

Summary
Descrevem-se os métodos de imagiologia que usamos para investigar a distribuição e a mobilidade das proteínas fluorescentes transfectadas residentes no retículo endoplasmático (ER), por meio da imagem confocal de células vivas. Também analisamos ultraestruturalmente o efeito de sua expressão na arquitetura deste compartimento subcelular.
Abstract
Os lípidos e proteínas em células eucarióticas são continuamente trocadas entre compartimentos celulares, embora estas mantêm a sua composição distinta e funções apesar do tráfego intenso interorganelle molecular. As técnicas descritas neste documento são poderosos meios de estudar proteínas e lipídios e mobilidade tráfico in vivo e em seu ambiente fisiológico. Recuperação de fluorescência após a fotodegradação (FRAP) e perda de fluorescência em fotodegradação (FLIP) são amplamente usadas técnicas de imagem em células vivas, para estudar o tráfico intracelular através da via endocítica-exo, a continuidade entre a organelos ou subcompartimentos, a formação de complexos de proteína, e proteína de localização em microdomínios lipídicos, todos os quais podem ser observados sob condições fisiológicas e patológicas. As limitações destas abordagens são principalmente devidos à utilização de proteínas de fusão fluorescentes, e as suas desvantagens potenciais incluem artefatual sobre-expressoíon nas células ea possibilidade de diferenças na dobradura e localização de proteínas marcadas e nativas. Finalmente, como o limite de resolução do microscópio óptico (cerca de 200 nm) não permite a investigação da estrutura fina do ER ou os subcompartimentos específicos que podem se originar nas células sob estresse (ie hipóxia, a administração da droga, a expressão excessiva de transmembrana proteínas residentes ER) ou em condições patológicas, combinamos de imagem de células vivas de células transfectadas cultivadas com ultraestrutural análises baseadas em microscopia eletrônica de transmissão.
Introduction
A descoberta da proteína verde fluorescente (GFP) e suas variantes espectrais, eo desenvolvimento paralelo de microscopia de fluorescência, abriram totalmente novos caminhos para a investigação do comportamento de proteínas nas células. Técnicas como a recuperação de fluorescência após a fotodegradação (FRAP) e perda de fluorescência em fotodegradação (FLIP), que é possível por causa da capacidade intrínseca de fluoróforos para extinguir a fluorescência, sob iluminação intensa, são baseados em imagens ao vivo de células confocal e o uso de transf As proteínas de fusão fluorescentes 1-3. Eles são amplamente utilizados para avaliar não só a localização de proteínas, mas também a sua mobilidade e transporte vesicular, o que pode revelar pistas importantes sobre a sua função 4.
A característica única de células eucarióticas é a presença de compartimentos intracelulares que têm composições de lípidos e de proteínas específicas. Embora organelas são fisicamente isolaed, eles precisam de se comunicar uns com os outros e partes componentes moleculares a fim de manter a homeostase celular. A via secretora garante que as proteínas e lípidos sintetizados no ER chegar ao destino final correcta em que exercem a sua função. Organelas intracelulares também pode ser conectado por sites de contacto dinâmicos que permitem que as moléculas (lipídios) a serem trocadas diretamente entre os compartimentos. Além disso, muitas proteínas têm de montados em grandes complexos heteroméricos ou associada com as espécies de lípidos específicos (jangadas lipídicas / microdomínios), a fim de se tornar funcionalmente activa ou pode ser transportado para o seu destino final. Todos estes aspectos biológicos influenciam grandemente as propriedades cinéticas das proteínas, e podem, portanto, ser adequadamente investigada por meio das técnicas descritas a seguir.
