

Summary
Vi beskriver avbildning vi använder för att undersöka spridning och rörlighet för de transfekterade fluorescerande proteiner bosatta i det endoplasmatiska retiklet (ER) med hjälp av den konfokala avbildning av levande celler. Vi också ultrastrukturellt analysera effekten av deras uttryck i arkitekturen i denna subcellulära fack.
Abstract
De lipider och proteiner i eukaryota celler ständigt byts ut mellan cell fack, även om dessa behåller sin distinkta sammansättning och funktioner trots det intensiva interorganelle molekylära trafik. De tekniker som beskrivs i detta dokument är kraftfulla medel för att studera protein och lipid rörlighet och handel in vivo och i deras fysiologiska miljö. Fluorescens återhämtning efter fotoblekning (FRAP) och fluorescens förlust i fotoblekning (FLIP) används ofta levande cellavbildningstekniker för att studera intracellulära människohandel genom exo-endocytic vägen, kontinuiteten mellan organeller eller underutrymmena, bildandet av proteinkomplex, och protein lokalisering i lipid mikroområden, som alla kan observeras under fysiologiska och patologiska tillstånd. Begränsningarna av dessa metoder är främst på grund av användningen av fluorescerande fusionsproteiner, och deras eventuella nackdelar inkluderar artefakter över expressjon i celler och möjligheten att skillnader i veckning och lokalisering av märkta och nativa proteiner. Slutligen, eftersom gränsen för upplösningen av optisk mikroskopi (ca 200 nm) inte tillåter undersökning av den fina strukturen hos ER eller de särskilda underutrymmena som kan ha sitt ursprung i celler under stress (t.ex. hypoxi, läkemedelsadministrering, överuttryck av membran ER bosatta proteiner) eller i enlighet med sjukdomstillstånd, kombinerar vi live-cell imaging av odlade transfekterade celler med ultraanalyser baserade på transmissionselektronmikroskop.
Introduction
Upptäckten av grönt fluorescerande protein (GFP) och dess spektrala varianter, och den parallella utvecklingen av fluorescensmikroskopi, har öppnat upp helt nya möjligheter för undersökning av protein beteende i celler. Tekniker såsom fluorescens återhämtning efter fotoblekning (FRAP) och fluorescensförlust i fotoblekning (FLIP), vilket är möjligt på grund av den inneboende förmågan av fluoroforer för att släcka sin fluorescens under intensiv belysning, är baserade på konfokal live-cell imaging och användningen av transfekterade fluorescerande fusionsproteiner 1-3. De används för att bedöma inte endast lokaliseringen av proteiner, men även deras rörlighet och vesikulär transport, som kan avslöja viktiga ledtrådar beträffande deras funktion 4.
Det unika med eukaryota celler är närvaron av intracellulära fack som har specifika lipid-och proteinkompositioner. Även om organeller är fysiskt isolated, de behöver för att kommunicera med varandra och dela med molekylära komponenter för att upprätthålla cellulär homeostas. Den sekretoriska vägen garanterar att proteiner och lipider syntetiseras i ER nå rätt slutdestination där de utövar sin funktion. Intracellulära organeller kan också anslutas genom en dynamisk kontakt webbplatser som tillåter molekyler (lipider) vara direkt utbytas mellan fack. Dessutom har många proteiner har att monteras i stora heteromera komplex eller förknippad med specifika lipid arter (lipid flottar / mikroområden) för att bli funktionellt aktiva eller transporteras till slutdestinationen. Alla dessa biologiska aspekter kraftigt påverka de kinetiska egenskaperna hos proteiner, och kan därför på lämpligt sätt undersöktes med hjälp av de tekniker som beskrivs nedan.
Vår grupp har i stor utsträckning används FRAP och FLIP kombination med elektronmikroskop för att studera strukturen för ER och dess olika underdomäner. ER är den första stationen i den sekretoriska vägen och spelar en viktig roll i protein och lipid sortering 5. Det är en mycket dynamisk organell vars distinkta subdomäner reflekterar dess många olika funktioner (dvs. protein och lipid biosyntes och människohandel, proteinveckning, Ca2 + lagring och uttag, samt xenobiotiska ämnesomsättning). Emellertid, även om de är morfologiskt, rumsligt och funktionellt distinkt, dessa domäner är kontinuerliga med varandra, och deras relativa förekomst kan modifieras på celler under fysiologiska och patologiska tillstånd. Den mest kända och oftast rumsligt segregerade områden av ER är kärnhöljet, samt en harmonisk och grov ER, men har vi och andra visat att det finns ER strukturer med en mer utarbetad arkitektur och tredimensionell organisation i olika celltyper och vävnader under fysiologiska betingelser som också kan induceras med hjälp av stressande stimuli, såsom hypoxi, narkotikaadministration, eller överuttryck av ER-bosatt transmembranproteiner 2,6 (och referenser däri).
