

Summary
私たちは、生きた細胞の共焦点イメージングにより、小胞体(ER)に常駐トランスフェクションし、蛍光タンパク質の分布や移動性を調査するために使用して画像化のアプローチについて説明します。また、超微細構造、この細胞内区画のアーキテクチャにその発現の効果を分析する。
Abstract
これらは強烈interorganelle分子のトラフィックにもかかわらず、彼らの独特の構成と機能を保持しているが、真核細胞での脂質とタンパク質が連続的に、細胞区画間で交換される。このホワイトペーパーに記載されている技術は、 生体内とその生理的な環境で、タンパク質や脂質の移動と人身売買を研究する強力な手段である。 (FLIP)の光退色で(FRAP)の光退色後の蛍光回復および蛍光損失が広くエキソ - エンドサイトーシス経路、オルガネラまたはサブコンパートメント間の連続性、タンパク質複合体の形成、およびタンパク質局在化を介して細胞内輸送を研究するための生細胞イメージング技術を用いている脂質ミクロドメインに、それらの全ては生理学的および病理学的条件下で観察することができる。これらのアプローチの限界は、主に蛍光性融合タンパク質の使用によるものであり、それらの潜在的な欠点は、過剰発現アーチファクトを含む細胞内のイオンと、タグ付きおよび天然のタンパク質のフォールディングおよびローカリゼーションの違いの可能性。最後に、光学顕微鏡(約200nm)の解像度の限界としてストレス下の細胞に由来することができERまたは特定のサブコンパートメントの微細構造の調査を許可していない( すなわち低酸素症、薬物投与、膜貫通の過剰発現ER常在性タンパク質)または病理学的条件の下で、我々は超微細で培養されたトランスフェクトされた細胞の生細胞イメージングを組み合わせることは、透過型電子顕微鏡法に基づいて解析する。
Introduction
緑色蛍光タンパク質(GFP)およびそのスペクトル変異体、および蛍光顕微鏡の並行開発の発見は、細胞におけるタンパク質の挙動の調査のための完全に新しい道を開いた。なぜなら強烈な照明の下での蛍光を消火する蛍光体の本質的な能力のために可能であるような(FRAP)を光退色後蛍光回復と(フリップ)の光退色の蛍光損失などの技術は、共焦点生細胞イメージングトランスフェクトの使用に基づいている蛍光融合タンパク質1-3。彼らは広く、その機能4に関する重要な手がかりを明らかにすることができ、タンパク質の局在だけでなく、彼らの機動性と小胞輸送だけでなく、を評価するために使用されています。
真核細胞のユニークな特徴は、特定の脂質およびタンパク質の組成物を有する細胞内コンパートメントの存在である。細胞小器官は、物理的に絶縁のあるですがオピニオン、それらは互いに通信し、細胞の恒常性を維持するために、分子成分を共有する必要がある。分泌経路は、ERで合成されたタンパク質や脂質は、彼らがその機能を発揮する正しい最終目的地に到達することが保証されます。細胞内小器官はまた、分子(脂質)が直接コンパートメントとの間で交換することを可能にする動的接触部位に接続することができる。さらに、多くのタンパク質は大きなヘテロ複合体で組み立てまたは機能的に活性になるか、その最終的な目的地に輸送されるために特定の脂質種(脂質ラフト/マイクロドメイン)に関連している。これらの生物学的局面の全てが大きく、タンパク質の動力学的特性に影響を与えるため、適宜、以下に記載される技術を用いて調べることができる。
