

Summary
Nous décrivons des méthodes d'imagerie nous utilisons pour étudier la distribution et la mobilité des protéines fluorescentes transfectées résidant dans le réticulum endoplasmique (RE) par l'intermédiaire de l'imagerie confocale de cellules vivantes. Nous analysons également ultrastructuralement l'effet de leur expression sur l'architecture de ce compartiment subcellulaire.
Abstract
Les lipides et des protéines dans les cellules eucaryotes sont continuellement échangés entre compartiments cellulaires, bien que ceux-ci conservent leur composition et les fonctions distinctif malgré le trafic interorganelle moléculaire intense. Les techniques décrites dans le présent document sont des moyens puissants de l'étude des protéines et de la mobilité des lipides et le trafic in vivo et dans leur environnement physiologique. récupération de fluorescence après photoblanchiment (FRAP) et la perte de la fluorescence dans photoblanchiment (FLIP) sont largement utilisés techniques d'imagerie des cellules vivantes pour étudier le trafic intracellulaire par l'intermédiaire de la voie de l'exo-endocytose, la continuité entre les organelles ou des sous-compartiments, la formation de complexes protéiques, et la localisation des protéines en microdomaines lipidiques, qui peut être observée dans des conditions physiologiques et pathologiques. Les limites de ces approches sont principalement dues à l'utilisation de protéines de fusion fluorescente, et leurs inconvénients potentiels comprennent un artefact sur-expression dans les cellules et la possibilité de différences dans le repliement et la localisation des protéines marquées et natives. Enfin, comme la limite de résolution de la microscopie optique (environ 200 nm) ne permet pas d'enquête de la structure fine de l'ER ou les sous-compartiments spécifiques qui peuvent provenir de cellules en situation de stress (c'est à dire l'hypoxie, l'administration du médicament, l'expression trop de transmembranaire protéines ER résidents) ou dans des conditions pathologiques, nous combinons l'imagerie des cellules vivantes des cellules transfectées cultivées avec ultrastructurale analyse basée sur la microscopie électronique à transmission.
Introduction
La découverte de la protéine verte fluorescente (GFP) et ses variantes spectrales, et le développement parallèle de la microscopie à fluorescence, ont ouvert complètement nouvelles avenues pour la recherche sur le comportement des protéines dans les cellules. Des techniques telles que la récupération de fluorescence après photoblanchiment (FRAP) et la perte de fluorescence dans photoblanchiment (FLIP), qui sont possibles en raison de la capacité intrinsèque de fluorophores à éteindre leur fluorescence sous éclairage intense, sont basées sur l'imagerie des cellules vivantes confocale et l'utilisation de transfectées des protéines de fusion fluorescente 3.1. Ils sont largement utilisés pour évaluer non seulement la localisation des protéines, mais aussi leur mobilité et le transport vésiculaire, qui peut révéler des indices importants concernant leur fonction 4.
La caractéristique unique des cellules eucaryotes est la présence de compartiments intracellulaires qui ont des compositions lipidiques et protéiques spécifiques. Bien que les organites sont physiquement isolated, ils doivent communiquer entre eux et partager des composants moléculaires, afin de maintenir l'homéostasie cellulaire. La voie de sécrétion garantit que les protéines et les lipides synthétisés dans le RE atteindre la destination finale correcte dans laquelle ils exercent leur fonction. Organites intracellulaires peuvent également être reliés par des sites de contact dynamiques qui permettent aux molécules (lipides) pour être directement échangés entre les compartiments. En outre, de nombreuses protéines ont de grands complexes assemblés dans hétéromériques ou associé avec des espèces lipidiques spécifiques (radeaux lipidiques / microdomaines) afin de devenir fonctionnellement active ou être transportés vers leur destination finale. Tous ces aspects biologiques influencent fortement les propriétés cinétiques des protéines, et peuvent donc être étudiés de façon appropriée au moyen des techniques décrites ci-dessous.
