

Summary
Wir beschreiben die bildgebenden Verfahren verwenden wir, um die Verteilung und Mobilität der transfizierten fluoreszierenden Proteinen in das endoplasmatische Retikulum (ER) mittels des konfokalen Abbildung von lebenden Zellen resident untersuchen. Wir haben auch ultrastrukturell analysieren die Wirkung ihrer Expression auf die Architektur dieser subzellulären Kompartiment.
Abstract
Die Lipide und Proteine in eukaryotischen Zellen kontinuierlich zwischen Zellkompartimenten ausgetauscht, obwohl diese trotz der intensiven interorganelle Molekularverkehr behalten ihre unverwechselbare Zusammensetzung und Funktionen. Die in diesem Beitrag beschriebenen Techniken sind mächtige Mittel des Studiums Protein-und Lipid Mobilität und Menschenhandel in vivo und in ihrer physiologischen Umgebung. Fluorescence Recovery After Photobleaching (FRAP) und Fluoreszenzverlust in Photobleaching (FLIP) werden häufig für die Untersuchung intrazellulären Transport durch die exo-endocytischen, die Kontinuität zwischen den Organellen oder Unterkammern, die Bildung von Proteinkomplexen und Protein-Lokalisierung verwendet Live-Cell-Imaging-Techniken Lipidmikrodomänen, die alle unter physiologischen und pathologischen Bedingungen zu beachten. Die Grenzen dieser Ansätze sind hauptsächlich auf die Verwendung von fluoreszierenden Fusionsproteine und ihre möglichen Nachteile umfassen artifactual überexprimierenIonen in Zellen und die Möglichkeit, Unterschiede in der Faltung und Lokalisierung von markierten und nativen Proteinen. Schließlich, als die Auflösungsgrenze der Lichtmikroskopie (etwa 200 nm) keine Untersuchung der Feinstruktur des ER oder den spezifischen Unterkammern, die in Zellen unter Stress entstehen kann damit (dh Hypoxie, Medikamentengabe, die Überexpression von Transmembran basierend auf Transmissions-Elektronenmikroskopie ER ansässig Proteine) oder unter pathologischen Bedingungen, kombinieren wir Live-Cell-Imaging von kultivierten transfizierten Zellen mit ultrastrukturelle Analysen.
Introduction
Die Entdeckung des grün fluoreszierenden Proteins (GFP) und dessen Varianten Spektralbereich und die parallele Entwicklung der Fluoreszenzmikroskopie wurden völlig neue Möglichkeiten für die Untersuchung von Protein-Verhalten in den Zellen geöffnet. Techniken wie Fluoreszenz-Erholung nach Photobleaching (FRAP) und Fluoreszenzverlust Photobleaching (FLIP), die aufgrund der Eigenkapazität der Fluorophore ihrer Fluoreszenz unter intensiver Beleuchtung erlischt möglich sind, werden auf der konfokalen Abbildung lebender Zellen basiert und die Verwendung von transfizierten Fluoreszenz-Fusionsproteine 1-3. Sie sind weit verbreitet, nicht nur die Lokalisierung von Proteinen, sondern auch ihre Mobilität und vesikulären Transport, die wichtige Hinweise über ihre Funktion 4 offenbaren kann beurteilen.
Das einzigartige Merkmal von eukaryontischen Zellen ist die Anwesenheit von intrazellulären Kompartimenten, die spezifische Lipid-und Proteinzusammensetzungen. Obwohl Organellen sind körperlich Isolated, müssen sie miteinander kommunizieren und molekularen Komponenten, um die zelluläre Homöostase aufrechtzuerhalten. Die sekretorischen Weg garantiert, dass die Proteine und Lipide im ER synthetisiert erreichen den richtigen Bestimmungsort in dem sie ihre Funktion ausüben. Intrazellulären Organellen kann auch durch dynamische Kontaktstellen, die Moleküle (Lipide), direkt zwischen den Abteilungen ausgetauscht werden können angeschlossen werden. Darüber hinaus haben viele Proteine in großen heteromeren Komplexen zusammengesetzt oder mit bestimmten Lipidspezies (Lipid Rafts / Mikrodomänen), um sich funktionell aktiv oder an ihren Bestimmungsort transportiert werden verbunden. Alle diese biologischen Aspekte stark beeinflussen die kinetischen Eigenschaften der Proteine, und kann daher in geeigneter Weise mittels der unten beschriebenen Methoden untersucht werden.