Nosso grupo tem utilizado FRAP e FLIP combinado com microscopia eletrônica, a fim de estudar a arquitetura do ER e seus diferentes subdomínios. O ER é a primeira estação da via secretora e desempenha um papel chave na proteína e lípido de classificação 5. É uma organela altamente dinâmico cujos subdomínios distintos refletem suas muitas funções diferentes (ou seja, proteínas e biossíntese de lipídios e de tráfico, dobradura de proteínas, Ca 2 + armazenamento e liberação, e do metabolismo de xenobióticos). No entanto, embora sejam morfologicamente, espacialmente e funcionalmente distinto, estes domínios são contínuas umas com as outras, e a sua abundância relativa pode ser modificado em células em condições fisiológicas e patológicas. O mais conhecido e domínios geralmente espacialmente segregadas do ER são o envelope nuclear, eo RE liso e áspero, no entanto, nós e outros demonstraram que há estruturas de ER, com uma arquitetura mais elaborada e organização tridimensional em vários tipos de células e tecidos sob condições fisiológicas que podem ser induzidas por meio de estímulos de stress, tais como a hipoxia, a drogaadministração, ou a sobre-expressão de proteínas de transmembrana ER residente 2,6 (e suas referências).
Também se demonstrou recentemente a presença de tais estruturas em modelos celulares de doenças humanas 1,7. Originário da cisternas empilhadas de RE liso, eles receberam o nome coletivo de organizado retículo endoplasmático liso (OSER), em 2003, 6, embora eles também são conhecidos como karmellae, lamelas e cristalóide ER com base em sua arquitetura que, como seu tamanho, pode variar. Depois as células são transfectadas com GFP fundida com a região citosólica de proteínas de ER-residente (TA) (d EGFP-ER) ancorada-cauda, a tendência fracamente dimerização de GFP em trans altera drasticamente a organização e estrutura do ER. FRAP e FLIP experimentos mostraram que d EGFP-ER é livre para difundir dentro OSERs, eo fato de que ela se move a partir da ER reticular ao OSER e vice-versa </ Em> indica que os agregados são contínuas com o ER reticular circundante. Análise ultraestrutural nos permitiu correlacionar os dados de fluorescência com uma descrição detalhada da arquitetura OSER e organização em escala nanométrica: OSERs são sempre composta de pilhas de cisternas emparelhado de RE liso, mas pode ter diferentes formas de organização espacial, como regularmente organizadas sinusoidal arrays ou espirais, ou hexagonais "cristalóides" matrizes tubulares. Estes rearranjos levar a morfologias cúbicos 8 que, como eles foram encontrados em células em condições fisiológicas e 9 seguintes stress como a hipóxia 10, o tratamento de droga 11, e cancro 9, podem ter um potencial significativo como marcadores de ultra-estruturais.
Após esta primeira demonstração utilizando proteínas de fusão de GFP, foram utilizadas experiências de imagiologia para analisar a proliferação de domínios de ER, em resposta aos tratamentos farmacológicos 12, assess a tendência de proteínas fluorescentes para oligomerise nas células 13, e para investigar o papel de um mutante, proteína TA ALS-linked na formação de agregados intracelulares de origem ER que podem ser relevantes para a sua patogenicidade a 1,8. Tem sido sugerido que a formação de agregados intracelulares (que ocorre em muitas doenças neurodegenerativas, 14) pode ser um mecanismo de protecção concebido para evitar que as interacções entre proteínas mutantes tóxicos e os componentes celulares circundantes 15.
O que se segue é uma descrição de uma combinação de métodos de microscopia óptica e eletrônica para investigar construções cujo C-terminal domínios hidrofóbicos são inseridos na membrana do ER, e uma análise de seu comportamento dinâmico e os efeitos de sua sobre-expressão no domínio ER Arquitectura em células em cultura (ver Figura 1 para um fluxograma do protocolo experimental).
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Plasmídeo, cultura de células, e transfecção com ER fluorescentes Proteínas
- O plasmídeo utilizado no presente estudo consiste de uma versão melhorada de GFP fundida na sua extremidade C-terminal para a região da cauda da isoforma ER de rato do citocromo b (5) (abreviada aqui como B (5)), através de uma sequência ligante. A região de cauda contém toda a sequência (Pro94-Asp134) que permanece associada membrana após a clivagem de tripsina de nativo B (5), incluindo a DTM 17-resíduo (domínio transmembrana), flanqueado por sequências polares a montante e a jusante (UPS e DPS) . O ligante é constituído por o epitopo myc seguido por [(Gli) 4 Ser] 3, e todo o ADNc é inserido nos sítios Hind3-Xba1 do mamífero pCDNA3 vector de expressão. Os detalhes da construção deste plasmídeo foi descrito numa publicação anterior, em que é referido como a GFP-ER 16.