我々のグループは、ERのアーキテクチャとその異なるサブを研究するために、電子顕微鏡と組み合わせ広く使われているFRAPやFLIPを持ってドメイン。 ER、分泌経路の最初のステーションであり、タンパク質および脂質5選別において重要な役割を果たしている。それは、その個別のサブドメイン( すなわち 、タンパク質および脂質生合成および輸送、タンパク質の折り畳み、Ca 2 +の貯蔵と放出、および異物代謝)その多くの異なる機能を反映して非常にダイナミックな細胞小器官である。それらは形態学的に、空間的、および機能的に別個であるものの、これらのドメインは、互いに連続しており、それらの相対存在量は、生理学的および病理学的条件下で、細胞内で改変することができる。最もよく知られ、ERの通常空間的に分離されたドメインは、核膜、および平滑、粗ERであるが、我々及び他のER構造は、種々の細胞型におけるより精巧なアーキテクチャおよび三次元の組織に存在することが実証されているまた、低酸素症、薬物などのストレス刺激により誘導することができる、生理学的条件下での組織投与、またはERに存在する膜貫通タンパク質2,6の過剰発現(およびその中の参考文献)。
また、最近、ヒト疾患1,7の細胞モデルにおいて、このような構造の存在が実証されている。彼らはまた、彼らのような彼らのアーキテクチャに基づいてkarmellae、ラメラ 、及び晶質ERとして知られているが、滑らかなERの積み重ね嚢から発信さが、それらは、2003年6で編成滑面小胞体(OSER)の集合的な名前を与えられたサイズは、変えることができる。細胞が尾アンカー(TA)ERに存在するタンパク質(Dの EGFP-ER)の細胞質ゾル領域に融合GFPをトランスフェクトされた後、 トランスでGFPの弱い二量化傾向が劇的にERの組織や構造を変更します。 FRAPやFLIPの実験は、D EGFP-ERはOSERs内に拡散して自由であることを示した、それが<OSERに網状ERから移動し、 その逆もあるという事実/ em>は凝集体が周囲の網状のERと連続していることを示しています。超微細構造解析は、私たちは、ナノスケールレベルでのOSERアーキテクチャと組織の詳細な説明と蛍光データを相関させることができました。OSERsは、常に滑らかなERの対になった嚢のスタックで構成されていますが、このような規則的に正弦波に配置などの空間組織のさまざまな形態を有することができる配列や渦巻き、または六角形の「クリスタ」管状の配列。これらの再配列は、それらが生理学的条件下9および10低酸素症、薬物治療11、および癌9として以下の応力下で細胞中に見出されているように、超微細構造のマーカーとして大きな可能性を有していてもよい立方晶の形態8に導く。
GFP融合タンパク質を使用して、この最初の実証の後、我々は薬理学的治療12、ASSEに応答してERドメインの増殖を分析するためにイメージング実験を用いカテゴリーセル13にオリゴマー化するため、およびその病原1,8に関連し得るER由来の細胞内凝集体の形成における変異、ALS TA-結合タンパク質の役割を調べるために蛍光タンパク質の傾向。これは、(多くの神経変性疾患において生じる14)は、細胞内凝集物の形成が毒性変異体タンパク質及び周辺セル構成要素15との間の相互作用を防止するように設計された保護メカニズムであり得ることが示唆されている。
以下は、そのC末端疎水性ドメインはERの膜に挿入され、それらの動的挙動の解析およびERドメインに対するそれらの過剰発現の効果を調査するための構築物の光学および電子顕微鏡法の組合せの説明である培養細胞におけるアーキテクチャ(実験プロトコルのフローについては図1を参照)。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1。 ER蛍光タンパク質を用いて、プラスミド、細胞培養、およびトランスフェクション
- この研究で使用したプラスミドは、ラットシトクロムbのERアイソフォームのテール領域へのC-末端で融合GFPの拡張バージョンで構成され(5)リンカー配列を介して(bと(5)ここでは略す)。テール領域(5)は、アップストリームおよびダウンストリームの極性配列が隣接する17残基のTMD(膜貫通ドメイン)、(UPSとDPS)などのネイティブBのトリプシン切断の後に結合した膜のまま、全配列(Pro94-Asp134)が含まれています。リンカーは、[(グリシン)4セリン] 3、続いてmycエピトープから構成され、全体のcDNAを哺乳動物発現ベクターのpCDNA3-Hind3でののXba1部位に挿入される。このプラスミドの構造の詳細は、それがGFP-ER 16とも呼ばれる、以前の刊行物に記載されている。