Notre groupe a largement utilisé FRAP et FLIP combinée avec la microscopie électronique pour étudier l'architecture de l'urgence et de ses différents sousdomaines. L'ER est la première station de la voie de sécrétion et joue un rôle clé dans les protéines et les lipides de tri 5. Il est un organite très dynamique dont les sous-domaines distincts refléter ses nombreuses fonctions différentes (c'est à dire des protéines et des lipides de biosynthèse et de la traite, le repliement des protéines, Ca 2 + stockage et la libération, et le métabolisme des xénobiotiques). Cependant, même si elles sont morphologiquement, spatialement et fonctionnellement distinctes, ces domaines sont continus les uns avec les autres, et leur abondance relative peuvent être modifiées dans des cellules dans des conditions physiologiques et pathologiques. Les plus connus et les domaines généralement spatialement distincts de l'ER sont l'enveloppe nucléaire, et le RE lisse et rugueux, mais nous et d'autres avons démontré qu'il existe des structures ER avec une architecture plus élaborée et organisation tridimensionnelle dans divers types de cellules et les tissus dans des conditions physiologiques qui peuvent également être induites par les moyens de stimuli stressants tels que l'hypoxie, la drogueadministration, ou la sur-expression de protéines transmembranaires 2,6 ER-résident (et les références qui y sont).
Nous avons récemment démontré la présence de telles structures dans des modèles cellulaires de maladies humaines 1,7. Originaire de la citernes empilées de RE lisse, ils ont reçu le nom collectif de réticulum endoplasmique lisse organisé (OSER) en 2003 6, mais ils sont également connus comme karmellae, lamelles, et cristalloïde ER sur la base de leur architecture qui, comme leur taille, peut varier. Après que les cellules sont transfectées avec la GFP fusionnée à la région de la queue cytosolique ancrée (TA) des protéines ER-résident (d EGFP-RE), la tendance à la dimérisation de la GFP dans faiblement trans altère considérablement l'organisation et de la structure de l'ER. FRAP et FLIP expériences ont montré que d EGFP-ER est libre de se diffuser à l'intérieur OSER, et le fait qu'il se déplace de l'ER réticulaire à la OSER et vice versa </ Em> indique que les agrégats sont en continuité avec l'ER réticulaire environnante. L'analyse ultrastructurale nous a permis de corréler les données de fluorescence avec une description détaillée de l'architecture OSER et l'organisation au niveau de l'échelle nanométrique: OSER sont toujours constitués d'empilements de citernes paires de RE lisse mais peuvent avoir différentes formes d'organisation spatiale, tels que disposés régulièrement sinusoïdale tableaux ou des spirales, ou des tableaux tubulaires "cristalloïdes" hexagonaux. Ces réarrangements conduisent à des morphologies cubes 8 qui, comme ils l'ont été trouvés dans les cellules dans des conditions physiologiques 9 et contraintes suivants tels que l'hypoxie 10, traitement de la toxicomanie 11, et le cancer 9, peuvent avoir un potentiel important en tant que marqueurs ultrastructuraux.
Après cette première démonstration en utilisant des protéines de fusion GFP, nous avons utilisé des expériences d'imagerie pour analyser la prolifération des domaines de l'urgence en réponse aux traitements pharmacologiques 12, assess la tendance des protéines fluorescentes pour oligomériser dans des cellules 13, et d'enquêter sur le rôle d'un mutant, protéines TA SLA liés à la formation d'agrégats intracellulaires d'origine ER qui peuvent être pertinents à son 1,8 de pathogénicité. Il a été suggéré que la formation d'agrégats intracellulaires (ce qui se produit dans de nombreuses maladies neurodégénératives 14) peut être un mécanisme de protection destinée à empêcher les interactions entre les protéines mutantes toxiques et les composants de la cellule autour de 15.
Ce qui suit est une description d'une combinaison de méthodes de microscopie optique et électronique pour enquêter sur des constructions dont les C-terminal domaines hydrophobes sont insérés dans la membrane du RE, et une analyse de leur comportement dynamique et les effets de leur surexpression sur le domaine de l'urgence l'architecture à des cellules en culture (voir la figure 1 pour un organigramme du protocole expérimental).
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Une. Plasmide, culture cellulaire, et transfection avec ER protéines fluorescentes
- Le plasmide utilisé dans cette étude est constitué d'une version améliorée de la GFP fusionnée à son extrémité C-terminale de la région de la queue de l'isoforme d'ER de rat cytochrome b (5) (abrégé ici en tant que b (5)) par l'intermédiaire d'une séquence de liaison. La région de queue contient la séquence entière (Pro94-Asp134) qui reste associée à la membrane après le clivage par la trypsine de b natif (5), y compris la DMT de 17 résidus (domaine transmembranaire), flanqué par des séquences polaires amont et aval (UPS et DPS) . Le lieur est constitué de l'épitope myc suivi de [(Gly) 4 Ser] 3, et l'ensemble de l'ADNc est inséré dans les sites Hind3-Xba1 du vecteur d'expression de mammifère pcDNA3. Les détails de la construction de ce plasmide ont été décrites dans une publication antérieure dans laquelle elle est désignée comme la GFP-ER 16.