Unsere Gruppe hat sehr stark genutzt FRAP und FLIP mit Elektronenmikroskopie kombiniert, um die Architektur des ER und ihrer verschiedenen studierenDomänen. Das ER ist die erste Station des sekretorischen Weg und spielt eine Schlüsselrolle in der Protein-und Lipidsortierung 5. Es ist eine sehr dynamische Organellen, deren unterschiedliche Teilbereiche spiegeln die viele verschiedene Funktionen (zB Protein-und Lipid Biosynthese und Menschenhandel, Proteinfaltung, Ca 2 +-Speicher-und Freigabe und Fremdstoffmetabolismus). Obwohl sie morphologisch räumlich und funktionell verschieden sind diese Bereiche kontinuierlich zueinander und ihre relative Häufigkeit in Zellen unter physiologischen und pathologischen Bedingungen modifiziert werden. Die bekannteste und meist räumlich getrennter Domänen des ER sind die Kernhülle und die glatten und rauen ER, allerdings haben wir und andere gezeigt, dass es ER Strukturen mit aufwendiger Architektur und dreidimensionale Organisation in verschiedenen Zelltypen und Geweben unter physiologischen Bedingungen, die auch durch Stressreize wie Hypoxie, Drogen induziert werden kannVerwaltung, oder die Überexpression von ER-residenten Transmembranproteine 2,6 (und darin).
Wir haben vor kurzem auch die Anwesenheit solcher Strukturen in Zellmodellen menschlicher Krankheiten 1,7 nachgewiesen. Hervorgegangen aus dem gestapelten Zisternen des glatten ER, sie den gemeinsamen Namen der organisierten glatten endoplasmatischen Retikulum (OSER) im Jahr 2003 6 angegeben wurden, obwohl sie auch als karmellae, Lamellen und kristalloiden ER auf der Grundlage ihrer Architektur bekannt, die, wie ihre Größe kann variieren. Nachdem die Zellen mit GFP zu cytosolischen Schwanz-Bereich verankert (TA) ER fremde Proteine (d EGFP-ER), die schwach Dimerisierung Tendenz von GFP in trans dramatisch verändert die Organisation und Struktur der ER fusioniert transfiziert. FRAP und FLIP-Experimente zeigten, dass d EGFP-ER ist kostenlos innerhalb Osers diffundieren, und die Tatsache, dass es von der retikulären ER bewegt sich nach OSER und umgekehrt </ Em> zeigt an, dass die Aggregate sind kontinuierlich mit der umgebenden retikulären ER. Ultrastrukturelle Analyse hat uns erlaubt, die Fluoreszenzdaten mit einer detaillierten Beschreibung der OSER Architektur und Organisation auf Nanoebene korrelieren: Osers werden immer von Stapeln von gepaarten Zisternen des glatten ER gemacht, sondern kann verschiedene Formen der räumlichen Organisation, wie regelmäßig angeordneten Sinus haben Arrays oder Quirlen oder sechseckigen "kristalloiden" Rohr Arrays. Diese Umlagerungen führen zu kubischen Morphologien 8, die, wie sie in Zellen unter physiologischen Bedingungen 9 und folgenden Belastungen wie Hypoxie 10. Arzneimittelbehandlung 11, 9 und Krebs gefunden worden, erhebliche Potential als ultra Marker aufweisen kann.
Nach dieser ersten Demonstration mit GFP-Fusionsproteine verwendeten wir Abbildungsexperimente, die Proliferation von ER-Domänen analysiert als Reaktion auf pharmakologische Behandlungen 12, assess der Neigung von fluoreszierenden Proteinen in Zellen 13 oligomerisieren, und die Rolle eines mutierten ALS-verknüpften TA Protein an der Bildung von intrazellulären Aggregaten von ER Herkunft, die relevant für seine Pathogenität 1,8 sein kann, zu untersuchen. Es wurde vorgeschlagen, daß die Bildung von intrazellulären Aggregaten (die in vielen neurodegenerativen Krankheiten 14 auftritt) kann eine Schutzmechanismus, der die Wechselwirkungen zwischen toxischen mutierten Proteinen und den umgebenden Zellkomponenten 15 zu verhindern.
Was folgt, ist eine Beschreibung einer Kombination optischer und Elektronenmikroskopie Verfahren zur Untersuchung Konstrukte, deren C-terminale hydrophobe Domäne in der Membran des ER eingeführt, und eine Analyse ihrer dynamischen Verhalten und die Wirkungen der Überexpression von ER-Domäne Architektur in kultivierten Zellen (siehe Abbildung 1 ein Ablaufdiagramm des experimentellen Protokolls).
