

Summary
Descriviamo gli approcci di imaging usiamo per indagare la distribuzione e la mobilità delle proteine fluorescenti trasfettate residenti nel reticolo endoplasmatico (ER) mediante la microscopia confocale di cellule viventi. Abbiamo anche ultrastrutturalmente di analizzare l'effetto della loro espressione sulla architettura di questo comparto subcellulare.
Abstract
I lipidi e proteine in cellule eucariotiche sono continuamente scambiati tra compartimenti cellulari, anche se questi mantengono la loro composizione distintivo e funzioni nonostante il traffico intenso interorganelle molecolare. Le tecniche descritte in questo documento sono potenti mezzi di studiare proteine e lipidi mobilità e il traffico in vivo e nel loro ambiente fisiologico. Recupero di fluorescenza dopo photobleaching (FRAP) e perdita di fluorescenza in photobleaching (FLIP) sono ampiamente utilizzati tecniche di live-cell imaging per studiare traffico intracellulare attraverso la via eso-endocitosi, la continuità tra organelli o sottocompartimenti, la formazione di complessi proteici, proteine e localizzazione in microdomini lipidici, ognuno dei quali può essere osservato in condizioni fisiologiche e patologiche. I limiti di questi approcci sono principalmente dovute all'uso di proteine di fusione fluorescenti, e loro potenziali svantaggi includono artefatta over-esprimonoione nelle cellule e la possibilità di differenze nella piegatura e localizzazione delle proteine marcate e native. Infine, come il limite di risoluzione della microscopia ottica (200 nm) non consente di indagine della struttura fine del ER o le sottocompartimenti specifici che possono provenire cellule sotto stress (cioè ipossia, la somministrazione del farmaco, l'espressione eccessiva di transmembrana proteine ER residenti) o in condizioni patologiche, uniamo live-cell imaging di cellule trasfettate coltivate con ultrastrutturali analisi sulla base di microscopia elettronica a trasmissione.
Introduction
La scoperta della proteina fluorescente verde (GFP) e le sue varianti spettrali, e lo sviluppo parallelo di microscopia a fluorescenza, hanno aperto completamente nuove strade per lo studio del comportamento delle proteine nelle cellule. Tecniche come recupero di fluorescenza dopo photobleaching (FRAP) e perdita di fluorescenza in photobleaching (FLIP), che sono possibili per la capacità intrinseca di fluorofori per estinguere la loro fluorescenza sotto illuminazione intensa, si basano su confocale live-cell imaging e l'uso di trasfettati proteine di fusione fluorescenti 1-3. Essi sono ampiamente utilizzati per valutare non solo la localizzazione delle proteine, ma anche la loro mobilità e del trasporto vescicolare, che può rivelare indizi importanti riguardanti la loro funzione 4.
La caratteristica unica di cellule eucariotiche è la presenza di compartimenti intracellulari con specifici composizioni lipidiche e proteiche. Anche se organelli sono fisicamente isolatEd, hanno bisogno di comunicare tra loro e condividere componenti molecolari al fine di mantenere l'omeostasi cellulare. La via secretoria garantisce che le proteine e lipidi sintetizzati in ER raggiungere la destinazione finale corretto in cui esercitano la loro funzione. Organelli intracellulari possono anche essere collegati da siti di contatto dinamici che permettono molecole (lipidi) da scambiare direttamente tra compartimenti. Inoltre, molte proteine devono assemblati in grandi complessi eteromerici o associati a determinate specie di lipidi (zattere lipidiche / microdomini) per diventare funzionalmente attivo o ad essere trasportati alla loro destinazione finale. Tutti questi aspetti biologici influenzare fortemente le proprietà cinetiche di proteine, e possono quindi essere opportunamente studiato mediante tecniche di seguito descritte.