- 10%フェタを補充したダルベッコ改変イーグル培地(DMEM)中でCOS-7細胞を増殖させるLのウシ血清、2mMのL-グルタミン、37℃でインキュベーター中で1%ペニシリン/ストレプトマイシンおよび10%CO 2。
- トランスフェクション。製造者によって記載されているように、プレート3×10 5 6ウェルプレート中の丸いガラスカバースリップ上の細胞とは、次の日に、JetPEIシステムとトランスフェクト。我々の場合に、JetPEI:2:1のDNA比は、70〜80%のトランスフェクション効率をもたらす最適なJetPEI / DNA比は、使用されるプラスミドおよび細胞株に応じて、最大のトランスフェクション効率を確立するためにテストされていることに留意されたい。
2。ライブ蛍光走査型共焦点顕微鏡
- 生細胞イメージング。トランスフェクトされた細胞を10%FBS、2mMのL-グルタミン、1%ペニシリン/ストレプトマイシン、25mMのHEPESを補充したW / OフェノールレッドDMEMで充填された24mmカバースリップ用スチール培養細胞チャンバ内に播種したカバースリップを置く退色からサンプルを防ぐために、50μg/ mlのシクロヘキシミドおよび1:100 OxyFluor。 SP5共焦点microscopd個の GFP-ERは、488nmレーザー及び50分の525バンドパス発光フィルターを用いて可視化した状態で温度制御されたCO 2インキュベーター(37℃、5%CO 2)を備えた電子は、生細胞イメージング実験のために使用される。
- (FRAP)を光退色後蛍光回復。 OSER構造に対応する、関心領域(ROI)を描き、それは(試料で5.5〜6μWに対応する、30 mWのアルゴンレーザーの100%)の20回の反復と488nmの組み合わせを使用し、405nmの漂白(30 mWのダイオード405のレーザーの60%が、サンプルで11.6μWに相当する)我々の経験では、効率的かつ迅速な光退色につながる、レーザーが。
- 10分(画素時間= 1.61秒/ピクセル)のための単一フレームごとに10秒を取ることによって、漂白されたROI内の蛍光の回復を記録します。
- (フリップ)の光退色の蛍光損失。 OSER構造に対応するROI、上述したように漂白剤を描く。漂白は、30秒毎に繰り返される、とポスト漂白画像は30分(画素時間= 1.61秒/ピクセル)ごとに10秒を記録している。
- FRAPやFLIP分析。すべての画像は、ImageJソフトウェア(使用して分析するhttp://rsbweb.nih.gov/ij/download.htmlを )。 FRAP実験では、漂白されたROIの蛍光回復を経時的に測定され、常に経時的に一定であることがチェックされる漂白細胞の総蛍光に対して標準化。
- FLIP実験のために、漂白OSER外と細胞全体をカバーするROIを描画します。経時的に蛍光強度を測定し、撮像自体による蛍光の低下を補正するために前記未漂白のセルに描かれたROIの蛍光レベルに正規化する。
- 全ての実験では、ROIの蛍光強度から(細胞の外側の領域で決定した)バックグラウンドシグナルを差し引く。最後に、Grのを使用して結果をプロットaphPad Prismソフトウェア。
3。透過型電子顕微鏡を用いて超微細構造解析
試薬の多くの毒性を考えると、すべての手順は、適切なラボを着て行われるべきである。ヒュームフードの下のコートと手袋。
- ペトリ皿からカバースリップを除去した後、室温で10分間、0.1 Mカコジル酸緩衝液、pH7.4中で濾過し、2%グルタルアルデヒドを用いて単層として皿の底の上に成長した残りの細胞を固定する。
- テフロンスクレーパーを用いて細胞をこすり落とし、1.5ミリリットルエッペンドルフチューブに転送します。 10分間9,000 gでの遠心分離により細胞をペレット化。上清を除去し、新鮮な固定を追加し、4℃で一晩のまま