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Plasmid, Zellkultur und Transfektion mit ER Fluoreszierende Proteine
- Das in dieser Studie verwendete Plasmid besteht aus einer verbesserten Version des GFP am C-Terminus mit dem Heckbereich des ER-Isoform von Ratten-Cytochrom b fusioniert (5) über eine Linkersequenz (als B (5), hier abgekürzt). Der Heckbereich die gesamte Sequenz (Pro94-Asp134), die Membran nach der Trypsin-Spaltung der nativen b (5), einschließlich der 17-Rückstand TMD (Transmembran-Domäne), von vor-und nachgelagerten Polar Sequenzen flankiert (USV und DPS) zugeordnet bleibt, enthält . Der Linker besteht aus dem myc-Epitop, gefolgt von [(Gly) 4 Ser] 3, und die gesamte cDNA wird in die Hind3-Xba1-Stellen des Säugetier-Expressionsvektor pcDNA3. Die Einzelheiten der Konstruktion dieses Plasmids wurden in einer früheren Veröffentlichung, in der sie als GFP-ER 16 bezeichnet wird beschrieben.
- Wachsen COS-7-Zellen in Dulbeccos modifiziertem Eagle Medium (DMEM) mit 10% Feta ergänztl Rinderserum, 2 mM L-Glutamin, 1% Penicillin / Streptomycin in einem Inkubator bei 37 ° C und 10% CO 2.
- Transfektion. Platte 3 x 10 5 Zellen auf einer runden Deckglas in einer 6-Well-Platte, und am folgenden Tag, Transfektion mit dem jetPEI System, wie vom Hersteller beschrieben. Beachten Sie, dass die optimale jetPEI / DNA-Verhältnis hat, um die maximale Effizienz der Transfektion in Abhängigkeit von der Plasmid-und Zelllinie zu etablieren getestet: in unserem Fall ein jetPEI: DNA-Verhältnis von 2:1 führt zu einer 70-80% Transfektionseffizienz.
2. Live-Fluoreszenzmikroskop-Mikroskopie
- Live-Cell-Imaging. Das Deckglas gelegt, auf dem die transfizierten Zellen wurden in einer Zellkulturkammer Stahl 24 mm Deckgläsern mit DMEM gefüllt w / o Phenolrot, mit 10% FBS, 2 mM L-Glutamin, 1% Penicillin / Streptomycin, 25 mM HEPES ausgesät , 50 ug / ml Cycloheximid und 1:100 OxyFluor, um die Proben von Photobleichung zu verhindern. Ein SP5 konfokalen mikroskopische mit einem temperaturgesteuerten CO 2-Inkubator (37 ° C und 5% CO 2) ausgestattet ist zum lebenden Zellen Experimente verwendet, mit d-GFP-ER unter Verwendung eines 488-nm-Laser und einen 525/50 Bandpass-Emissionsfilter visualisiert.
- Fluorescence Recovery After Photobleaching (FRAP). Ziehung einer Region von Interesse (ROI), entsprechend einer OSER Struktur und entfärben mit 20 Iterationen und eine Kombination von 488 nm (100% eines 30 mW Argonlaser, entsprechend 5,5-6 &mgr; W in der Probe) und 405 nm (60% der 30 mW Laserdiode 405, das entspricht 11,6 uW bei der Probe)-Laser, die in unserer Erfahrung, führt zu einer effizienten und schnellen Photobleaching.
- Notieren Sie sich die Erholung der Fluoreszenz in den gebleichten ROIs, indem sie einen einzelnen Frame alle 10 s für 10 min (Pixelzeit = 1,61 Mikrosekunden / Pixel).
- Fluoreszenzverlust in Photobleaching (FLIP). Ziehung einer ROI entsprechend einem OSER Struktur und Bleichmittel, wie oben beschrieben. Das Bleichen wird alle 30 Sekunden wiederholt,und Post-Bleich Bilder werden alle 10 Sekunden für 30 min (Pixelzeit = 1,61 Mikrosekunden / Pixel) aufgezeichnet.
- FRAP und FLIP-Analyse. Alle Bilder sind mit ImageJ Software (analysiert http://rsbweb.nih.gov/ij/download.html ). In den FRAP Experimenten wird die Wiederherstellung der Fluoreszenz des gebleichten ROI über die Zeit und normiert auf die Gesamtfluoreszenz der gebleichten Zell, das immer aktiviert ist über die Zeit konstant ist, gemessen.
- Für die FLIP-Experimenten ziehen eine ROI außerhalb des gebleichten OSER und für die gesamte Zelle. Messung ihrer Fluoreszenzintensität über die Zeit zu normalisieren und die Fluoreszenzniveaus eines ROI auf einem ungebleichten Zell um für jede Abnahme der Fluoreszenz von der Abbildungs selbst zu korrigieren gezogen.
- In allen Experimenten, subtrahiert das Hintergrundsignal (in einem Bereich außerhalb der Zellen bestimmt) von den Fluoreszenzintensitäten der ROIs. Schließlich zeichnen die Ergebnisse mit GraphPad Prism Software.
3. Ultrastrukturelle Analyse mittels Transmissionselektronenmikroskopie
Angesichts der Toxizität vieler der Reagenzien sollten alle den Verfahren trägt einen geeigneten Labor durchgeführt werden. Mantel und Handschuhe unter einer Abzugshaube.