Il nostro gruppo ha ampiamente utilizzato FRAP e FLIP combinato con microscopia elettronica per studiare l'architettura del ER e la sua diversa subdomini. La ER è la prima stazione della via secretoria e svolge un ruolo chiave nel proteine e lipidi ordinamento 5. Si tratta di un organello altamente dinamica la cui sottodomini distinti riflettere le sue diverse funzioni (ad esempio proteine e lipidi biosintesi e il traffico, proteine pieghevoli, Ca 2 + stoccaggio e il rilascio, e il metabolismo xenobiotici). Tuttavia, anche se sono morfologicamente, spazialmente e funzionalmente distinte, questi domini sono continue tra loro, e la loro numerosità possono essere cambiate in cellule in condizioni fisiologiche e patologiche. Il più conosciuto e domini di solito spazialmente separate della ER sono la membrana nucleare, e la ER liscio e ruvido, tuttavia, noi ed altri abbiamo dimostrato che non ci sono strutture ER con un'architettura più elaborata e l'organizzazione tridimensionale in vari tipi di cellule e tessuti in condizioni fisiologiche che può anche essere indotta mediante stimoli stressanti quali ipossia, farmacoamministrazione, o l'over-espressione di ER-residente proteine transmembrana 2,6 (e riferimenti).
Abbiamo recentemente dimostrato la presenza di tali strutture in modelli cellulari di malattie umane 1,7. Provenienti dalla cisterne pila di ER liscio, sono stati dati il nome collettivo di reticolo endoplasmatico liscio organizzata (OSER) nel 2003 6, anche se sono noti anche come karmellae, lamelle, e cristalloide ER sulla base della loro architettura che, come loro dimensione, può variare. Dopo che le cellule sono trasfettate con GFP fusa alla regione citosolica di coda ancorata (TA) proteine ER-residente (d EGFP-ER), la tendenza debolmente dimerizing di GFP in trans altera drasticamente l'organizzazione e la struttura del ER. FRAP e FLIP esperimenti hanno mostrato che d EGFP-ER è libero di diffondere all'interno OSERs, e il fatto che si muove dal ER reticolare alla OSER e viceversa </ Em> indica che gli aggregati sono continui con il reticolare circostante ER. Analisi ultrastrutturale ha permesso di correlare i dati di fluorescenza con una descrizione dettagliata di architettura OSER e di organizzazione a livello di nanoscala: OSERs sono sempre costituito da pile di cisterne abbinato di ER liscia, ma possono avere diverse forme di organizzazione spaziale, come regolarmente organizzato sinusoidale matrici o spirali, o esagonali "cristalloidi" matrici tubolari. Questi riarrangiamenti portano a morfologie cubi 8 che, così come sono stati trovati in cellule in condizioni fisiologiche 9 e seguenti sollecitazioni quali ipossia 10, il trattamento farmacologico 11, e cancro 9, possono avere un potenziale significativo come marcatori ultrastrutturali.
Dopo questa prima dimostrazione utilizzando proteine di fusione GFP, abbiamo usato esperimenti di imaging per analizzare la proliferazione di domini ER in risposta a trattamenti farmacologici 12, assess la tendenza delle proteine fluorescenti a oligomerise in celle 13, e per studiare il ruolo di un mutante proteina TA ALS-linked nella formazione di aggregati intracellulari di origine ER che possono essere rilevanti per la sua 1,8 patogenicità. È stato suggerito che la formazione di aggregati intracellulari (che si verifica in molte malattie neurodegenerative 14) può essere un meccanismo protettivo progettato per prevenire le interazioni tra proteine mutanti tossiche e le componenti cellulari circostanti 15.
Quello che segue è una descrizione di una combinazione di metodi di microscopia ottica ed elettronica per lo studio costrutti cui C-terminale domini idrofobici sono inseriti nella membrana del ER, e l'analisi del loro comportamento dinamico e gli effetti della loro iper-espressione di dominio ER architettura in cellule in coltura (vedere la Figura 1 per un diagramma di flusso del protocollo sperimentale).
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Plasmide, coltura cellulare, e Transfezione con ER proteine fluorescenti
- Il plasmide utilizzato in questo studio consiste di una versione migliorata del GFP fusa al suo C-terminale della regione di coda della isoforma di ER ratto citocromo b (5) (abbreviato qui come b (5)) tramite una sequenza linker. La regione coda contiene l'intera sequenza (Pro94-Asp134) che rimane membrana associato dopo la scissione di tripsina nativo b (5), compreso il TMD 17 residui (dominio transmembrana), fiancheggiata da sequenze polari a monte ea valle (UPS e DPS) . Il linker consiste nella epitopo myc seguito da [(Gly) 4 Ser] 3, e l'intero cDNA viene inserito nei siti Hind3-Xba1 del vettore di espressione di mammifero pCDNA3. I dettagli della costruzione di questo plasmide sono stati descritti in una precedente pubblicazione in cui è indicato come GFP-ER 16.