- 次いで、室温で1時間カコジル酸緩衝液中の1%四酸化オスミウム溶液で後固定し、緩衝液でペレットを洗浄する。
- ミリQ水で洗い流し、とした一括染色エン20〜60の間分間蒸留水中1%酢酸ウラニル。
- エタノールシリーズ(70%、80%、90%、100%、10分ごとに、100%)の増加でサンプルを脱水し、プロピレンオキシド(15分毎)で二回簡単に洗浄する。
- (2時間から一晩)、プロピレンオキシド+エポン(1:1)の混合物中にサンプルを浸潤する。
- 少なくとも24時間60℃で硬化さエポンエポキシ樹脂に埋め込む。
- セクション60から70ナノメートルの厚さの部分を取得するために45°のダイヤモンドナイフを装備したウルトラライカUC6を使用して手動でトリミングされた樹脂圏。 300メッシュ銅グリッド上のセクションを収集します。
- 酢酸ウラニル(20分)の飽和溶液でグリッド上のセクションを染色し、クエン酸鉛(7分)、徹底的に再蒸留ろ過された水でそれらを浸漬することによってグリッドを洗浄し、それらを室温で乾燥させます。
- 染色されたグリッドはTECNAI G2透過型電子顕微鏡を用いて観察し、画像がボットを使用して捕捉される(一般的には6,000-39,000 Xの範囲)異なる最終倍率でOMマウントされたCCDカメラ。
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
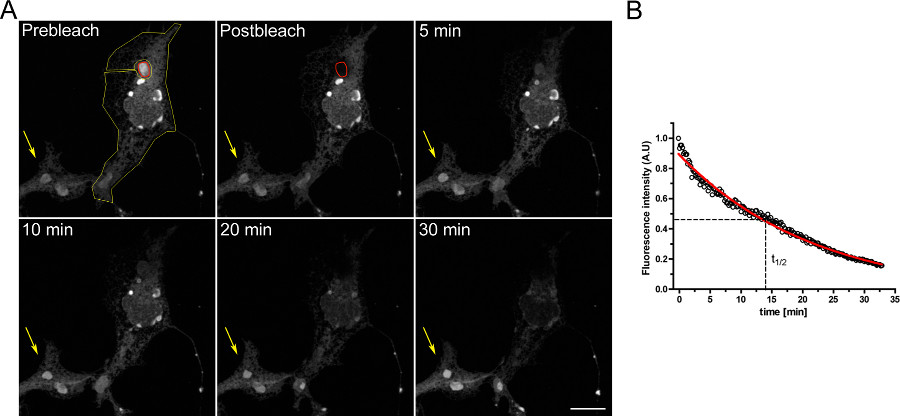
図2は、タンパク質の移動の例FRAP研究を示しています。 dは EGFP-ERタンパク質の移動度は、漂白OSERsに光退色後蛍光回復急速によって実証される。定量分析のために、半分の時間、モバイル画分は、以下の単一指数の式を当てはめることによって実験的に測定されたデータから導出された。
F(T)= F ポスト +(F REC-Fポスト )(1-E-T /τ)
F ポストは 、光退色後の蛍光シグナルであり、F rec視聴、登録時トン 、漂白後に到達し、時定数τ最大蛍光回復値である。
蛍光回復と、結果的に、タンパク質の移動度分析を変える可能性が飽和画素せずに画像を取得することの重要性に注意してください。それI常に焦点面での画像取得又は小さな変化時に脱色による蛍光強度の変化を考慮するために、同じ細胞の総蛍光に漂白ROIにおける蛍光シグナルを正規化するためにも不可欠だ。
細胞内区画間の連続性を研究するためのFLIP実験の例を図3に示す。 OSERドメインが連続的に漂白されるERの進行に空にすることによって実証されるように物理的OSERs ERの残りの部分と接続されている。
適切な分析のために、飽和画素の買収は、(上記参照)は避けなければならない、さらに、収集パラメータは、画像取得のために光退色を避けるために、可能な限り低いレーザー出力を使用して設定する必要があります。この理由は、非常に漂白された画像cは蛍光シグナルを正規化するために使用される同じフィールド内の未漂白のセルをお勧めしますエル。
全ての実験は、タンパク質生合成の任意のER蛍光シグナルの増加(その結果、総蛍光)を回避するために、シクロヘキシミド、翻訳阻害剤の存在下で実施されなければならない。
透過型電子顕微鏡は、D、EGFP-ERをトランスフェクションした培養細胞で観察された蛍光凝集体がスムーズなのパッチを表していることを実証し、空間的にそのパターンに基づいて分類し、明確に定義された3Dジオメトリに身を組織し、ER嚢フラット化:多くの場合、線状または湾曲したスタックを( 、核膜に関連付けられて示されていない)( 図4AおよびB)正弦ERの領域( 図4A)と連続していてもよいこと、一部の地域では、膜を正方形または六角形の対称性(クリスタERを、図示していないとの格子に編成されています)。隣接嚢はやや電子の薄い層によって分離されている密な細胞質約11ナノメートルの厚さの凝集体を取り巻く細胞質と連続していること。