- Nach dem Entfernen des Deckglases aus der Petrischale, fixieren die restlichen Zellen am Boden der Schale als eine Monoschicht mit gefilterter 2% Glutaraldehyd in 0,1 M Cacodylatpuffer, pH 7,4, für 10 Minuten bei Raumtemperatur gezüchtet.
- Kratzen Sie die Zellen mit einer Teflon Schaber und übertragen sie in 1,5-ml-Eppendorf-Röhrchen. Pelletieren Sie die Zellen durch Zentrifugation bei 9000 g für 10 min. Entfernen Sie den Überstand, fügen Sie frisches Fixiermittel, und lassen Sie über Nacht bei 4 ° C
- Waschen der Pellets mit Puffer, dann post-fix mit einer Lösung von 1% Osmiumtetroxid in Cacodylat-Puffer für 1 Stunde bei Raumtemperatur.
- Spülen Sie mit MilliQ Wasser und en bloc Fleck mit1% Uranylacetat in destilliertem Wasser für 20-60 min.
- Entwässern der Proben in steigenden Ethanolreihe (70%, 80%, 90%, 100% und 100% für jeweils 10 Minuten) und kurz waschen zweimal in Propylenoxid (jeweils 15 min).
- Infiltrieren der Proben in einem Gemisch aus Propylenoxid + Epon (1:1) (2 h bis über Nacht).
- In Epon Epoxidharz bei 60 ° C für mindestens 24 Stunden gehärtet einzubetten.
- § die manuell getrimmt Harzblöcke mit Hilfe eines Ultramikrotoms LEICA UC6 mit einem 45 °-Diamantmesser ausgestattet, um Abschnitte mit einer Dicke von 60-70 nm zu erhalten. Sammeln Sie die Abschnitte über 300 mesh Kupfernetze.
- Färben Sie die Abschnitte in der Startaufstellung mit einer gesättigten Lösung von Uranylacetat (20 min) und Blei-Citrat (7 min) gründlich waschen, in die Netze von bi-destilliertem gefiltertes Wasser eingetaucht werden, und es ihnen ermöglichen, bei Raumtemperatur trocknen.
- Die gefärbten Gitter G2 TECNAI mit einem Transmissionselektronenmikroskop beobachtet, und die Bilder werden unter Verwendung eines erfassten Bottom-CCD-Kamera montiert in verschiedenen Vergrößerungen letzten (in der Regel zwischen 6,000-39,000 X).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Figur 2 zeigt ein Beispiel FRAP Untersuchung von Protein-Mobilität. Die Mobilität von d EGFP-ER-Protein wird durch die schnelle Erholung nach Fluoreszenzphotobleaching in gebleicht Osers demonstriert. Für die quantitative Analyse wurden die Halbzeit-und Mobil Fraktion aus experimentell gemessenen Daten durch Einsetzen der folgenden monoexponentiellen Gleichung abgeleitet:
F (t) = F post + (F-F rec post) (1-e-t / τ)
wobei F Beitrag ist das Fluoreszenzsignal nach Photobleaching, F rec der maximale Wert, der Wiederherstellung der Fluoreszenz nach dem Bleichen zu erreichen ist, t zum Zeitpunkt der Registrierung und τ die Zeitkonstante.
Bitte beachten Sie die Bedeutung des Erwerbs von Bildern ohne gesättigten Pixeln, die die Wiederherstellung der Fluoreszenz verändern könnten und damit das Protein Mobilitätsanalyse. Es iist auch wichtig, um das Fluoreszenzsignal im gebleichten ROI immer normalisiert auf die Gesamtfluoreszenz der gleichen Zelle, um die Fluoreszenzintensität aufgrund von Variationen Bleichen während der Bildaufnahme oder kleine Veränderungen in der Fokusebene zu berücksichtigen.
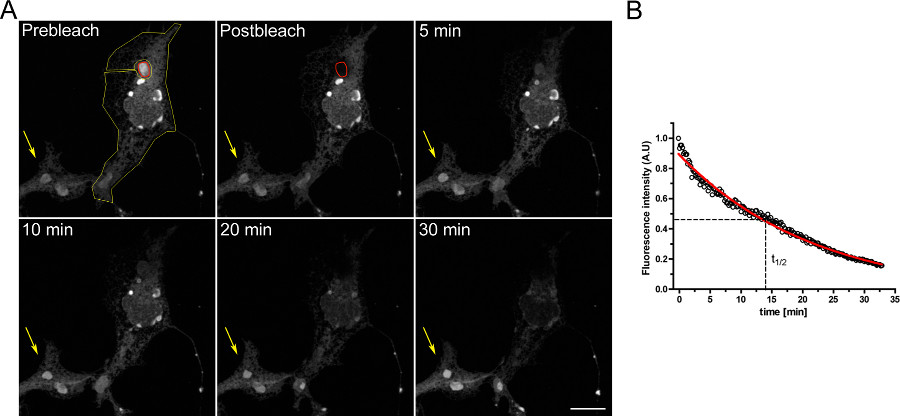
Ein Beispiel eines FLIP-Experiment, um die Kontinuität zwischen den intrazellulären Kompartimenten zu untersuchen, ist in Fig. 3 gezeigt. Osers physikalisch mit dem Rest des ER verbunden ist, wie der progressive Entleerung des ER bei OSER Domäne kontinuierlich gebleicht wird demonstriert.