- Crescer células COS-7 em Meio Eagle Modificado por Dulbecco (DMEM) suplementado com 10% de fetal de soro de bovino, 2 mM de L-glutamina, 1% de penicilina / estreptomicina, numa incubadora a 37 ° C e 10% de CO 2.
- Transfecção. Placa de 3 x 10 5 células sobre uma lamela de vidro redondo de uma placa de 6 poços e, no dia seguinte, com o sistema de transfecção JetPEI como descrito pelo fabricante. Note-se que o rácio de JetPEI / ADN optimizada foi testada a fim de estabelecer a máxima eficiência de transfecção de acordo com o plasmídeo e linha celular utilizada: no nosso caso, uma JetPEI: proporção de ADN de 2:01 conduz a 70-80% de eficiência de transfecção.
2. Vivo Fluorescência Scanning microscopia confocal
- De imagem de células vivas. Coloque a lamela em que as células transfectadas foram semeadas numa câmara de cultura de células de aço de 24 mm lamelas cheios com DMEM w / o vermelho de fenol, suplementado com 10% de FBS, 2 mM de L-glutamina, 1% de pen / strep, 25 mM de HEPES , 50 ug / ml de cicloheximida e 1:100 OxyFluor para evitar as amostras de fotodegradação. Um microscop SP5 confocale equipado com uma incubadora de CO 2, com temperatura controlada (37 ° C e 5% de CO 2) é utilizado nas experiências de imagem de células vivas, com d GFP-ER ser visualizada usando um laser de 488 nm e um filtro 525/50 de passagem de banda de emissão.
- Recuperação de fluorescência após a fotodegradação (FRAP). Desenhar uma região de interesse (ROI), correspondente a uma estrutura OSER, e descora usando 20 iterações e uma combinação de 488 nm (100% de um laser de Árgon de 30 mW, correspondendo a 5.5-6 μW na amostra) e 405 nm (60% de 30 mW de diodo laser de 405, o que corresponde a 11,6 μW na amostra) lasers que, em nossa experiência, leva a fotodegradação rápida e eficiente.
- Grave a recuperação de fluorescência nas ROIs branqueada, tomando um único quadro a cada 10 segundos por 10 minutos (tempo de pixel = 1,61 ms / px).
- Perda de fluorescência na fotodegradação (FLIP). Desenhar um ROI correspondente a uma estrutura OSER, e lixívia, como descrito acima. O branqueamento é repetido a cada 30 seg,e as imagens pós-clareamento são registrados a cada 10 segundos por 30 minutos (tempo de pixel = 1,61 ms / px).
- FRAP e análise FLIP. Todas as imagens são analisadas utilizando o software ImageJ ( http://rsbweb.nih.gov/ij/download.html ). Nas experiências de FRAP, a recuperação de fluorescência do ROI branqueada é medida ao longo do tempo e normalizados para a fluorescência total de células branqueadas, que é sempre verificado para ser constante ao longo do tempo.
- Para os experimentos FLIP, desenhar um ROI fora do OSER branqueada e abrangendo toda a célula. Medir a intensidade de fluorescência ao longo do tempo e normalizar os níveis de fluorescência de um ROI desenhados em uma célula não branqueada, a fim de corrigir qualquer diminuição na fluorescência provocada pela própria imagem.
- Em todas as experiências, subtraído do sinal de fundo (determinado numa zona fora das células) a partir das intensidades de fluorescência das ROI. Finalmente, plotar os resultados usando GraphPad software Prism.
3. Ultraestrutural Análise por Microscopia Eletrônica de Transmissão
Dada a toxicidade de muitos dos reagentes, todos os procedimentos devem ser efectuados usando um laboratório adequado. casaco e luvas sob uma coifa.
- Depois de retirar a lamela do prato de Petri, fixar as restantes células cultivadas na parte inferior do prato como uma monocamada utilizando filtrada glutaraldeído a 2% em tampão cacodilato 0,1 M, pH 7,4, durante 10 min à temperatura ambiente.