図1。実験手順のフローチャートを。培養細胞を最初に、目的の蛍光融合タンパク質を発現する過剰のために、jetPEI(プロトコルを参照のこと)でトランスフェクトされる。 24時間後、生きたトランスフェクトされた細胞は可視化され、FRAPおよびFLIPの実験は、温度制御されたCO 2インキュベーターを備えた共焦点顕微鏡を用いて行われ、記録された画像をエクスポートし、適切なソフトウェア( 例えば、ImageJの)を使用して分析する。超微細構造分析のために、トランスフェクトされた細胞は、固定されたペレット化し、エポンエポキシ樹脂圏に埋め込まれている。超薄切片は、c上に集め、ダイヤモンドナイフを使用して得られるopperグリッド、透過型電子顕微鏡下で観察した。 大きな画像を見るにはここをクリックしてください 。

図2。一過Dの EGFP-ERトランスフェクトしたCOS-7細胞を用いたFRAP実験。 A)二OSER構造(赤色のROIが)漂白し、そして蛍光回復を経時的に記録した。鮮明な蛍光回復は1分後に漂白し、4分後(スケールバーは10μm)信号がさらに上昇を検出することができ、B):回復ハーフタイムを示すFRAP実験の定量分析とD EGFPのモバイル割合-ERタンパク質。 Clで大きな画像を表示するには、こちらICK

図3。一過Dの EGFP-ERトランスフェクトしたCOS-7細胞を用いたフリップ実験。 A)赤のROIで示さOSER()の連続漂白は、ERの残りの部分と黄色のROIが示す同一セル内の他のOSER構造()内の蛍光の漸進的な低下を引き起こす。黄色の矢印は、蛍光シグナルが経時的に一定である、前記未漂白のセルの部分を示す。 (スケールバーは10μm)。B)FLIP実験の定量分析。 大きな画像を見るにはここをクリックしてください
図4。固定および包埋した後、OSER構造は蛍光光学顕微鏡法によって検出することができたd個の EGFP-ERを高レベルで発現する細胞を、透過型電子顕微鏡で観察した。積み重ね嚢と起伏の正弦波の膜からなるOSERを含む細胞の細胞質の一部のA)の低倍率ビュー。リボソームが最も外側の嚢(矢頭および挿入図)の唯一の膜を飾る一方で、ミトコンドリア(M)は、OSER構造の周りに集まっ見ることができます。膜間の11 nmの厚さの電子密度の高い空間は、細胞質(矢印および挿入図)(L =リソソーム/(オート)ファゴソーム)と連続している(スケールバー1.5μmで、挿入図0.25ミクロン)、B)AN OSERを形成することができる。連続またはfragmeことができ、平坦化、ER嚢すなわちスタック:ラメラERによる薄い部分での外観nted。スタックの最嚢から出芽小胞は時折(アスタリスク)(PM、原形質膜)(スケールバーは150nm)を観察することができる。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
このホワイトペーパーで説明プロトコルおよびイメージングのアプローチは、生細胞の小胞体内に常駐トランスフェクションしたTAの蛍光タンパク質の分布や移動性を調査するために使用されてきた。我々はまた、超微細構造解析により、この細胞内区画のアーキテクチャにこれらのタンパク質の過剰発現の効果を分析した。
生細胞の共焦点イメージングおよび電子顕微鏡法の組み合わせが表すタンパク質の動的な特性を調査する非常に強力な手段であり、タンパク質機能に関する重要な情報を提供することができる。記載された方法は時間がかかり(仕事の典型的には3日間)ではなく、画像収集及び分析のための多くのユーザーフレンドリーなソフトウェアアプリケーションの開発は、比較的単純な、生細胞イメージングベースの光退色ができる。