- Grow COS-7 cellule di Dulbecco Modified Eagle Medium (DMEM) supplementato con 10% fetasiero bovino l, 2 mM L-glutammina, 1% di penicillina / streptomicina in un incubatore a 37 ° C e 10% di CO 2.
- Trasfezione. Piastra 3 x 10 5 cellule su un vetrino di vetro rotondo in una piastra da 6 pozzetti e, il giorno seguente, trasfezione con il sistema JetPEI come descritto dal produttore. Si noti che il rapporto ottimale JetPEI / DNA è stato testato per stabilire la massima efficienza di trasfezione seconda del plasmide e linea cellulare utilizzata: nel nostro caso, un JetPEI: DNA rapporto di 2:1 conduce al 70-80% efficienza di trasfezione.
2. Diretta a fluorescenza Microscopia confocale a scansione
- Live-cell imaging. Mettere il vetrino in cui le cellule transfettate sono state seminate in una camera di coltura cellulare in acciaio per 24 millimetri coprioggetto riempiti con DMEM w / o rosso fenolo, supplementato con 10% FBS, 2 mM L-glutammina, 1% pen / strep, 25 HEPES mM , 50 pg / ml cicloeximide e 1:100 OxyFluor per prevenire i campioni da photobleaching. Un microscop SP5 confocalee dotato di CO 2 incubatore a temperatura controllata (37 ° C e 5% CO 2) è utilizzato per esperimenti di imaging cellulare dal vivo, con d GFP-ER essere visualizzata utilizzando un laser 488 nm ed un filtro 525/50 emissione passa banda.
- Recupero di fluorescenza dopo photobleaching (FRAP). Prelievo di una regione di interesse (ROI), corrispondente ad una struttura OSER, e decolorazione con 20 iterazioni e una combinazione di 488 nm (100% di un laser 30 mW Argon, corrispondente a 5,5-6 μW al campione) e 405 nm (60% di 30 mW diodo laser 405, corrispondente a 11,6 μW al campione) laser che, nella nostra esperienza, porta a fotoscolorimento efficiente e rapido.
- Registrare il recupero della fluorescenza nei ROI sbiancate prendendo un singolo fotogramma ogni 10 secondi per 10 minuti (tempo di pixel = 1.61 msec / px).
- Perdita di fluorescenza in photobleaching (FLIP). Disegnare un ROI corrispondente ad una struttura OSER, e candeggina come descritto sopra. Lo sbiancamento si ripete ogni 30 sec,e le immagini post-sbiancanti vengono registrati ogni 10 secondi per 30 minuti (tempo di pixel = 1.61 msec / px).
- FRAP e FLIP analisi. Tutte le immagini sono analizzate utilizzando il software ImageJ ( http://rsbweb.nih.gov/ij/download.html ). Negli esperimenti FRAP, il recupero di fluorescenza della ROI sbiancato è misurata nel tempo e normalizzati alla fluorescenza totale di cellule sbiancato, che è sempre verificata sia costante nel tempo.
- Per gli esperimenti FLIP, disegnare un ROI al di fuori della OSER sbiancato e che coprono tutta la cella. Misurare l'intensità di fluorescenza nel tempo e normalizzare i livelli di fluorescenza di una ROI tratti su una cella greggi per correggere in caso di riduzione della fluorescenza causata dalla rappresentazione stessa.
- In tutti gli esperimenti, sottratto il segnale di fondo (determinato in una zona diversa da cellule) dalle intensità di fluorescenza delle ROI. Infine, tracciare i risultati utilizzando Grsoftware aphPad Prism.
3. Ultrastrutturali analisi mediante microscopia elettronica a trasmissione
Data la tossicità di molti dei reagenti, tutte le procedure devono essere effettuate indossando un laboratorio appropriato. cappotto e guanti sotto una cappa aspirante.
- Dopo aver rimosso il vetrino dalla piastra di Petri, fissare le restanti cellule cresciute sul fondo del piatto come un monostrato utilizzando filtrata 2% glutaraldeide in 0,1 M tampone cacodilato, pH 7,4, per 10 min a temperatura ambiente.