Vi har också nyligen påvisat sådana strukturer i cellmodeller för mänskliga sjukdomar 1,7. Med ursprung från den staplade cisternae av släta ER, fick de samlingsnamnet organiserade släta endoplasmatiska nätverket (OSER) 2003 6, även om de är även känd som karmellae, lameller, och kristalloid ER utifrån sin arkitektur som, liksom deras storlek kan variera. När cellerna transfekterade med GFP smält till cytosoliska regionen i svansen förankrade (TA) ER-bosatt proteiner (d EGFP-ER), den svagt dimerisering av tendensen av GFP i trans förändrar dramatiskt organisation och struktur av ER. FRAP och FLIP experiment visade att d EGFP-ER är fri att diffundera inom OSERs, och det faktum att den rör sig från det retikulära ER till OSER och omvänt </ Em> indikerar att aggregaten är kontinuerlig med den omgivande retikulära ER. Ultra analys har gett oss möjlighet att korrelera fluorescens data med en detaljerad beskrivning av OSER arkitektur och organisation på nanonivå: OSERs alltid består av högar av parade cisternae av släta ER, men kan ha olika former för fysisk planering, som regelbundet arrangeras sinus arrayer eller virvlar, eller sexkantiga "kristalloida" rörformiga arrayer. Dessa omlagringar leda till kubiska morfologier 8 vilket, som de har visat i celler under fysiologiska betingelser 9 och följande spänningar såsom hypoxi 10, läkemedelsbehandling 11, och cancer 9, kan ha en betydande potential som ultrastrukturella markörer.
Efter denna första demonstrationen med hjälp av GFP fusionsproteiner, använde vi imaging experiment för att analysera spridningen av ER-domäner som svar på farmakologisk behandling 12, Assess tendens fluorescerande proteiner att oligomerise i celler 13, samt att undersöka rollen av en mutant, ALS-kopplade TA protein i bildandet av intracellulära aggregat av ER ursprung som kan vara relevanta för dess patogenicitet 1,8. Det har föreslagits att bildandet av intracellulära aggregat (som förekommer i många neurodegenerativa sjukdomar 14) kan vara en skyddsmekanism som syftar till att förhindra interaktioner mellan giftiga muterade proteiner och de omgivande cellkomponenter 15.
Vad som följer är en beskrivning av en kombination av optiska och elektronmikroskopiska metoder för att undersöka konstruktioner vars C-terminal hydrofoba domäner in i membranet av ER, och en analys av deras dynamiska beteende och effekterna av deras överuttryck på ER domän arkitektur i odlade celler (se figur 1 för ett flödesschema över det experimentella protokollet).
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Plasmid, cellodling, och transfektion med ER fluorescerande proteiner
- Den plasmid som användes i denna studie består av en förbättrad version av GFP fusionerad vid sin C-terminal till den bakre regionen av ER-isoformen av rått-cytokrom-b (5) (förkortat här som b (5)) via en länksekvens. Svansen region innehåller hela sekvensen (Pro94-Asp134) som förblir membranassocierat efter trypsin klyvning av nativ B (5), inklusive 17-rest TMD (transmembran domän), flankerad av uppströms och nedströms polära sekvenser (UPS och DPS) . Linkern består av myc-epitopen, följt av [(Gly) 4 Ser] 3, och hela cDNA sätts in i Hind3-Xba1 ställena i däggdjursuttrycksvektorn pCDNA3. Detaljerna i konstruktionen av denna plasmid har beskrivits i en tidigare publikation, i vilken det hänvisas till som GFP-ER 16.
- Grow COS-7-celler i Dulbeccos modifierade Eagles medium (DMEM) kompletterat med 10% fetal bovint serum, 2 mM L-glutamin, 1% penicillin / streptomycin i en inkubator vid 37 ° C och 10% CO2.
- Transfektion. Tallrik 3 x 10 5 celler på en rund täckglas i en 6-brunnsplatta och, följande dag, transfektera med JetPEI system som beskrivs av tillverkaren. Observera att den optimala JetPEI / DNA kvoten har testats för att fastställa maximal transfektion effektivitet beroende på plasmiden och cellinje som används: i vårt fall, en JetPEI: DNA-förhållande på 2:1 leder till 70-80% transfektion effektivitet.
2. Live Fluorescens Scanning konfokalmikroskopi
- Live-cell imaging. Sätt täckglas på vilka de transfekterade cellerna såddes i en stål kultur cellkammare för 24 mm täckglas fylld med DMEM w / o-fenolrött, kompletterad med 10% FBS, 2 mM L-glutamin, 1% pen / strep, 25 mM HEPES , 50 | ig / ml cykloheximid och 1:100 OxyFluor att förhindra proverna från fotoblekning. En SP5 konfokal microscope utrustad med en temperaturstyrd CO2 inkubator (37 ° C och 5% CO2) används för levande cell imaging experiment, med d GFP-ER som visualiserades med användning av en 488 nm laser och en 525/50 bandpassemissionsfilter.