これらの技術の主な限界は、蛍光融合タンパク質の使用であるため、蛍光タグは、目的のタンパク質の適切な折り畳みおよび/またはアセンブリに影響を与えることができる。また、過剰発現は、トランスフェクション、蛍光タグ化タンパク質の挙動を変更することができ、したがって、内因性タンパク質の実際の特性を反映しないことがあるが、これは、発現レベルが可能な誘導性および安定にトランスフェクトされた細胞を用いることによって克服することができる正確内因性タンパク質の1,7のものと同等のレベルを得るために変調される。オリゴマー化に対するFPの傾向は広く文書化されており、著しくキメラタンパク質の挙動( すなわち、動力学、望ましくないタンパク質-タンパク質相互作用及び凝集体の形成)を変化させることができる。最適化された単量体の蛍光タンパク質の使用は、17考慮されるべきである。
蛍光および光退色を使用して動的画像化研究の別の重要な態様は、効率的に蛍光漂白剤、蛍光再測定するために必要な時間であるcovery(したがって、タンパク質の移動度)を正確に、またROIおよびローカルセル厚の面積に依存する。所与のGFPタグ付きタンパク質は、高い拡散係数を有する場合、拡散が漂白中に発生するので、回復時間の測定を妨害し得る。迅速かつ効率的な漂白を得るためには、強い機能(利用可能な場合)、「ズームイン」し、複数のレーザ線を用いることが推奨される。高速スキャンモジュール( すなわちレゾナントスキャナ)の使用が大幅に実験の回復期イメージングの速度を向上させることができますが、私たちの手で、それはまた、かなりの漂白効率が低下する。しかしながら、(例えば、専用の光退色装置を備えたスピニングディスクのような)別の走査システムと、より強力なレーザは、漂白効率及び取得速度の両方を向上させることができる。
FRAPおよびFLIPの実験で使用されるほとんどの蛍光タンパク質は、可逆的光退色およびBLIある程度を示す定量的な分析を行う際に考慮しなければならないnking。蛍光と暗い状態の間の変動は微小な時間スケール第二に起こる。 EGFPのために、それは、漂白実験の間、蛍光の変化は、このように、本プロトコルでは、この現象が無視できる、分子の10%未満が存在するおそれのあることが示されている。全ての条件が一定に保たれている場合、これは結果に一定のバイアスを導入する。他の蛍光タンパク質は、可逆画分( すなわち YFP)有意に高い、または光退色可逆性を検出し、評価するためにここで使用される場合、これは全体生細胞における光退色後蛍光回復を測定することによって行うことができ、回復が観察される場合、これは缶唯一の可逆性18を光退色の結果である。
実験中の光の潜在的な毒性は、光退色が強い照明を必要とするため、特に、別の重要な要因である。それはWELですlで励起されたフルオロフォアは、様々な細胞内プロセス、さらには細胞生存率19に影響を与えることができるフリーラジカルを生成するために酸素と反応する傾向があることが知られているので、それは、効率的な漂白および最小毒性との間のバランスを確立する必要がある、さらに、細胞生存率は、常に、チェックされるべき生細胞イメージング実験た。短い記録時間を考えると、本稿で説明した例では、短波長光(405 nm)での遺伝毒性影響を考慮していないが、長い実験が必要な場合は、405 nmのレーザーラインを使用するべきではありません。
我々は、理由OSERアーキテクチャの不均一な性質と我々はできるだけ多くの細胞(および構造)を観察したかったという事実を、透過型電子顕微鏡の相関アプローチを使用しないことを選んだ。細胞におけるタンパク質凝集体の微細構造の多様性は、種々の疾患の重要な特徴であってもよいし、我々はsaの広い範囲を得ることに興味があったmples、相関アプローチは、同期間の時間の間に少数のイベントの観察を可能にするのに対し。しかし、相関光電子顕微鏡(クレム)を容易に(このような目立たERサブドメインとして)識別できない構造でイベントを調査する際に最初の選択肢であるか、(このようなミクロ注入された細胞など)は、細胞の限られた数の必要があります。それ以外noncorrelatively OSER構造を観察する可能性は非常に限られ、(細胞の少なくとも30%がトランスフェクトされた)我々の実験は、トランスフェクション効率の高い程度によって特徴付けられたことは注目に値する。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