- Cultivez cellules COS-7 en milieu de Eagle modifié par Dulbecco (DMEM) additionné de 10% fetal de sérum bovin, 2 mM de L-glutamine, 1% de pénicilline / streptomycine dans un incubateur à 37 ° C et 10% de CO 2.
- Transfection. La plaque 3 x 10 5 cellules sur une lamelle de verre ronde dans une plaque à 6 puits et, le jour suivant, transfecter avec le système JetPEI comme décrit par le fabricant. A noter que le rapport optimal JetPEI / ADN a été testé afin de déterminer l'efficacité de transfection maximale en fonction du plasmide et la lignée cellulaire utilisée: dans notre cas, une JetPEI: rapport d'ADN de 2:1 conduit à 70-80% de l'efficacité de transfection.
2. Direct fluorescence microscopie confocale à balayage
- Imagerie des cellules vivantes. Placer la lamelle sur laquelle les cellules transfectées ont été ensemencées dans une chambre de cellule de la culture de l'acier pour 24 mm lamelles remplis avec du DMEM w / o rouge de phénol, complété avec 10% de FBS, 2 mM de L-glutamine, 1% de pénicilline / streptomycine, 25 mM d'HEPES , 50 ug / ml de cycloheximide et 1:100 OxyFluor pour empêcher les échantillons de photoblanchiment. Un microscop SP5 confocalee équipé d'un incubateur à CO2 à température contrôlée (37 ° C et 5% CO 2) est utilisé pour des expériences d'imagerie des cellules vivantes, avec d GFP-ER étant visualisé en utilisant un laser à 488 nm et d'un filtre 525/50 passe de la bande d'émission.
- récupération de fluorescence après photoblanchiment (FRAP). Dessiner une région d'intérêt (ROI), correspondant à une structure OSER, et blanchir à l'aide de 20 itérations et une combinaison de 488 nm (100% d'un laser Argon 30 mW, ce qui correspond à 5,5-6 mW à l'échantillon) et 405 nm (60% de 30 mW Diode laser 405, correspondant à 11,6 mW à l'échantillon) lasers qui, dans notre expérience, conduit à photoblanchiment efficace et rapide.
- Enregistrer la reprise de la fluorescence dans les régions d'intérêt blanchis en prenant une seule image toutes les 10 secondes pendant 10 minutes (temps de pixel = 1,61 ps / px).
- perte de fluorescence dans photoblanchiment (FLIP). Dessinez un retour sur investissement correspondant à une structure OSER, et l'eau de Javel comme décrit ci-dessus. Le blanchiment est répété toutes les 30 secondes,et les images post-blanchiment sont enregistrées toutes les 10 secondes pendant 30 minutes (temps de pixel = 1,61 ps / px).
- FRAP et FLIP analyse. Toutes les images sont analysées en utilisant le logiciel ImageJ ( http://rsbweb.nih.gov/ij/download.html ). Dans les expériences de FRAP, la récupération de la fluorescence de la ROI blanchie est mesurée au cours du temps et normalisées par rapport à la fluorescence totale des cellules blanchie, qui est toujours contrôlée pour être constante dans le temps.
- Pour les expériences de FLIP, tirer un retour sur investissement en dehors de l'OSER blanchi et couvrant l'ensemble de la cellule. Mesurer l'intensité de la fluorescence au cours du temps et à normaliser les niveaux de fluorescence d'un retour sur investissement tirés sur une cellule non blanchie afin de corriger toute diminution de la fluorescence provoquée par la formation d'image lui-même.
- Dans toutes les expériences, soustrait le signal de fond (déterminé dans une zone à l'extérieur des cellules) à partir des intensités de fluorescence des régions d'intérêt. Enfin, tracer les résultats en utilisant Gr.aphPad logiciel Prism.