- Fluorescens återhämtning efter fotoblekning (FRAP). Rita en region av intresse (ROI), vilket motsvarar en OSER struktur, och bleker den med 20 iterationer och en kombination av 488 nm (100% av en 30 mW argonlaser, motsvarande 5,5-6 ^ W på provet) och 405 nm (60% av en 30 mW Diod 405 laser, vilket motsvarar 11,6 pW vid provet) lasrar som, enligt vår erfarenhet, leder till effektiv och snabb fotoblekning.
- Spela återvinning av fluorescens i de blekta ROI genom att ta en enda bild var 10 sekund i 10 min (pixel tid = 1,61 ^ sek / px).
- Fluorescens förlust i fotoblekning (FLIP). Rita en ROI motsvarande en OSER struktur och blekmedel såsom beskrivits ovan. Blekningen upprepas varje 30 sekund,och efter bleknings bilder registreras varje 10 sek 30 min (pixel tid = 1,61 ^ sek / px).
- FRAP och FLIP analys. Alla av bilder analyseras med ImageJ programvara ( http://rsbweb.nih.gov/ij/download.html ). I FRAP experiment fluorescensen återvinning av den blekta ROI mäts över tid och normaliseras till den totala fluorescensen av blekt cell, som alltid skall kontrolleras för att vara konstanta över tiden.
- För FLIP experiment, rita en ROI utanför blekt OSER och täcker hela cellen. Mät dess fluorescensintensitet med tiden och normalisera till fluorescensnivåer av en ROI som dras från ett oblekt cellen för att korrigera för eventuell minskning i fluorescens orsakad av avbildnings självt.
- I alla de experiment subtraherades bakgrundssignalen (bestämd i ett område utanför de celler) från de fluorescerande intensiteten hos ROI. Slutligen, plotta resultaten med hjälp av GraphPad Prism mjukvara.
3. Ultra Analys genom transmissionselektronmikroskopi
Med tanke på toxiciteten hos många av reagensen, bör alla förfaranden utföras bär ett lämpligt laboratorium. kappa och handskar under en huv.
- Efter avlägsnande av skyddsremsan från petriskålen, fastställa de återstående cellerna odlas på botten av skålen som ett monoskikt med användning av filtrerad 2% glutaraldehyd i 0,1 M kakodylatbuffert, pH 7,4, under 10 min vid rumstemperatur.
- Skrapa cellerna med användning av en Teflon-skrapa och överföra dem in i 1,5 ml Eppendorf-rör. Pelletera cellerna genom centrifugering vid 9000 g under 10 min. Avlägsna supernatanten, tillsätt färskt fixativ, och lämna över natt vid 4 ° C.
- Tvätta pelleten med buffert, varefter efter fixa med en lösning av 1% osmiumtetroxid i kakodylatbuffert under 1 timme vid rumstemperatur.
- Skölj med MilliQ vatten, och i klump fläcken med1% uranylacetat i destillerat vatten i mellan 20 till 60 min.
- Torka proven i ökande etanolserie (70%, 80%, 90%, 100% och 100% efter 10 min var), och tvätta kortvarigt två gånger i propenoxid (15 min vardera).
- Infiltrera proverna i en blandning av propenoxid + Epon (01:01) (från 2 h till över natten).
- Bädda in i Epon epoxihartset härdades vid 60 ° C under minst 24 timmar.
- § de manuellt trimmade harts block med hjälp av en ultramicrotome LEICA UC6 utrustad med en 45 ° diamant kniv för att erhålla sektioner med en tjocklek av 60-70 nm. Samla avsnitten om 300 mesh koppargaller.
- Fläck avsnitten på rutnätet med en mättad lösning av uranylacetat (20 min) och blycitrat (7 min), grundligt tvätta gallren genom att nedsänka dem i bi-destillerat filtrerat vatten, och tillåta dem att torka vid rumstemperatur.
- De färgade galler observeras med hjälp av en Tecnai G2 transmissionselektronmikroskop, och bilderna har tagits med en Bottom-monterade CCD-kamera vid olika slut förstoringar (i allmänhet sträcker sig från 6,000-39,000 X).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Figur 2 visar ett exempel FRAP studie av protein rörlighet. Rörligheten för d EGFP-ER-proteinet visas av den snabba fluorescens återhämtning efter fotoblekning i blekta OSERs. För den kvantitativa analysen, var det halvtid och mobildelen från experimentellt uppmätta data genom att montera följande monoexponentiellt ekvation:
F (t) = F inlägget + (F rec-F post) (1-e-t / τ)
där F inlägget är fluorescenssignal efter fotoblekning, är F rec maximal fluorescens återhämtning värde som når efter blekning, t registreringstillfället och τ tidskonstanten.
