: the biggest story in diabetes genetics since HLA. Diabetologia. 50, 1-4 (2007).
- Weedon, M. N. The importance of TCF7L2. Diabet. Med. 24, 1062-1066 (2007).
- Hattersley, A. T. Prime suspect: the TCF7L2 gene and type 2 diabetes risk. The Journal of Clinical Investigation. 117, 2077-2079 (2007).
- Yochum, G. S., et al. Serial analysis of chromatin occupancy identifies beta-catenin target genes in colorectal carcinoma cells. Proceedings of the National Academy of Sciences of the United States of America. 104, 3324-3329 (2007).
- Duval, A., Busson-Leconiat, M., Berger, R., Hamelin, R. Assignment of the TCF-4 gene (TCF7L2) to human chromosome band 10q25.3. Cytogenet. Cell Genet. 88, 264-265 (2000).
- Pomerantz, M. M., et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nature Genetics. 41, 882-884 (2009).
- Tuupanen, S., et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nature Genetics. 41, 885-890 (2009).
- Zhao, J., Schug, J., Li, M., Kaestner, K. H., Grant, S. F. Disease-associated loci are significantly over-represented among genes bound by transcription factor 7-like 2 (TCF7L2) in vivo. Diabetologia. 53, 2340-2346 (2010).
- Benjamini, Y., Yekutieli, D. Quantitative trait Loci analysis using the false discovery rate. Genetics. 171, 783-790 (2005).