3. Ultrastructurale Analyse par microscopie électronique à transmission
Compte tenu de la toxicité de la plupart des réactifs, toutes les procédures devraient être effectuées portant un laboratoire approprié. manteau et des gants sous une hotte.
- Après élimination de la lamelle de la boîte de Pétri, fixer les cellules cultivées restantes sur le fond de la cuvette en monocouche en utilisant filtré 2% de glutaraldéhyde dans 0,1 M de tampon cacodylate, pH 7,4, pendant 10 min à température ambiante.
- Grattez les cellules en utilisant un grattoir en téflon et les transférer dans 1,5 ml tubes Eppendorf. Sédimenter les cellules par centrifugation à 9000 g pendant 10 min. Retirer le surnageant, ajouter du fixateur frais, et laisser une nuit à 4 ° C.
- Laver les pastilles avec le tampon, puis post-fixation avec une solution de tétroxyde d'osmium à 1% dans du tampon cacodylate pendant 1 heure à température ambiante.
- Rincer à l'eau MilliQ, et en bloc tache avec1% d'acétate d'uranyle dans de l'eau distillée pendant entre 20 à 60 min.
- Déshydrater les échantillons en série croissante d'éthanol (70%, 80%, 90%, 100% et 100% pendant 10 min chacun), et le laver deux fois brièvement dans de l'oxyde de propylène (15 min chacun).
- Infiltrer les échantillons dans un mélange d'oxyde de propylene + Epon (1:1) (à partir de 2 h à une nuit).
- Incorporer dans une résine époxy Epon durci à 60 ° C pendant au moins 24 h.
- Section des blocs de résine taillés manuellement à l'aide d'un ultramicrotome LEICA UC6 équipé d'un couteau de diamant de 45 ° pour obtenir sections d'une épaisseur de 60-70 nm. Recueillir les sections sur les 300 mailles des grilles de cuivre.
- Colorer les sections sur la grille avec une solution saturée d'acétate d'uranyle (20 min) et du citrate de plomb (7 min), bien se laver les grilles en les plongeant dans de l'eau filtrée bi-distillée, et laissez-les sécher à température ambiante.
- Les grilles colorées sont observées en utilisant un microscope électronique à transmission TECNAI G2, et les images sont capturées en utilisant un Bottcaméra CCD om-monté à différents grossissements finales (allant généralement de 6,000-39,000 X).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
La figure 2 montre une étude d'FRAP d'exemple de la mobilité de la protéine. La mobilité de la protéine EGFP d-ER est démontrée par la récupération rapide de la fluorescence après photoblanchiment dans OSER blanchies. Pour l'analyse quantitative, la mi-temps et fraction mobile ont été calculées à partir des données mesurées expérimentalement par ajustement de l'équation monoexponentielle suivante:
F (t) = F + poste (poste F rec-F) (1-e-t / τ)
où après F est le signal de fluorescence après photoblanchiment, F rec est la valeur maximale de récupération de fluorescence qui est atteint après le blanchiment, t le temps d'enregistrement et τ la constante de temps.
S'il vous plaît noter l'importance de l'acquisition d'images sans pixels saturés qui pourraient modifier le recouvrement de fluorescence et, par conséquent, l'analyse de la mobilité de la protéine. Il is également essentiel de toujours normaliser le signal de fluorescence dans la ROI blanchie à la fluorescence totale de la même cellule afin de tenir compte des variations d'intensité de fluorescence en raison de blanchiment au cours de l'acquisition d'image ou de petits changements dans le plan de mise au point.
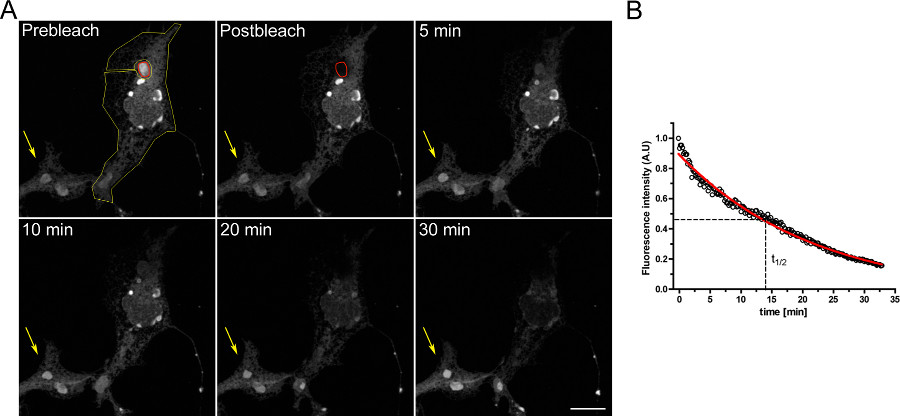
Un exemple d'un essai de FLIP pour étudier la continuité entre les compartiments intracellulaires est représenté sur la figure 3. OSER sont physiquement connectés avec le reste de l'ER comme en témoigne la désertion progressive des ER lorsque le domaine OSER est continuellement blanchi.