- Raschiare le cellule utilizzando un raschietto Teflon e trasferirli in provette da 1,5 ml Eppendorf. Agglomerare le cellule mediante centrifugazione a 9000 g per 10 min. Rimuovere il surnatante, aggiungere fissativo fresco, e lasciare per una notte a 4 ° C.
- Lavare il pellet con il tampone, poi post-fissare con una soluzione di 1% tetrossido di osmio in tampone cacodilato per 1 ora a temperatura ambiente.
- Risciacquare con acqua MilliQ, e bagno macchia blocco con1% acetato di uranile in acqua distillata per tra 20-60 min.
- Disidratare i campioni ascendente serie di etanolo (70%, 80%, 90%, 100% e 100% per 10 min ciascuno), e lavare rapidamente due volte in ossido di propilene (15 min ciascuna).
- Infiltrati i campioni in una miscela di ossido di propilene + Epon (1:1) (da 2 ore a tutta la notte).
- Incorpora in resina epossidica Epon indurito a 60 ° C per almeno 24 ore.
- Sezione blocchi resina tagliati manualmente utilizzando un ultramicrotomo LEICA UC6 dotato di una lama di diamante 45 ° per ottenere sezioni dello spessore di 60-70 nm. Raccogliere le sezioni su 300 griglie di rame maglia.
- Macchiare le sezioni sulla griglia di partenza con una soluzione satura di acetato di uranile (20 min) e citrato di piombo (7 min), lavare accuratamente le griglie immergendole in acqua filtrata bi-distillata e lasciare asciugare a temperatura ambiente.
- Le griglie colorate vengono osservate con un microscopio elettronico a trasmissione Tecnai G2, e le immagini sono state acquisite utilizzando una bottcamera CCD om-montato in diversi ingrandimenti finali (in genere vanno da 6,000-39,000 X).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
La Figura 2 mostra un esempio di studio FRAP mobilità proteina. La mobilità dei d proteina EGFP-ER è dimostrata dal rapido recupero di fluorescenza dopo photobleaching in OSERs sbiancato. Per l'analisi quantitativa, il primo tempo e la frazione cellulare risultano dai dati misurati sperimentalmente inserendo la seguente equazione monoesponenziale:
F (t) = F postale + (F rec-F post) (1-e-t / τ)
dove F post è il segnale di fluorescenza dopo photobleaching, F rec è il valore massimo recupero di fluorescenza che si raggiungono dopo lo sbiancamento, t il tempo di registrazione e τ la costante di tempo.
Si prega di notare l'importanza di acquisire immagini senza pixel saturi che potrebbero alterare il recupero di fluorescenza e, di conseguenza, l'analisi mobilità proteina. E is anche essenziale per normalizzare sempre il segnale di fluorescenza nella ROI sbiancato alla fluorescenza totale della stessa cella per considerare variazioni di intensità di fluorescenza a causa di sbiancamento durante l'acquisizione dell'immagine o piccoli cambiamenti nel piano focale.
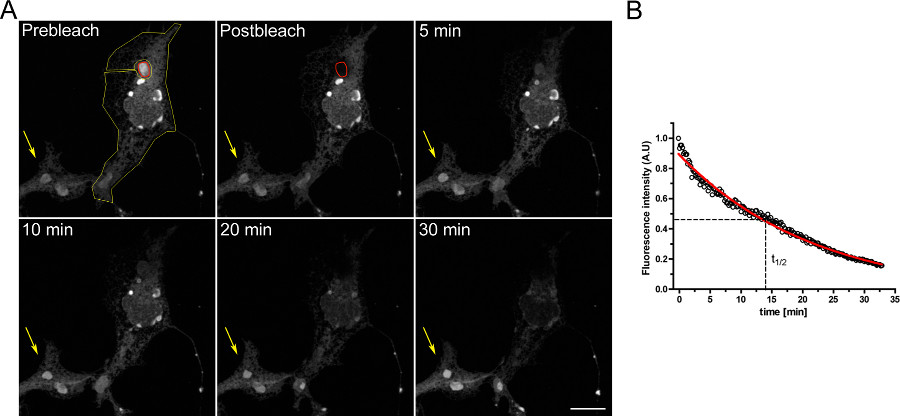
Un esempio di un esperimento FLIP per studiare la continuità tra compartimenti intracellulari è mostrato in Figura 3. OSERs sono fisicamente collegati con il resto del ER come dimostra il progressivo svuotamento della ER quando il dominio OSER continuamente sbiancato.