著者らは、開示することは何もありません。
Acknowledgments
著者らは、この記事の出版物でのヘルプとサポートのための伊Filareteに感謝しています。またTECNAI G2、透過型電子顕微鏡を使用するためのセントロEuropeoのディNanomedicinaに感謝したいと思います。
Materials
| Name | Company | Catalog Number | Comments |
| Dulbecco’s Modified Eagle Medium (DMEM) | Invitrogen | 41966029 | |
| Dulbecco’s Modified Eagle Medium (DMEM) w/o phenol red | Invitrogen | 31053028 | |
| Fetal Bovine Serum (FBS) | Invitrogen | 10270106 | |
| Pen/Strep | Invitrogen | 15140-122 | |
| L-Glutamine 200 mM solution | Invitrogen | 25030-024 | |
| jetPEI | Polyplus Transfection | PP10110 | |
| OxyFluor | Oxyrase Inc. | OF-0005 | |
| Glutaraldehyde Grade I | Sigma Aldrich | G5882 | |
| Sodium Cacodylate Trihydrate | Sigma Aldrich | C0250 | |
| Osmium Tetroxide 4% solution | Electron Microscopy Science | 19150 | |
| Uranyl Acetate Dihydrate | Sigma Aldrich | 73943 | slightly radioactive |
| Propylene Oxide | Sigma-Aldrich | 82320 | |
| EPON embedding medium kit | Sigma-Aldrich | 45359-1EA-F | |
| Lead Citrate | Electron Microscopy Science | 17800 | |
| Bench top centrifuge | Eppendorf | 5415 D | |
| Spectral Confocal Microscope | Leica Microsystems | TCS SP5 | |
| CO2 Microscope Cage Incubation System | OkoLab | ||
| Ultramicrotome | Leica Microsystems | UC6 | |
| Diamond knife | Diatome | Ultra 45 ° | |
| Transmission Electron Microscope | FEI | Tecnai G2 | |
| GraphPad Prism Software | GraphPad Software, Inc | ||
| Steel culture cell chamber for 24 mm coverslip | Bioscience Tools | CSC-25 | |
| Electron Microscopy grids | Electron Microscopy Science | G300Cu |
References
- Fasana, E., et al. A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 24, 1419-1430 (2010).