Pour une bonne analyse, l'acquisition de pixels saturés doit être évitée (voir ci-dessus), de plus, les paramètres d'acquisition doivent être mis en place avec des puissances laser aussi bas que possible afin d'éviter photoblanchiment en raison de l'acquisition de l'image. Pour cette raison, il est fortement recommandé à l'image d'une cellule non blanchie dans le même domaine qui sera utilisé pour normaliser le signal de fluorescence de la c blanchieell.
Toutes les expériences doivent être réalisées en présence de cycloheximide, un inhibiteur de traduction, afin d'éviter toute augmentation du signal de fluorescence de ER (et par conséquent la fluorescence totale) en raison de la biosynthèse des protéines.
microscopie électronique à transmission a montré que les agrégats fluorescents observés dans les cellules cultivées transfectées avec d EGFP-ER représentent des taches de lisse et aplatie ER citernes que l'espace se sont organisés en géométries 3D bien définies, classées sur la base de leurs habitudes: linéaires ou courbes piles (souvent associé à l'enveloppe nucléaire, non représenté) (figures 4A et B) qui peut être continue avec les régions de ER sinusoïdal (figure 4A), les membranes, dans certaines régions sont organisés en treillis avec une symétrie carrée ou hexagonale (ER cristalloïde, non représentés ). Citernes adjacentes sont séparées par une fine couche d'un peu d'électronscytoplasme dense d'environ 11 nm d'épaisseur qui est en continuité avec le cytoplasme entourant les agrégats.

Figure 1. Diagramme de la procédure expérimentale. Les cellules en culture sont tout d'abord transfectées avec jetPEI (voir le protocole) afin de surexprimer la protéine de fusion fluorescente d'intérêt. Après 24 heures, les cellules transfectées sont visualisés en direct et des expériences de FRAP et FLIP sont effectuées à l'aide d'un microscope confocal équipé d'une température contrôlée de CO 2 incubateur, et les images enregistrées sont exportées et analysées avec le logiciel approprié (par exemple ImageJ). Pour l'analyse ultrastructural, les cellules transfectées sont fixées, noyées dans un culot et EPON blocs de résine époxy. Des coupes ultrafines sont obtenues en utilisant un couteau à diamant, collectés sur copper grilles, et observée au microscope électronique à transmission. Cliquez ici pour agrandir la figure .

Figure 2. Expérience FRAP utilisant cellules COS-7 transfectées de façon transitoire avec d EGFP-ER. A) Deux structures OSER (de ROI en rouge) ont été blanchis, et la récupération de fluorescence ont été enregistrées au cours du temps. Nette reprise de la fluorescence peut être détectée 1 min post-blanchiment, et le signal de nouvelles augmentations 4 min plus tard (barre d'échelle 10 pm) B):. Analyse quantitative de l'expérience de FRAP montrant la reprise à mi-temps et la fraction mobile du d EGFP ER-protéine. Click ici pour agrandir la figure

Figure 3. Expérience FLIP utilisant cellules COS-7 transfectées de façon transitoire avec d EGFP-ER. A) Le blanchiment en continu d'une OSER (indiqué par le rouge ROI) entraîne une diminution progressive de la fluorescence dans le reste de l'ER et dans d'autres structures de OSER au sein de la même cellule (indiqué par la ROI jaune). La flèche jaune indique une partie d'une cellule non blanchie dans lequel le signal de fluorescence est constante dans le temps. (Barre d'échelle de 10 um). B) Analyse quantitative de l'expérience de la FLIP. Cliquez ici pour agrandir la figure
Figure 4. Après fixation et l'incorporation, les cellules exprimant des niveaux élevés de d EGFP-ER dans lequel les structures de OSER pouvaient être détectées au moyen de la microscopie optique de fluorescence ont été observées à travers un microscope électronique à transmission. A) vue à faible grossissement d'une partie du cytoplasme d'une cellule contenant un OSER constitué de citernes empilées et membranes ondulantes sinusoïdales. Les mitochondries (M) peut être vu regroupés autour des structures de OSER, alors que seuls les ribosomes décorent les membranes de la plus extérieure des citernes (têtes de flèches et encart). L'espace nm d'épaisseur 11 dense aux électrons entre les membranes est en continuité avec le cytoplasme (flèche et encadré) (L = lysosomes / (auto-) phagosomes) (barre d'échelle de 1,5 um; encart 0,25 um). B) Un OSER peut être formé par lamellaire ER: c'est à dire des piles de aplati ER citernes qui peuvent être continus ou fragmented dans leur apparence dans les sections minces. Vésicules bourgeonnant à l'citernes extérieure de la pile peuvent parfois être observées (astérisque) (PM, membrane plasmique) (barre d'échelle de 150 nm).