Für eine korrekte Analyse, muss der Erwerb von gesättigten Pixel vermieden werden (siehe oben), außerdem müssen die Erfassungsparameter mit Laserleistungen so gering wie möglich, um zu vermeiden Bleichen durch Bildaufnahme eingestellt werden. Aus diesem Grund ist es dringend empfohlen, um Bild eines ungebleichten Zell im gleichen Feld, das verwendet wird, um das Fluoreszenzsignal des gebleichten c normalisiert werdenell.
Alle Experimente wurden in Gegenwart von Cycloheximid, ein Inhibitor Übersetzung durchgeführt werden, um eine Erhöhung des Fluoreszenzsignals ER (und folglich Gesamtfluoreszenz) aufgrund der Proteinbiosynthese zu vermeiden.
Transmissionselektronenmikroskopie zeigte, daß die kultivierten Zellen mit EGFP-d ER transfiziert beobachteten Fluoreszenz Aggregate darstellen Flecken glatt und flach ER-Zisternen, die sich räumlich in definierte 3D-Geometrien klassifiziert auf der Grundlage der Muster organisiert: linear oder gekrümmt Stapel (oft mit der Kernhülle, nicht gezeigt) (Fig. 4A und B), die kontinuierlich mit Bereichen Sinus ER sein kann (4A) verbunden ist; die Membranen in einigen Regionen sind in Gittern mit quadratischen oder hexagonalen Symmetrie (kristalloiden ER organisiert nicht dargestellten ). Neben Zisternen sind durch eine dünne Schicht von etwas getrennt elektronendichte Zytoplasma etwa 11 nm dick, dass ist kontinuierlich mit dem Zytoplasma rund um die Aggregate.

Fig. 1 ist. Flussdiagramm des experimentellen Verfahrens. Die kultivierten Zellen werden zuerst mit jetPEI (siehe Protokoll), um überexprimieren das fluoreszierende Fusionsprotein von Interesse transfiziert. Nach 24 Stunden werden die Live-transfizierten Zellen visualisiert und FRAP-und FLIP-Experimente werden unter Verwendung eines konfokalen Mikroskops mit einer kontrollierten Temperatur CO 2-Inkubator ausgestattet war, und die aufgezeichneten Bilder ausgeführt werden, unter Verwendung geeigneter Software (zB ImageJ) analysiert. Für die ultrastrukturelle Analyse, werden die transfizierten Zellen fixiert, pelletiert und in Epon Epoxidharz Blöcke eingebettet. Ultradünne Schnitte werden unter Verwendung eines Diamantmessers erhalten wird, auf C gewonnenopper Gitter, und unter einem Transmissionselektronenmikroskop beobachtet. Klicken Sie hier, um eine größere Abbildung anzuzeigen .