- Raspar as células com um raspador de Teflon e transferi-los para tubos de 1,5 ml de Eppendorf. Agregar as células por meio de centrifugação a 9.000 g durante 10 min. Retirar o sobrenadante, adicionar fixador fresco, e deixar durante a noite a 4 ° C.
- Lavar os sedimentos com o tampão, então pós-corrigir com uma solução de 1% de tetróxido de ósmio em tampão cacodilato, durante 1 hora à temperatura ambiente.
- Enxágüe com água MilliQ, e em bloco mancha com1% acetato de uranila em água destilada por entre 20-60 min.
- Desidratar as amostras em série crescente de etanol (70%, 80%, 90%, 100% e 100% durante 10 min cada), e lava-se rapidamente duas vezes em óxido de propileno (15 min cada).
- Infiltrar as amostras em uma mistura de óxido de propileno + Epon (1:1) (a partir de 2 horas durante a noite).
- Incorporar na resina epoxi Epon curada a 60 ° C durante pelo menos 24 horas.
- Seção dos blocos de resina aparadas manualmente usando um ultramicrótomo LEICA UC6 equipado com um diamante faca de 45 ° para obtenção de cortes com espessura de 60-70 nm. Colete as seções 300 grades de cobre de malha.
- Corar as secções sobre a grelha com uma solução saturada de acetato de uranilo (20 min) e citrato de chumbo (7 min), lavar as grades por imersão em água bi-destilada, filtrada, e deixar secar à temperatura ambiente.
- As grades manchadas são observados usando um Tecnai G2 microscópio eletrônico de transmissão, e as imagens foram captadas com uma bottcâmera CCD om-montado em diferentes ampliações finais (geralmente variando de 6,000-39,000 X).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
A Figura 2 mostra um estudo FRAP exemplo de mobilidade da proteína. A mobilidade da proteína d EGFP-ER é demonstrado pela recuperação de fluorescência após a fotodegradação rápida em OSERs branqueadas. Para a análise quantitativa, a meia hora e fração móvel foram obtidos a partir de dados medidos experimentalmente ajustando a seguinte equação monoexponencial:
F (t) = F pós + (F rec-F post) (1-e-t / τ)
onde F post é o sinal de fluorescência após a fotodegradação, F rec é o valor máximo de recuperação de fluorescência que é chegar após o clareamento, t no momento da inscrição e τ a constante de tempo.
Por favor, note a importância de adquirir imagens sem pixels saturadas que podem alterar a recuperação de fluorescência e, conseqüentemente, a análise da mobilidade de proteínas. É is também essencial para normalizar sempre o sinal de fluorescência na ROI branqueada para a fluorescência total da mesma célula, a fim de considerar as variações de intensidade de fluorescência devido ao branqueamento durante a aquisição de imagem ou de pequenas alterações no plano de foco.
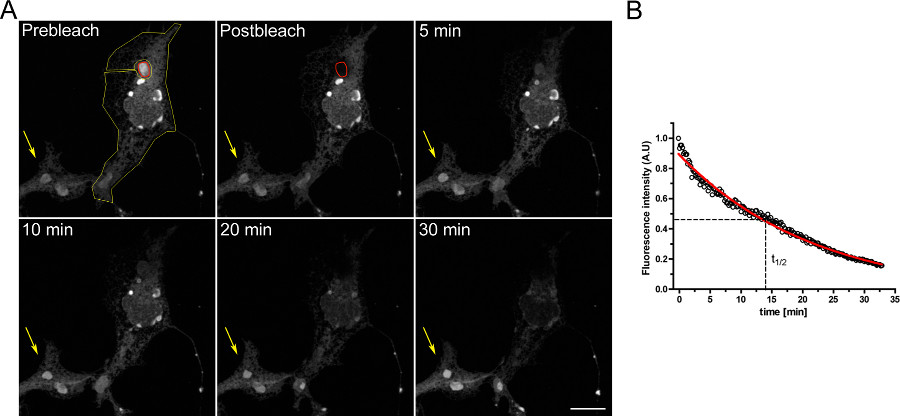
Um exemplo de um ensaio FLIP para estudar a continuidade entre os compartimentos intracelulares é mostrado na Figura 3. OSERs está fisicamente ligado com o resto do ER como demonstrado pelo esvaziamento progressivo do ER, quando o domínio está continuamente OSER branqueada.