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Les protocoles et les méthodes d'imagerie décrites dans le présent document ont été utilisées pour étudier la distribution et la mobilité des transfectées TA protéines fluorescentes résidents dans le RE de cellules vivantes. Nous avons également analysé l'effet de l'expression excessive de ces protéines sur l'architecture de ce compartiment sous-cellulaire à l'aide d'analyses ultrastructurales.
La combinaison de cellules vivantes imagerie confocale et la microscopie électronique représente est un moyen très puissant de l'étude des propriétés dynamiques des protéines, et peut fournir des informations importantes concernant la fonction des protéines. Les méthodes décrites ne sont pas de temps (généralement trois jours de travail), et le développement de nombreuses applications logicielles conviviales pour l'acquisition et l'analyse d'images fait à base de photoblanchiment, imagerie des cellules vivantes relativement simple.
La principale limitation de ces techniques est l'utilisation de protéines de fusion fluorescente, car l'marqueur fluorescent peut affecter le repliement et / ou l'assemblage correct de la protéine d'intérêt. En outre, la sur-expression peut modifier le comportement des, des protéines par fluorescence taggés transfectées, et peut donc ne pas refléter les propriétés réelles de protéines endogènes, mais cela peut être surmonté en utilisant des cellules inductibles et transfectées de façon stable dans lequel le niveau d'expression peut être modulée avec précision afin d'obtenir des niveaux comparables à ceux de la protéine endogène de 1,7. La tendance des PC à oligomériser a été largement documentée et pourrait modifier sensiblement le comportement (c.-à-cinétique, les interactions protéine-protéine non désirées et la formation d'agrégats) des protéines chimériques. L'utilisation de protéines fluorescentes monomères optimisés devrait donc être considéré 17.
Un autre aspect critique des études d'imagerie dynamiques en utilisant la fluorescence et de photoblanchiment est le temps nécessaire pour blanchir la fluorescence de manière efficace et mesurer la fluorescence redécouverte (et ainsi la mobilité de la protéine) avec précision, qui dépend également de la zone de la ROI et de l'épaisseur locale de la cellule. Si une protéine GFP-étiqueté donné a un fort coefficient de diffusion, la diffusion peut se produire pendant le blanchiment et donc interférer avec des mesures de temps de récupération. Pour obtenir blanchiment rapide et efficace, il est fortement recommandé d'utiliser un "zoom" fonction (si disponible) et plus d'une ligne de laser utilisés. Bien que l'utilisation d'un module de balayage rapide (ie un scanner de résonance) peut grandement améliorer la vitesse de l'imagerie au cours de la phase de récupération d'une expérience, dans nos mains, il réduit aussi considérablement l'efficacité de blanchiment. Cependant, d'autres systèmes de balayage (par exemple un disque rotatif muni d'un dispositif de photo-blanchiment dédié), et les lasers les plus puissants peuvent améliorer à la fois l'efficacité du blanchiment et de la vitesse d'acquisition.