- Borgese, N., Francolini, M., Snapp, E. Endoplasmic reticulum architecture: structures in flux. Curr. Opin. Cell Biol. 18, 358-364 (2006).
- Ronchi, P., Colombo, S., Francolini, M., Borgese, N. Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J. Cell Biol. 181, 105-118 (2008).
- Lippincott-Schwartz, J., Snapp, E., Kenworthy, A. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2, 444-456 (2001).
- Lee, M. C., Miller, E. A., Goldberg, J., Orci, L., Schekman, R. Bi-directional protein transport between the ER and. 20, 87-123 (2004).
- Snapp, E. L., et al. Formation of stacked ER cisternae by low affinity protein interactions. J. Cell Biol. 163, 257-269 (2003).
- Papiani, G., et al. Restructured endoplasmic reticulum generated by mutant amyotrophic lateral sclerosis-linked VAPB is cleared by the proteasome. J. Cell Sci. 125, 3601-3611 (2012).
- Almsherqi, Z. A., Kohlwein, S. D., Deng, Y. Cubic membranes: a legend beyond the Flatland* of cell membrane organization. J. Cell Biol. 173, 839-844 (2006).
- Federovitch, C. M., Ron, D., Hampton, R. Y. The dynamic ER: experimental approaches and current questions. Curr. Opin. Cell Biol. 17, 409-414 (2005).
- Takei, K., Mignery, G. A., Mugnaini, E., Sudhof, T. C., De Camilli, P. Inositol 1,4,5-trisphosphate receptor causes formation of ER cisternal stacks in transfected fibroblasts and in cerebellar Purkinje cells. Neuron. 12, 327-342 (1994).
- Feldman, D., Swarm, R. L., Becker, J. Ultrastructural study of rat liver and liver neoplasms after long-term treatment with phenobarbital. Cancer Res. 41, 2151-2162 (1981).
- Sprocati, T., Ronchi, P., Raimondi, A., Francolini, M., Borgese, N. Dynamic and reversible restructuring of the endoplasmic reticulum induced by PDMP in cultured cells. J. Cell Sci. 119, 3249-3260 (2006).
- Costantini, L. M., Fossati, M., Francolini, M., Snapp, E. L. Assessing the tendency of fluorescent proteins to oligomerize under physiologic conditions. Traffic. 13, 643-649 (2012).
- Pyszniak, A. M., Welder, C. A., Takei, F. Cell surface distribution of high-avidity LFA-1 detected by soluble ICAM-1-coated microspheres. J. Immunol. 152, 5241-5249 (1994).
- Taylor, J. P., Hardy, J., Fischbeck, K. H. Toxic proteins in neurodegenerative disease. Science. 296, 1991-1995 (2002).
- Winklhofer, K. F., Tatzelt, J., Haass, C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 27, 336-349 (2008).
- Borgese, N., Gazzoni, I., Barberi, M., Colombo, S., Pedrazzini, E. Targeting of a tail-anchored protein to endoplasmic reticulum and mitochondrial outer membrane by independent but competing pathways. Mol. Biol. Cell. 12, 2482-2496 (2001).
- Chudakov, D. M., Matz, M. V., Lukyanov, S., Lukyanov, K. A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 90, 1103-1163 (2010).
- Bancaud, A., Huet, S., Rabut, G., Ellenberg, J. Fluorescence perturbation techniques to study mobility and molecular dynamics of proteins in live cells FRAP, photoactivation, photoconversion, and FLIP. Cold Spring Harb. Protoc. 2010, (2010).
- Michida, T., et al. Role of endothelin 1 in hemorrhagic shock-induced gastric mucosal injury in rats. Gastroenterology. 106, 988-993 (1994).

{kind=link}