Para uma análise adequada, a aquisição de pixels saturadas devem ser evitadas (veja acima), além disso, os parâmetros de aquisição deve ser configurado com poderes de laser o mais baixo possível, a fim de evitar a fotodegradação, devido à aquisição de imagem. Por esta razão, recomenda-se fortemente a imagem de uma célula não branqueada no mesmo campo que vai ser usada para normalizar o sinal de fluorescência do c branqueadaell.
Todas as experiências devem ser realizadas na presença de cicloheximida, um inibidor da tradução, a fim de evitar qualquer aumento no sinal de fluorescência do ER (e, consequentemente, a fluorescência total) devido a biossíntese de proteínas.
A microscopia electrónica de transmissão demonstrou que os agregados fluorescentes observados em células de cultura transfectadas com EGFP d-ER representa manchas de lisa e achatada ER cisternas que se organizam espacialmente em geometrias bem definidas 3D classificadas com base no seu padrão: pilhas lineares ou curvos (frequentemente associada com o envelope nuclear, não mostrado) (Figuras 4A e B), que pode ser contínuo com regiões de ER sinusoidal (Figura 4A); as membranas em algumas regiões estão organizados em reticulados com uma simetria quadrada ou hexagonal (cristalóide ER, não mostrado ). Cisternas adjacentes são separados por uma fina camada de um pouco de electrõescitoplasma denso cerca de 11 nm de espessura, que é contínua com o citoplasma, os agregados.

Figura 1. Fluxograma do procedimento experimental. As células de cultura são primeiro transfectadas com jetPEI (ver protocolo), de modo a sobre-expressar a proteína de fusão de interesse fluorescente. Após 24 horas, as células transfectadas ao vivo são visualizadas e FRAP e FLIP experimentos são realizados usando um microscópio confocal equipado com uma temperatura controlada incubadora de CO 2, e as imagens gravadas são exportados e analisadas utilizando software apropriado (por exemplo, o ImageJ). Para a análise ultra-estrutural, as células transfectadas são fixos, sedimentadas e embebidos em EPON epoxi blocos de resina. Cortes ultrafinos são obtidos utilizando uma faca de diamante, coletados em copper grades, e observados em microscópio eletrônico de transmissão. Clique aqui para ver maior figura .

Figura 2. FRAP experimento utilizando células COS-7 transfectadas transitoriamente com d EGFP-ER. A) Duas estruturas OSER (ROIs vermelho) foram branqueados e recuperação de fluorescência foi gravado ao longo do tempo. Limpar recuperação de fluorescência pode ser detectada 1 min pós-branqueamento, e o sinal de novos aumentos de 4 minutos mais tarde (barra de escala de 10 um) B):. Análise quantitativa do experimento FRAP que mostra o semi-tempo de recuperação e a fracção celular do d EGFP ER-proteína. Click aqui para ver maior figura

Figura 3. FLIP experimento utilizando células COS-7 transfectadas transitoriamente com d EGFP-ER. A) O branqueamento contínuo de um OSER (indicado pela ROI vermelho) provoca uma diminuição progressiva na fluorescência no resto do ER e em outras estruturas OSER dentro da mesma célula (indicado pela ROI amarelo). A seta amarela indica uma parte de uma célula não branqueada, em que o sinal de fluorescência é constante ao longo do tempo. (Barra de escala de 10 mm). B) Análise quantitativa do experimento FLIP. Clique aqui para ver maior figura
Figura 4. Após a fixação e a incorporação, as células que expressam altos níveis de d EGFP-ER, em que as estruturas OSER poderiam ser detectados por meio de microscopia óptica de fluorescência foram observados através de um microscópio electrónico de transmissão. A) Vista de baixa ampliação de uma parte do citoplasma de uma célula contendo um OSER consistindo de cisternas empilhadas e membranas sinusoidais onduladas. As mitocôndrias (M) pode ser visto agrupados em torno das estruturas OSER, enquanto que os ribossomos decorar apenas as membranas das cisternas mais externa (pontas de seta e inserção). O espaço 11 de electrões denso nm de espessura entre as membranas é contínuo com o citoplasma (seta e inserção) (L = lisossomas / (auto-) phagosomes) (barra de escala de 1,5 mM; inserir 0,25 mM). B) Uma OSER pode ser formado por lamelar ER: ou seja, pilhas de cisternas achatada ER que pode ser contínua ou Fragmented em sua aparência em seções finas. As vesículas brotando do cisternas mais externa da pilha pode ocasionalmente ser observada (asterisco) (PM, membrana plasmática) (Barra de escala de 150 nm).