La plupart des protéines fluorescentes utilisées dans les expériences de FRAP et FLIP montrent un certain degré de photoblanchiment et bli réversibleNking qui doivent être considérés lors de l'exécution des analyses quantitatives. Les fluctuations entre les Etats fluorescentes et sombres apparaissent dans la seconde à l'échelle de temps minute. Pour EGFP, il a été montré que lors des expériences de blanchiment, des variations de fluorescence peut impliquer moins de 10% des molécules, donc dans le présent protocole de ce phénomène est négligeable. Si toutes les conditions sont maintenues constantes, ce qui introduirait un biais constant dans les résultats. Si d'autres protéines fluorescentes sont utilisées, dans lequel la fraction réversible est sensiblement plus élevé (c.-à-YFP), ou pour détecter et évaluer la réversibilité de photoblanchiment, cela peut être fait par la mesure de la récupération de fluorescence après photoblanchiment dans la cellule vivante entière, si la récupération est observée cette boîte seulement être le résultat de photoblanchiment réversibilité 18.
La toxicité potentielle de la lumière au cours des expériences est un autre facteur critique, en particulier parce que photoblanchiment nécessite un fort éclairage. Il est well sait que fluorophores excités ont tendance à réagir avec l'oxygène pour produire des radicaux libres qui peuvent affecter divers processus intracellulaires et même la viabilité des cellules 19, et il est donc nécessaire d'établir un équilibre entre le blanchiment efficace et la phototoxicité minimal, de plus, la viabilité cellulaire doit toujours être vérifiée après des expériences d'imagerie en direct de la cellule. Compte tenu de la courte durée d'enregistrement, nous n'avons pas tenu compte de l'effet génotoxique de lumière de courte longueur d'onde (405 nm) dans l'exemple décrit dans le présent document, mais, si une expérience plus longue est nécessaire, une ligne laser à 405 nm doit pas être utilisé.
Nous avons choisi de ne pas utiliser une approche corrélative à la microscopie électronique à transmission en raison de la nature hétérogène de l'architecture OSER et le fait que nous voulions observer que de nombreuses cellules (et les structures) que possible. La diversité de la structure fine des agrégats de protéines dans les cellules peut être un élément clé de différentes maladies et nous nous sommes intéressés à l'obtention d'un large éventail de samples, alors qu'une approche corrélative permet l'observation de moins d'événements au cours de la même période de temps. Cependant, la microscopie électronique lumière corrélative (CLEM) devrait être le premier choix quand enquêter sur des événements dans des structures qui ne peuvent pas être facilement identifiés (tels que les sous-domaines ER moins importants) ou dans un nombre limité de cellules (comme les cellules de micro-injection). Il est à noter que nos expériences ont été caractérisées par un degré élevé d'efficacité de la transfection (au moins 30% des cellules ont été transfectées), sinon, la possibilité d'observer les structures de OSER noncorrelatively est assez limité.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Les auteurs n'ont rien à révéler.
Acknowledgments
Les auteurs sont reconnaissants à Fondazione Filarète pour son aide et son appui à la publication de cet article. Nous tenons également à remercier Centro Europeo di Nanomedicina pour l'utilisation du microscope électronique à transmission TECNAI G2.
Materials
| Name | Company | Catalog Number | Comments |
| Dulbecco’s Modified Eagle Medium (DMEM) | Invitrogen | 41966029 | |
| Dulbecco’s Modified Eagle Medium (DMEM) w/o phenol red | Invitrogen | 31053028 | |
| Fetal Bovine Serum (FBS) | Invitrogen | 10270106 | |
| Pen/Strep | Invitrogen | 15140-122 | |
| L-Glutamine 200 mM solution | Invitrogen | 25030-024 | |
| jetPEI | Polyplus Transfection | PP10110 | |
| OxyFluor | Oxyrase Inc. | OF-0005 | |
| Glutaraldehyde Grade I | Sigma Aldrich | G5882 | |
| Sodium Cacodylate Trihydrate | Sigma Aldrich | C0250 | |
| Osmium Tetroxide 4% solution | Electron Microscopy Science | 19150 | |
| Uranyl Acetate Dihydrate | Sigma Aldrich | 73943 | slightly radioactive |
| Propylene Oxide | Sigma-Aldrich | 82320 | |
| EPON embedding medium kit | Sigma-Aldrich | 45359-1EA-F | |
| Lead Citrate | Electron Microscopy Science | 17800 | |
| Bench top centrifuge | Eppendorf | 5415 D | |
| Spectral Confocal Microscope | Leica Microsystems | TCS SP5 | |
| CO2 Microscope Cage Incubation System | OkoLab | ||
| Ultramicrotome | Leica Microsystems | UC6 | |
| Diamond knife | Diatome | Ultra 45 ° | |
| Transmission Electron Microscope | FEI | Tecnai G2 | |
| GraphPad Prism Software | GraphPad Software, Inc | ||
| Steel culture cell chamber for 24 mm coverslip | Bioscience Tools | CSC-25 | |
| Electron Microscopy grids | Electron Microscopy Science | G300Cu |
References
- Fasana, E., et al. A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 24, 1419-1430 (2010).