2. FRAP-Experiment unter Verwendung von COS-7-Zellen transient mit d EGFP-ER transfiziert. A) Zwei OSER Strukturen (rot ROIs) wurden gebleicht, und Fluoreszenz Erholung wurde im Laufe der Zeit aufgezeichnet. Klar Fluorescence Recovery erkannt werden 1 min nach dem Bleichen, und das Signal weiter zunimmt 4 Minuten später (Maßstab 10 um) B):. Quantitative Analyse der FRAP-Experiment, das die Erholung der Halbzeit und die mobile Bruchteil der d EGFP ER-Protein. Click hier, um eine größere Abbildung anzuzeigen

3. FLIP-Experiment unter Verwendung von COS-7-Zellen transient mit d EGFP-ER transfiziert. A) Die kontinuierliche Bleich eines OSER (rote ROI angezeigt) bewirkt eine fortschreitende Abnahme in der Fluoreszenz in den Rest des ER und anderen OSER Strukturen innerhalb der gleichen Zelle (durch den gelben ROI angezeigt). Der gelbe Pfeil zeigt einen Teil eines ungebleichten Zell in dem das Fluoreszenzsignal über die Zeit konstant ist. (Maßstabsbalken 10 um). B) Quantitative Analyse der FLIP-Experiment. Klicken Sie hier, um eine größere Abbildung anzuzeigen
4. Nach der Fixierung und Einbettung wurden die Zellen, die hohe Konzentrationen von d EGFP-ER in dem OSER Strukturen könnten mittels Fluoreszenzmikroskopie festgestellt werden durch ein Transmissionselektronenmikroskop beobachtet. A) Niedervergrößerungsansicht eines Teils des Zytoplasma einer Zelle, die ein OSER bestehend aus gestapelten Zisternen und sinuswellenförmigen Membranen. Mitochondrien (M) zu sehen rund um die OSER Strukturen gebündelt werden, während Ribosomen schmücken nur die Membranen der äußersten Zisternen (Pfeilspitzen und Kasten). Die 11 nm dicken elektronendichten Raum zwischen den Membranen ist kontinuierlich mit dem Zytoplasma (Pfeil und kleines Bild) (L = Lysosomen / (Auto-) Phagosomen) (Maßstab 1,5 um; Einsatz 0,25 um). B) Ein OSER gebildet werden von Lamellen ER: dh Stapel von abgeflachten ER-Zisternen, die kontinuierliche oder fragme seinin ihrem Aussehen in Dünnschnitten nted. Vesikel Knospung aus der äußersten Zisternen des Stapels (Sternchen) (PM, Plasmamembran) (Maßstab 150 nm) gelegentlich beobachtet werden.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Die in diesem Dokument beschriebenen Protokolle und bildgebenden Verfahren sind verwendet worden, um die Verteilung und Mobilität von transfizierten TA fluoreszierende Proteine im ER lebender Zellen resident untersuchen. Wir haben auch untersucht die Wirkung der Überexpression dieser Proteine auf die Architektur dieser subzellulären Kompartiment durch ultrastrukturelle Analyse.
Die Kombination aus Live-Zell-konfokale Bildgebung und Elektronenmikroskopie stellt ein sehr leistungsfähiges Mittel zur Untersuchung der dynamischen Eigenschaften von Proteinen und kann wichtige Informationen für die Proteinfunktion zu schaffen. Die beschriebenen Verfahren sind nicht zeitaufwendig (in der Regel drei Tage der Arbeit) und die Entwicklung von vielen benutzerfreundliche Software-Anwendungen für die Bildaufnahme und Analyse macht Photobleaching-basierte, Live-Cell-Imaging relativ einfach.
Die Haupteinschränkung dieser Techniken ist die Verwendung von fluoreszierenden Fusionsproteinen, da dieFluoreszenzmarkierung kann die richtige Faltung und / oder Montage des Proteins von Interesse zu beeinflussen. Darüber hinaus über-Ausdruck kann das Verhalten von transfizierten, fluoreszenzmarkierten Proteine verändern, und können daher nicht die tatsächlichen Eigenschaften der körpereigenen Proteinen widerspiegeln, jedoch kann dies durch die Verwendung von induzierbaren und stabil transfizierten Zellen, in denen das Expressionsniveau kann überwunden werden präzise moduliert, um ein Niveau vergleichbar mit denen des endogenen Proteins 1,7 erhalten. Die Tendenz der FPs oligomerisieren wurde ausführlich dokumentiert und können das Verhalten (dh Kinetik unerwünschte Protein-Protein-Wechselwirkungen und die Bildung von Aggregaten) von chimären Proteinen signifikant verändert. Die Verwendung von fluoreszierenden Proteinen optimiert monomeren sollte daher als 17 sein.
Ein weiterer kritischer Aspekt der dynamische Bildgebung Studien unter Verwendung von Fluoreszenz und Photobleaching ist die Zeit benötigt, um die Fluoreszenz effizient zu bleichen und zu messen FluoreszenzwiederErholung (und damit die Protein Mobilität) genau, die auch abhängig von der Fläche der ROI und lokale Zelldicke. Wenn eine gegebene GFP-markierten Protein einen hohen Diffusionskoeffizienten, könnte Diffusion während des Bleich auftreten und somit stören Erholungszeit-Messungen. Um eine schnelle und effiziente Bleich zu erhalten, ist es dringend empfohlen, dass ein "zoom in" Funktion (wenn verfügbar) und mehr als eine Laserlinie verwendet werden. Obwohl die Verwendung einer Fast-Scan-Modul (dh ein Resonanz-Scanner) kann stark die Geschwindigkeit der Bildgebung während der Erholungsphase des Experiments zu verbessern, in der Hand auch erheblich reduziert Bleicheffizienz. Allerdings können alternative Scansysteme (wie eine sich drehende Scheibe mit einem speziellen Gerät ausgestattet Photobleaching) und leistungsfähiger Laser sowohl Bleicheffizienz und Aufnahmegeschwindigkeit zu verbessern.