Per una corretta analisi, l'acquisizione di pixel saturi deve essere evitato (vedi sopra), inoltre, i parametri di acquisizione devono essere impostati con potenze laser più basso possibile per evitare photobleaching a causa di acquisizione delle immagini. Per questo motivo è fortemente consigliato immagine una cella greggi nello stesso campo che verrà utilizzato per normalizzare il segnale di fluorescenza del c sbiancatoell.
Tutti gli esperimenti devono essere eseguite in presenza di cicloesimide, un inibitore della traduzione, al fine di evitare qualsiasi aumento del segnale di fluorescenza ER (e conseguentemente fluorescenza totale) a causa di biosintesi delle proteine.
Microscopia elettronica a trasmissione dimostrato che gli aggregati fluorescenti osservati in cellule coltivate trasfettate con d EGFP-ER rappresentano macchie di liscia e appiattito ER cisterne che spazialmente si articola in geometrie 3D ben definite classificati sulla base dei loro modelli: pile lineari o curvi (spesso associata alla membrana nucleare, non mostrato) (Figure 4A e B) che può essere continuo con le regioni di ER sinusoidale (Figura 4A), le membrane in alcune regioni sono organizzati in reticoli aventi una simmetria quadrata o esagonale (cristalloidi ER, non illustrato ). Cisterne adiacenti sono separati da un sottile strato di un po 'elettrone-citoplasma denso circa 11 nm di spessore che è continua con il citoplasma circostante aggregati.

Figura 1. Diagramma di flusso della procedura sperimentale. Le cellule coltivate vengono prima trasfettate con jetPEI (vedi Protocol) per un eccesso di esprimere la proteina di fusione fluorescente di interesse. Dopo 24 ore, le cellule trasfettate live vengono visualizzati e FRAP e FLIP esperimenti vengono eseguiti utilizzando un microscopio confocale dotato di una temperatura controllata di CO 2 incubatore, e le immagini registrate vengono esportati e analizzati utilizzando il software appropriato (per esempio ImageJ). Per l'analisi ultrastrutturale, le cellule trasfettate sono fissi, pellet ed inclusi in Epon epossidiche blocchi di resina. Sezioni ultrafini sono ottenuti utilizzando un coltello di diamante, raccolta su copper griglie, e osservati sotto un microscopio elettronico a trasmissione. Clicca qui per ingrandire la figura .

Figura 2. Esperimento FRAP utilizzando cellule COS-7 trasfettate transitoriamente con d EGFP-ER. A) Due strutture Oser (Rois rossi) sono stati sbiancati, e il recupero di fluorescenza è stato registrato nel corso del tempo. Eliminare recupero di fluorescenza può essere rilevata 1 min post-candeggio, e il segnale di ulteriori aumenti dopo 4 min (barra della scala 10 micron) B):. Analisi quantitativa dell'esperimento FRAP mostrando il recupero del primo tempo e la frazione mobile del d EGFP ER-proteina. Click qui per ingrandire la figura

Figura 3. Esperimento FLIP utilizzando cellule COS-7 trasfettate transitoriamente con d EGFP-ER. A) Il candeggio in continuo di un OSER (indicato dal ROI rosso) provoca una progressiva diminuzione della fluorescenza nel resto del ER e in altre strutture Oser all'interno della stessa cella (indicato dal ROI giallo). La freccia gialla indica una porzione di una cellula greggi in cui il segnale di fluorescenza è costante nel tempo. (Barra Scala 10 micron). B) Analisi quantitativa dell'esperimento FLIP. Clicca qui per ingrandire la figura
Figura 4. Dopo la fissazione e l'inclusione, cellule che esprimono alti livelli di d EGFP-ER in cui le strutture Oser potrebbe essere rilevato mediante microscopia ottica in fluorescenza sono stati osservati attraverso un microscopio elettronico a trasmissione. A) vista dal basso ingrandimento di una porzione di citoplasma di una cellula contenente un OSER costituito cisterne impilati e membrane sinusoidali ondulate. I mitocondri (M) può essere visto raggruppati intorno alle strutture Oser, mentre ribosomi decorare solo le membrane delle cisterne più esterno (punte di freccia e riquadro). Lo spazio tra le membrane 11 nm di spessore elettrone-denso è continua con il citoplasma (freccia e riquadro) (L = lisosomi / (auto-) phagosomes) (scala bar 1,5 micron; inserto 0,25 micron). B) Un OSER può essere formato da lamellare ER: cioè pile di appiattito ER cisterne che può essere continuo o Fragmented nel loro aspetto in sezioni sottili. Vescicole erba dalla cisterne esterno della pila occasionalmente possono essere osservati (asterisco) (PM, membrana plasmatica) (bar Scale 150 nm).