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Os protocolos e abordagens de imagem descritos neste trabalho foram utilizados para investigar a distribuição e mobilidade das proteínas fluorescentes TA transfectadas residentes no ER de células vivas. Analisamos também o efeito da expressão sobre-destas proteínas na arquitetura deste compartimento subcelular por meio de análises ultra-estruturais.
A combinação de células vivas de imagem confocal e microscopia eletrônica representa é um poderoso meio de investigar as propriedades dinâmicas de proteínas, e pode fornecer informações importantes sobre a função da proteína. Os métodos descritos não são demoradas (normalmente de três dias de trabalho), e o desenvolvimento de muitas aplicações de software de fácil utilização para aquisição e análise de imagens faz com base em fotodegradação, imagens ao vivo de células relativamente simples.
A principal limitação destas técnicas é a utilização de proteínas de fusão fluorescentes, porque omarcador fluorescente pode afectar a dobragem e / ou a montagem adequada da proteína de interesse. Além disso, a sobre-expressão pode alterar o comportamento de transfectadas, proteínas marcadas por fluorescência, e não pode, portanto, reflectir as propriedades reais de proteínas endógenas, no entanto, este pode ser ultrapassada pela utilização de células de indutíveis e transfectadas estavelmente, em que o nível de expressão pode ser precisamente modulada para obter níveis comparáveis aos da proteína endógena 1,7. A tendência de FPs para oligomerise tem sido amplamente documentado e pode alterar significativamente o comportamento (ou seja, cinética, interações proteína-proteína indesejados e formação de agregados) de proteínas quiméricas. A utilização de proteínas fluorescentes monoméricos optimizados deve, portanto, ser considerado 17.
Outro aspecto crítico de estudos de imagens dinâmicas utilizando fluorescência e fotodegradação é o tempo necessário para branquear a fluorescência de forma eficiente e medir a fluorescência reberta (e, assim, a mobilidade da proteína), precisamente, o qual depende também da área da ROI e espessura celular local. Se uma dada proteína marcadas com GFP tem um alto coeficiente de difusão, a difusão pode ocorrer durante o branqueamento e, assim, interfere com as medições de tempo de recuperação. Para obter clareamento rápido e eficiente, é altamente recomendável que um "zoom" de função (se disponível) e mais do que uma linha de laser ser usado. Embora a utilização de um módulo de verificação rápida (ou seja, um scanner de ressonância) pode melhorar significativamente a velocidade da imagem durante a fase de recuperação de uma experiência, nas nossas mãos é também reduz consideravelmente a eficiência do branqueamento. No entanto, os sistemas alternativos de varrimento (tal como um disco giratório equipado com um dispositivo dedicado de fotodegradação), e os lasers mais potentes podem melhorar a eficiência do branqueamento e a velocidade de aquisição.