Die meisten fluoreszierenden Proteinen in FRAP und FLIP-Experimente verwendet zeigen einen gewissen Grad von reversiblen Photobleaching und blinking, dass muss bei der Durchführung quantitativer Analysen werden kann. Die Schwankungen zwischen den Leuchtstoff-und Dunkelzustände auftreten, in der zweiten Minute auf Zeitskala. Für EGFP, hat es sich gezeigt, dass beim Bleichen Experimente könnten Fluoreszenz-Variationen beinhalten weniger als 10% der Moleküle, also in der vorliegenden Protokoll dieses Phänomen ist vernachlässigbar. Wenn alle Bedingungen konstant gehalten werden, wird diese eine konstante Vorspannung in den Ergebnissen vor. Wenn andere fluoreszierende Proteine eingesetzt werden, bei denen der reversible Anteil ist wesentlich höher (dh YFP) oder zu erfassen und auszuwerten Bleichen Reversibilität kann dies durch Messen der Fluoreszenz Erholung nach Bleichen in der gesamten lebenden Zellen durchgeführt werden, wenn diese Rückgewinnung beobachtet kann nur das Ergebnis von Photobleichen Reversibilität 18 sein.
Die potentielle Toxizität von Licht während der Experimente ist ein weiterer kritischer Faktor, insbesondere weil Bleichen erfordert eine starke Beleuchtung. Es ist well bekannt, daß angeregte Fluorophore neigen dazu, mit Sauerstoff, um freie Radikale, die verschiedene intrazelluläre Prozesse und auch die Lebensfähigkeit der Zellen 19 beeinflussen können produzieren reagieren, und so ist es notwendig, ein Gleichgewicht zwischen effizienter Bleich und minimale Photo herzustellen, außerdem sollte die Lebensfähigkeit der Zellen immer überprüft werden, nach den Live-Cell-Imaging-Experimente. Angesichts der kurzen Aufnahmezeit, wir haben nicht die gentoxische Wirkung von Licht kurzer Wellenlänge (405 nm) in der in diesem Dokument beschriebenen Beispiel betrachten, aber, wenn eine längere Experiment erforderlich ist, sollte eine 405 nm Laserlinie nicht verwendet werden.
Wir haben uns nicht auf eine korrelative Ansatz für Transmissions-Elektronenmikroskopie wegen der Heterogenität der OSER Architektur und der Tatsache, dass wir so viele Zellen (und Strukturen) wie möglich zu beobachten wollte, zu verwenden. Die Vielfalt der Feinstruktur von Proteinaggregaten in den Zellen kann ein Schlüsselmerkmal der verschiedenen Krankheiten sein, und wir waren daran interessiert, eine breite Palette von samples, während ein Korrelat Ansatz ermöglicht die Beobachtung von weniger Veranstaltungen im gleichen Zeitraum der Zeit. Jedoch sollte korrelativen Lichtelektronenmikroskopie (Clem) die erste Wahl bei der Untersuchung von Ereignissen in Strukturen, die nicht leicht identifiziert werden kann (z. B. weniger prominent ER Domänen) oder in einer begrenzten Anzahl von Zellen (wie z. B. Mikro-injizierten Zellen) sein. Es ist erwähnenswert, dass unsere Experimente wurden mit einem hohen Grad an Transfektionseffizienz dadurch (zumindest 30% der Zellen transfiziert wurden), da sonst die Möglichkeit der Beobachtung OSER Strukturen noncorrelatively ist recht begrenzt.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Die Autoren haben nichts zu offenbaren.
Acknowledgments
Die Autoren sind dankbar, Fondazione Filarete für seine Hilfe und Unterstützung bei der Veröffentlichung dieses Artikels. Wir würden auch gerne Centro Europeo di Nanomedicina für die Nutzung der TECNAI G2 Transmissionselektronenmikroskop danken.