Subscription Required. Please recommend JoVE to your librarian.
Discussion
I protocolli e approcci di imaging descritti in questo documento sono stati utilizzati per studiare la distribuzione e la mobilità delle proteine fluorescenti TA trasfettate residenti nel ER di cellule viventi. Abbiamo anche analizzato l'effetto dell'espressione eccessiva di queste proteine sull'architettura di questo compartimento subcellulare mediante analisi ultrastrutturali.
La combinazione di live-cell imaging confocale e microscopia elettronica rappresenta è un potente mezzo di indagare le proprietà dinamiche delle proteine, e può fornire informazioni importanti riguardanti la funzione della proteina. I metodi descritti non sono molto tempo (tipicamente tre giorni di lavoro), e lo sviluppo di molte applicazioni software di facile utilizzo per l'acquisizione delle immagini e analisi rende basato photobleaching, live-cell imaging relativamente semplice.
Il principale limite di queste tecniche è l'uso di proteine di fusione fluorescenti perché l'tag fluorescente può influire sulla corretta piegatura e / o assemblaggio della proteina di interesse. Inoltre, la sovra-espressione può alterare il comportamento di trasfettate, proteine fluorescente con tag, e possono pertanto non riflettere le proprietà reali delle proteine endogene, ma questo può essere superato usando delle cellule inducibili e stabilmente trasfettate in cui il livello di espressione può essere precisamente modulato avere livelli comparabili a quelli della proteina endogena 1,7. La tendenza dei programmi quadro oligomerise è stato ampiamente documentato e potrebbe alterare significativamente il comportamento (cioè cinetica, interazioni indesiderate proteina-proteina e la formazione di aggregati) di proteine chimeriche. L'uso delle proteine fluorescenti monomeriche ottimizzate devono essere considerate 17.
Un altro aspetto critico di studi di imaging dinamici mediante fluorescenza e photobleaching è il tempo necessario per sbiancare la fluorescenza efficiente e misurare la fluorescenza recupero (e quindi mobilità proteina) appunto, che dipende anche l'area della ROI e spessore locale cellula. Se una data proteina GFP-tag ha un alto coefficiente di diffusione, diffusione potrebbe verificarsi durante la sbianca e quindi interferire con misure di tempo di ripristino. Al fine di ottenere una rapida ed efficace sbiancamento, si raccomanda vivamente di usare un "zoom in" funzione (se disponibile) e più di una riga laser. Sebbene l'uso di un modulo di scansione veloce (cioè uno scanner di risonanza) può migliorare notevolmente la velocità delle immagini durante la fase di recupero di un esperimento, nelle nostre mani si riduce considerevolmente l'efficienza sbianca. Tuttavia, i sistemi alternativi di scansione (come per esempio un disco rotante dotato di un dispositivo fotoscolorimento dedicato), e più potenti laser possono migliorare sia l'efficienza candeggio e velocità di acquisizione.