A maioria das proteínas fluorescentes usadas na FRAP e FLIP experimentos mostram algum grau de fotodegradação reversível e blinking que deve ser considerado quando se realiza análises quantitativas. As flutuações entre estados fluorescentes e escuras ocorrer no segundo a escala de tempo minuto. Para EGFP, Tem sido demonstrado que, durante as experiências de branqueamento, as variações de fluorescência podem envolver menos do que 10% das moléculas, assim, no presente protocolo este fenómeno é negligenciável. Se todas as condições são mantidas constantes, isto irá introduzir um desvio constante nos resultados. Se forem utilizadas outras proteínas fluorescentes, em que a fracção reversível é significativamente mais elevado (isto é, YFP), ou para detectar e avaliar a fotodegradação reversibilidade, isto pode ser feito medindo-se a recuperação de fluorescência após a fotodegradação em toda a célula viva, se a recuperação é observada esta pode apenas ser o resultado de fotodegradação reversibilidade 18.
A toxicidade potencial da luz durante os experimentos é outro fator crítico, principalmente porque fotodegradação exige iluminação forte. É well conhecido que fluoróforos excitados tendem a reagir com o oxigênio para produzir radicais livres que podem afetar vários processos intracelulares e até mesmo a viabilidade das células 19, e por isso, é necessário estabelecer um equilíbrio entre o branqueamento eficiente e fototoxicidade mínima, além disso, a viabilidade celular deve ser sempre verificado depois de experimentos com imagens de células ao vivo. Dado o tempo de gravação curto, não consideramos o efeito genotóxico de luz de comprimento de onda curto (405 nm) no exemplo descrito neste artigo, mas, se for necessária uma experiência mais longa, não deve ser usada uma linha de laser 405 nm.
Optamos por não usar uma abordagem correlativa à microscopia eletrônica de transmissão, devido à natureza heterogênea da arquitetura OSER eo fato de que nós queríamos observar o maior número de células (e estruturas) possível. A diversidade da estrutura fina de agregados de proteínas nas células pode ser uma característica fundamental de diferentes doenças e estávamos interessados em obter uma ampla gama de samples, ao passo que uma abordagem correlativa permite a observação de menos eventos durante o mesmo período de tempo. No entanto, a microscopia de elétrons luz correlativo (CLEM) deve ser a primeira escolha quando se investiga eventos em estruturas que não podem ser facilmente identificados (como subdomínios ER menos proeminentes), ou em um número limitado de células (como células de injeção de micro). Vale a pena notar que as nossas experiências foram caracterizados por um elevado grau de eficiência da transfecção (no mínimo 30% das células foram transfectadas), caso contrário, a possibilidade de observar estruturas OSER noncorrelatively é bastante limitado.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Os autores não têm nada a revelar.
Acknowledgments
Os autores são gratos a Fondazione Filarete por sua ajuda e apoio na publicação deste artigo. Gostaríamos também de agradecer Centro Europeo di Nanomedicina para o uso do Tecnai G2 microscópio eletrônico de transmissão.
Materials
| Name | Company | Catalog Number | Comments |
| Dulbecco’s Modified Eagle Medium (DMEM) | Invitrogen | 41966029 | |
| Dulbecco’s Modified Eagle Medium (DMEM) w/o phenol red | Invitrogen | 31053028 | |
| Fetal Bovine Serum (FBS) | Invitrogen | 10270106 | |
| Pen/Strep | Invitrogen | 15140-122 | |
| L-Glutamine 200 mM solution | Invitrogen | 25030-024 | |
| jetPEI | Polyplus Transfection | PP10110 | |
| OxyFluor | Oxyrase Inc. | OF-0005 | |
| Glutaraldehyde Grade I | Sigma Aldrich | G5882 | |
| Sodium Cacodylate Trihydrate | Sigma Aldrich | C0250 | |
| Osmium Tetroxide 4% solution | Electron Microscopy Science | 19150 | |
| Uranyl Acetate Dihydrate | Sigma Aldrich | 73943 | slightly radioactive |
| Propylene Oxide | Sigma-Aldrich | 82320 | |
| EPON embedding medium kit | Sigma-Aldrich | 45359-1EA-F | |
| Lead Citrate | Electron Microscopy Science | 17800 | |
| Bench top centrifuge | Eppendorf | 5415 D | |
| Spectral Confocal Microscope | Leica Microsystems | TCS SP5 | |
| CO2 Microscope Cage Incubation System | OkoLab | ||
| Ultramicrotome | Leica Microsystems | UC6 | |
| Diamond knife | Diatome | Ultra 45 ° | |
| Transmission Electron Microscope | FEI | Tecnai G2 | |
| GraphPad Prism Software | GraphPad Software, Inc | ||
| Steel culture cell chamber for 24 mm coverslip | Bioscience Tools | CSC-25 | |
| Electron Microscopy grids | Electron Microscopy Science | G300Cu |
References
- Fasana, E., et al. A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 24, 1419-1430 (2010).