Materials
| Name | Company | Catalog Number | Comments |
| Dulbecco’s Modified Eagle Medium (DMEM) | Invitrogen | 41966029 | |
| Dulbecco’s Modified Eagle Medium (DMEM) w/o phenol red | Invitrogen | 31053028 | |
| Fetal Bovine Serum (FBS) | Invitrogen | 10270106 | |
| Pen/Strep | Invitrogen | 15140-122 | |
| L-Glutamine 200 mM solution | Invitrogen | 25030-024 | |
| jetPEI | Polyplus Transfection | PP10110 | |
| OxyFluor | Oxyrase Inc. | OF-0005 | |
| Glutaraldehyde Grade I | Sigma Aldrich | G5882 | |
| Sodium Cacodylate Trihydrate | Sigma Aldrich | C0250 | |
| Osmium Tetroxide 4% solution | Electron Microscopy Science | 19150 | |
| Uranyl Acetate Dihydrate | Sigma Aldrich | 73943 | slightly radioactive |
| Propylene Oxide | Sigma-Aldrich | 82320 | |
| EPON embedding medium kit | Sigma-Aldrich | 45359-1EA-F | |
| Lead Citrate | Electron Microscopy Science | 17800 | |
| Bench top centrifuge | Eppendorf | 5415 D | |
| Spectral Confocal Microscope | Leica Microsystems | TCS SP5 | |
| CO2 Microscope Cage Incubation System | OkoLab | ||
| Ultramicrotome | Leica Microsystems | UC6 | |
| Diamond knife | Diatome | Ultra 45 ° | |
| Transmission Electron Microscope | FEI | Tecnai G2 | |
| GraphPad Prism Software | GraphPad Software, Inc | ||
| Steel culture cell chamber for 24 mm coverslip | Bioscience Tools | CSC-25 | |
| Electron Microscopy grids | Electron Microscopy Science | G300Cu |
References
- Fasana, E., et al. A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 24, 1419-1430 (2010).
- Borgese, N., Francolini, M., Snapp, E. Endoplasmic reticulum architecture: structures in flux. Curr. Opin. Cell Biol. 18, 358-364 (2006).
- Ronchi, P., Colombo, S., Francolini, M., Borgese, N. Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J. Cell Biol. 181, 105-118 (2008).
- Lippincott-Schwartz, J., Snapp, E., Kenworthy, A. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2, 444-456 (2001).
- Lee, M. C., Miller, E. A., Goldberg, J., Orci, L., Schekman, R. Bi-directional protein transport between the ER and. 20, 87-123 (2004).
- Snapp, E. L., et al. Formation of stacked ER cisternae by low affinity protein interactions. J. Cell Biol. 163, 257-269 (2003).
- Papiani, G., et al. Restructured endoplasmic reticulum generated by mutant amyotrophic lateral sclerosis-linked VAPB is cleared by the proteasome. J. Cell Sci. 125, 3601-3611 (2012).
- Almsherqi, Z. A., Kohlwein, S. D., Deng, Y. Cubic membranes: a legend beyond the Flatland* of cell membrane organization. J. Cell Biol. 173, 839-844 (2006).
- Federovitch, C. M., Ron, D., Hampton, R. Y. The dynamic ER: experimental approaches and current questions. Curr. Opin. Cell Biol. 17, 409-414 (2005).
- Takei, K., Mignery, G. A., Mugnaini, E., Sudhof, T. C., De Camilli, P. Inositol 1,4,5-trisphosphate receptor causes formation of ER cisternal stacks in transfected fibroblasts and in cerebellar Purkinje cells. Neuron. 12, 327-342 (1994).
- Feldman, D., Swarm, R. L., Becker, J. Ultrastructural study of rat liver and liver neoplasms after long-term treatment with phenobarbital. Cancer Res. 41, 2151-2162 (1981).
- Sprocati, T., Ronchi, P., Raimondi, A., Francolini, M., Borgese, N. Dynamic and reversible restructuring of the endoplasmic reticulum induced by PDMP in cultured cells. J. Cell Sci. 119, 3249-3260 (2006).
- Costantini, L. M., Fossati, M., Francolini, M., Snapp, E. L. Assessing the tendency of fluorescent proteins to oligomerize under physiologic conditions. Traffic. 13, 643-649 (2012).
- Pyszniak, A. M., Welder, C. A., Takei, F. Cell surface distribution of high-avidity LFA-1 detected by soluble ICAM-1-coated microspheres. J. Immunol. 152, 5241-5249 (1994).
- Taylor, J. P., Hardy, J., Fischbeck, K. H. Toxic proteins in neurodegenerative disease. Science. 296, 1991-1995 (2002).
- Winklhofer, K. F., Tatzelt, J., Haass, C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 27, 336-349 (2008).
- Borgese, N., Gazzoni, I., Barberi, M., Colombo, S., Pedrazzini, E. Targeting of a tail-anchored protein to endoplasmic reticulum and mitochondrial outer membrane by independent but competing pathways. Mol. Biol. Cell. 12, 2482-2496 (2001).
- Chudakov, D. M., Matz, M. V., Lukyanov, S., Lukyanov, K. A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 90, 1103-1163 (2010).
- Bancaud, A., Huet, S., Rabut, G., Ellenberg, J. Fluorescence perturbation techniques to study mobility and molecular dynamics of proteins in live cells FRAP, photoactivation, photoconversion, and FLIP. Cold Spring Harb. Protoc. 2010, (2010).
- Michida, T., et al. Role of endothelin 1 in hemorrhagic shock-induced gastric mucosal injury in rats. Gastroenterology. 106, 988-993 (1994).

{kind=link}