La maggior parte delle proteine fluorescenti utilizzati nei FRAP e FLIP esperimenti mostrano un certo grado di fotoscolorimento reversibile e blinking che devono essere considerati quando si eseguono analisi quantitative. Le fluttuazioni tra stati fluorescenti e scure si verificano nel secondo tempo-scala dei minuti. Per EGFP, E 'stato dimostrato che durante gli esperimenti sbiancanti, variazioni di fluorescenza potrebbe coinvolgere meno del 10% delle molecole, così nel presente protocollo questo fenomeno è trascurabile. Se tutte le condizioni sono mantenute costanti, questo introdurrà una polarizzazione costante nei risultati. Se si utilizzano altre proteine fluorescenti, in cui la frazione reversibile è sensibilmente più elevata (YFP), o per rilevare e valutare photobleaching reversibilità, questo può essere fatto misurando recupero di fluorescenza dopo photobleaching nell'intera cellula vivente; se il recupero si osserva questo può solo essere il risultato di photobleaching reversibilità 18.
La potenziale tossicità della luce durante gli esperimenti è un altro fattore critico, soprattutto perché fotoscolorimento richiede forte illuminazione. E 'well noto che fluorofori eccitati tendono a reagire con l'ossigeno per produrre radicali liberi che possono interessare vari processi intracellulari e anche la vitalità cellulare 19, e quindi è necessario stabilire un equilibrio tra sbiancamento efficace e fototossicità minimale, inoltre, la vitalità cellulare deve essere sempre controllato dopo gli esperimenti di imaging live-cell. Dato il tempo di registrazione breve, non abbiamo considerato l'effetto genotossico della luce di lunghezza d'onda corta (405 nm) nell'esempio descritto in questo documento, ma, se è necessaria una sperimentazione più lunga, non deve essere utilizzato una linea di laser a 405 nm.
Abbiamo scelto di non utilizzare un approccio correlativo a trasmissione microscopia elettronica a causa della natura eterogenea dell'architettura OSER e il fatto che volevamo osservare come molte cellule (e strutture) possibile. La diversità della struttura fine di aggregati proteici nelle cellule può essere una caratteristica fondamentale di diverse malattie ed eravamo interessati ad ottenere una vasta gamma di samples, considerando che un approccio correlativo permette l'osservazione di un minor numero di eventi durante lo stesso periodo di tempo. Tuttavia, correlativo microscopio ottico-elettronico (CLEM) dovrebbe essere la prima scelta quando indagare eventi in strutture che non possono essere facilmente identificati (come sottodomini ER meno prominenti) o in un numero limitato di cellule (come le cellule in microiniezione). Vale la pena notare che i nostri esperimenti sono stati caratterizzati da un elevato grado di efficienza di trasfezione (almeno il 30% delle cellule sono state trasfettate), altrimenti la possibilità di osservare strutture Oser noncorrelatively è piuttosto limitata.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Gli autori non hanno nulla da rivelare.
Acknowledgments
Gli autori sono grati alla Fondazione Filarete per il suo aiuto e il sostegno nella pubblicazione di questo articolo. Vorremmo anche ringraziare Centro Europeo di Nanomedicina per l'uso del microscopio elettronico a trasmissione Tecnai G2.
Materials
| Name | Company | Catalog Number | Comments |
| Dulbecco’s Modified Eagle Medium (DMEM) | Invitrogen | 41966029 | |
| Dulbecco’s Modified Eagle Medium (DMEM) w/o phenol red | Invitrogen | 31053028 | |
| Fetal Bovine Serum (FBS) | Invitrogen | 10270106 | |
| Pen/Strep | Invitrogen | 15140-122 | |
| L-Glutamine 200 mM solution | Invitrogen | 25030-024 | |
| jetPEI | Polyplus Transfection | PP10110 | |
| OxyFluor | Oxyrase Inc. | OF-0005 | |
| Glutaraldehyde Grade I | Sigma Aldrich | G5882 | |
| Sodium Cacodylate Trihydrate | Sigma Aldrich | C0250 | |
| Osmium Tetroxide 4% solution | Electron Microscopy Science | 19150 | |
| Uranyl Acetate Dihydrate | Sigma Aldrich | 73943 | slightly radioactive |
| Propylene Oxide | Sigma-Aldrich | 82320 | |
| EPON embedding medium kit | Sigma-Aldrich | 45359-1EA-F | |
| Lead Citrate | Electron Microscopy Science | 17800 | |
| Bench top centrifuge | Eppendorf | 5415 D | |
| Spectral Confocal Microscope | Leica Microsystems | TCS SP5 | |
| CO2 Microscope Cage Incubation System | OkoLab | ||
| Ultramicrotome | Leica Microsystems | UC6 | |
| Diamond knife | Diatome | Ultra 45 ° | |
| Transmission Electron Microscope | FEI | Tecnai G2 | |
| GraphPad Prism Software | GraphPad Software, Inc | ||
| Steel culture cell chamber for 24 mm coverslip | Bioscience Tools | CSC-25 | |
| Electron Microscopy grids | Electron Microscopy Science | G300Cu |
References
- Fasana, E., et al. A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 24, 1419-1430 (2010).