- Borgese, N., Francolini, M., Snapp, E. Endoplasmic reticulum architecture: structures in flux. Curr. Opin. Cell Biol. 18, 358-364 (2006).
- Ronchi, P., Colombo, S., Francolini, M., Borgese, N. Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J. Cell Biol. 181, 105-118 (2008).
- Lippincott-Schwartz, J., Snapp, E., Kenworthy, A. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2, 444-456 (2001).
- Lee, M. C., Miller, E. A., Goldberg, J., Orci, L., Schekman, R. Bi-directional protein transport between the ER and. 20, 87-123 (2004).
- Snapp, E. L., et al. Formation of stacked ER cisternae by low affinity protein interactions. J. Cell Biol. 163, 257-269 (2003).
- Papiani, G., et al. Restructured endoplasmic reticulum generated by mutant amyotrophic lateral sclerosis-linked VAPB is cleared by the proteasome. J. Cell Sci. 125, 3601-3611 (2012).
- Almsherqi, Z. A., Kohlwein, S. D., Deng, Y. Cubic membranes: a legend beyond the Flatland* of cell membrane organization. J. Cell Biol. 173, 839-844 (2006).
- Federovitch, C. M., Ron, D., Hampton, R. Y. The dynamic ER: experimental approaches and current questions. Curr. Opin. Cell Biol. 17, 409-414 (2005).
- Takei, K., Mignery, G. A., Mugnaini, E., Sudhof, T. C., De Camilli, P. Inositol 1,4,5-trisphosphate receptor causes formation of ER cisternal stacks in transfected fibroblasts and in cerebellar Purkinje cells. Neuron. 12, 327-342 (1994).
- Feldman, D., Swarm, R. L., Becker, J. Ultrastructural study of rat liver and liver neoplasms after long-term treatment with phenobarbital. Cancer Res. 41, 2151-2162 (1981).
- Sprocati, T., Ronchi, P., Raimondi, A., Francolini, M., Borgese, N. Dynamic and reversible restructuring of the endoplasmic reticulum induced by PDMP in cultured cells. J. Cell Sci. 119, 3249-3260 (2006).
- Costantini, L. M., Fossati, M., Francolini, M., Snapp, E. L. Assessing the tendency of fluorescent proteins to oligomerize under physiologic conditions. Traffic. 13, 643-649 (2012).
- Pyszniak, A. M., Welder, C. A., Takei, F. Cell surface distribution of high-avidity LFA-1 detected by soluble ICAM-1-coated microspheres. J. Immunol. 152, 5241-5249 (1994).
- Taylor, J. P., Hardy, J., Fischbeck, K. H. Toxic proteins in neurodegenerative disease. Science. 296, 1991-1995 (2002).
- Winklhofer, K. F., Tatzelt, J., Haass, C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 27, 336-349 (2008).
- Borgese, N., Gazzoni, I., Barberi, M., Colombo, S., Pedrazzini, E. Targeting of a tail-anchored protein to endoplasmic reticulum and mitochondrial outer membrane by independent but competing pathways. Mol. Biol. Cell. 12, 2482-2496 (2001).
- Chudakov, D. M., Matz, M. V., Lukyanov, S., Lukyanov, K. A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 90, 1103-1163 (2010).
- Bancaud, A., Huet, S., Rabut, G., Ellenberg, J. Fluorescence perturbation techniques to study mobility and molecular dynamics of proteins in live cells FRAP, photoactivation, photoconversion, and FLIP. Cold Spring Harb. Protoc. 2010, (2010).
- Michida, T., et al. Role of endothelin 1 in hemorrhagic shock-induced gastric mucosal injury in rats. Gastroenterology. 106, 988-993 (1994).

{kind=link}