- Borgese, N., Francolini, M., Snapp, E. Endoplasmic reticulum architecture: structures in flux. Curr. Opin. Cell Biol. 18, 358-364 (2006).
- Ronchi, P., Colombo, S., Francolini, M., Borgese, N. Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J. Cell Biol. 181, 105-118 (2008).
- Lippincott-Schwartz, J., Snapp, E., Kenworthy, A. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2, 444-456 (2001).
- Lee, M. C., Miller, E. A., Goldberg, J., Orci, L., Schekman, R. Bi-directional protein transport between the ER and. 20, 87-123 (2004).
- Snapp, E. L., et al. Formation of stacked ER cisternae by low affinity protein interactions. J. Cell Biol. 163, 257-269 (2003).
- Papiani, G., et al. Restructured endoplasmic reticulum generated by mutant amyotrophic lateral sclerosis-linked VAPB is cleared by the proteasome. J. Cell Sci. 125, 3601-3611 (2012).
- Almsherqi, Z. A., Kohlwein, S. D., Deng, Y. Cubic membranes: a legend beyond the Flatland* of cell membrane organization. J. Cell Biol. 173, 839-844 (2006).
- Federovitch, C. M., Ron, D., Hampton, R. Y. The dynamic ER: experimental approaches and current questions. Curr. Opin. Cell Biol. 17, 409-414 (2005).
- Takei, K., Mignery, G. A., Mugnaini, E., Sudhof, T. C., De Camilli, P. Inositol 1,4,5-trisphosphate receptor causes formation of ER cisternal stacks in transfected fibroblasts and in cerebellar Purkinje cells. Neuron. 12, 327-342 (1994).
- Feldman, D., Swarm, R. L., Becker, J. Ultrastructural study of rat liver and liver neoplasms after long-term treatment with phenobarbital. Cancer Res. 41, 2151-2162 (1981).
- Sprocati, T., Ronchi, P., Raimondi, A., Francolini, M., Borgese, N. Dynamic and reversible restructuring of the endoplasmic reticulum induced by PDMP in cultured cells. J. Cell Sci. 119, 3249-3260 (2006).
- Costantini, L. M., Fossati, M., Francolini, M., Snapp, E. L. Assessing the tendency of fluorescent proteins to oligomerize under physiologic conditions. Traffic. 13, 643-649 (2012).
- Pyszniak, A. M., Welder, C. A., Takei, F. Cell surface distribution of high-avidity LFA-1 detected by soluble ICAM-1-coated microspheres. J. Immunol. 152, 5241-5249 (1994).
- Taylor, J. P., Hardy, J., Fischbeck, K. H. Toxic proteins in neurodegenerative disease. Science. 296, 1991-1995 (2002).
- Winklhofer, K. F., Tatzelt, J., Haass, C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 27, 336-349 (2008).
- Borgese, N., Gazzoni, I., Barberi, M., Colombo, S., Pedrazzini, E. Targeting of a tail-anchored protein to endoplasmic reticulum and mitochondrial outer membrane by independent but competing pathways. Mol. Biol. Cell. 12, 2482-2496 (2001).
- Chudakov, D. M., Matz, M. V., Lukyanov, S., Lukyanov, K. A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 90, 1103-1163 (2010).
- Bancaud, A., Huet, S., Rabut, G., Ellenberg, J. Fluorescence perturbation techniques to study mobility and molecular dynamics of proteins in live cells FRAP, photoactivation, photoconversion, and FLIP. Cold Spring Harb. Protoc. 2010, (2010).
- Michida, T., et al. Role of endothelin 1 in hemorrhagic shock-induced gastric mucosal injury in rats. Gastroenterology. 106, 988-993 (1994).

{kind=link}