- Borgese, N., Francolini, M., Snapp, E. Endoplasmic reticulum architecture: structures in flux. Curr. Opin. Cell Biol. 18, 358-364 (2006).
- Ronchi, P., Colombo, S., Francolini, M., Borgese, N. Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J. Cell Biol. 181, 105-118 (2008).
- Lippincott-Schwartz, J., Snapp, E., Kenworthy, A. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2, 444-456 (2001).
- Lee, M. C., Miller, E. A., Goldberg, J., Orci, L., Schekman, R. Bi-directional protein transport between the ER and. 20, 87-123 (2004).
- Snapp, E. L., et al. Formation of stacked ER cisternae by low affinity protein interactions. J. Cell Biol. 163, 257-269 (2003).
- Papiani, G., et al. Restructured endoplasmic reticulum generated by mutant amyotrophic lateral sclerosis-linked VAPB is cleared by the proteasome. J. Cell Sci. 125, 3601-3611 (2012).
- Almsherqi, Z. A., Kohlwein, S. D., Deng, Y. Cubic membranes: a legend beyond the Flatland* of cell membrane organization. J. Cell Biol. 173, 839-844 (2006).
- Federovitch, C. M., Ron, D., Hampton, R. Y. The dynamic ER: experimental approaches and current questions. Curr. Opin. Cell Biol. 17, 409-414 (2005).
- Takei, K., Mignery, G. A., Mugnaini, E., Sudhof, T. C., De Camilli, P. Inositol 1,4,5-trisphosphate receptor causes formation of ER cisternal stacks in transfected fibroblasts and in cerebellar Purkinje cells. Neuron. 12, 327-342 (1994).
- Feldman, D., Swarm, R. L., Becker, J. Ultrastructural study of rat liver and liver neoplasms after long-term treatment with phenobarbital. Cancer Res. 41, 2151-2162 (1981).
- Sprocati, T., Ronchi, P., Raimondi, A., Francolini, M., Borgese, N. Dynamic and reversible restructuring of the endoplasmic reticulum induced by PDMP in cultured cells. J. Cell Sci. 119, 3249-3260 (2006).
- Costantini, L. M., Fossati, M., Francolini, M., Snapp, E. L. Assessing the tendency of fluorescent proteins to oligomerize under physiologic conditions. Traffic. 13, 643-649 (2012).
- Pyszniak, A. M., Welder, C. A., Takei, F. Cell surface distribution of high-avidity LFA-1 detected by soluble ICAM-1-coated microspheres. J. Immunol. 152, 5241-5249 (1994).
- Taylor, J. P., Hardy, J., Fischbeck, K. H. Toxic proteins in neurodegenerative disease. Science. 296, 1991-1995 (2002).
- Winklhofer, K. F., Tatzelt, J., Haass, C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 27, 336-349 (2008).
- Borgese, N., Gazzoni, I., Barberi, M., Colombo, S., Pedrazzini, E. Targeting of a tail-anchored protein to endoplasmic reticulum and mitochondrial outer membrane by independent but competing pathways. Mol. Biol. Cell. 12, 2482-2496 (2001).
- Chudakov, D. M., Matz, M. V., Lukyanov, S., Lukyanov, K. A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 90, 1103-1163 (2010).
- Bancaud, A., Huet, S., Rabut, G., Ellenberg, J. Fluorescence perturbation techniques to study mobility and molecular dynamics of proteins in live cells FRAP, photoactivation, photoconversion, and FLIP. Cold Spring Harb. Protoc. 2010, (2010).
- Michida, T., et al. Role of endothelin 1 in hemorrhagic shock-induced gastric mucosal injury in rats. Gastroenterology. 106, 988-993 (1994).

{kind=link}