Observera vikten av att få bilder utan mättade bildpunkter som skulle kunna ändra den fluorescens återhämtning och därmed protein rörlighet analysen. Det jagär också viktigt att alltid normalisera fluorescenssignalen i blekt ROI till den totala fluorescens i samma cell för att överväga fluorescensintensitetsvariationer på grund av blekning under bildtagning eller små förändringar i fokusplanet.
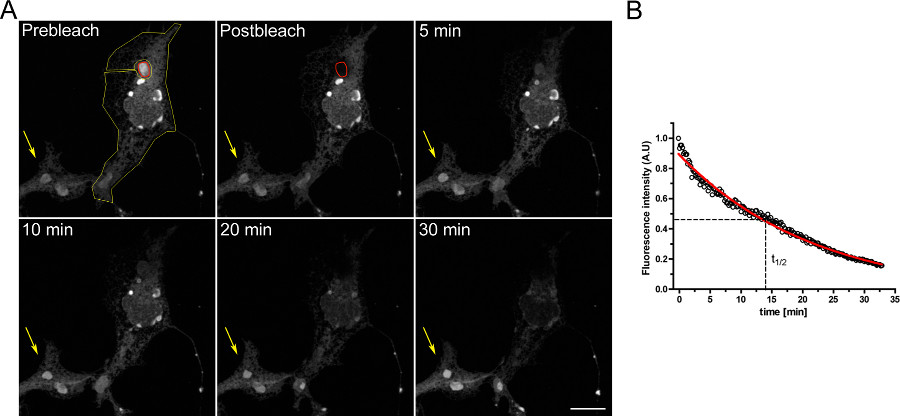
Ett exempel på en FLIP experiment för att studera kontinuitet mellan intracellulära fack visas i Figur 3. OSERs är fysiskt förbunden med resten av ER som visat av den progressiva tömningen av ER när OSER domän kontinuerligt blekas.
För en ordentlig analys, skall förvärvet av mättade pixlar undvikas (se ovan), och dessutom måste de förvärvsparametrarna ställas in med lasereffekter så låga som möjligt för att undvika fotoblekning på grund av bildtagning. Därför är det starkt rekommenderat att bilden en oblekt cell inom samma område som kommer att användas för att normalisera fluorescenssignalen av den blekta cell.
Alla experiment måste utföras i närvaro av cykloheximid, en översättning-inhibitor, i syfte att undvika varje ökning i ER fluorescenssignalen (och följaktligen den totala fluorescens) på grund av proteinsyntes.
Transmissionselektronmikroskopi visade att de fluorescerande aggregat observerades i odlade celler transfekterade med d EGFP-ER representerar fläckar av jämn och tillplattad ER cisternae att spatialt organiserat sig i väl definierade 3D-geometrier som klassificeras på grundval av sina mönster: linjära eller krökta stackar (ofta förknippade med kärnhöljet, visas inte) (fig. 4A och B) som kan vara kontinuerlig med regioner av sinus ER (Figur 4A), membranen i vissa regioner är organiserade i gitter med en offentlig hexagonal symmetri (kristalloid ER, visas inte ). Intilliggande cisternae är åtskilda av ett tunt lager av något elektrontät cytoplasma cirka 11 nm tjockt som är kontinuerlig med cytoplasman omger aggregaten.

Figur 1. Flödesschema för den experimentella proceduren. De odlade celler först transfekteras med jetPEI (se protokoll) för att överuttrycka den fluorescenta fusionsproteinet av intresse. Efter 24 timmar är de levande transfekterade cellerna visualiseras och FRAP och FLIP experiment utförs med hjälp av en konfokalmikroskop utrustad med en kontrollerad temperatur CO2 inkubator, och de inspelade bilderna exporteras och analyseras med hjälp av lämplig programvara (t.ex. ImageJ). För ultra analysen, är de transfekterade cellerna fast, pelleteras och inbäddade i Epon epoxiharts blocken. Ultratunna sektioner erhålls med hjälp av en diamant kniv, samlas på cOpper galler, och observerades under ett transmissionselektronmikroskop. Klicka här för att visa en större bild .

Figur 2. FRAP experiment med COS-7 celler övergående transfekterade med d EGFP-ER. A) Två Oser strukturer (röda ROI) blektes och fluorescens återhämtning registrerades över tiden. Rensa fluorescens återhämtning kan detekteras en min efter blekning, och signalen ökar ytterligare 4 min senare (skala bar 10 pm) B):. Kvantitativ analys av FRAP Försök som visar återhämtning halv-tid och den mobila fraktion av d EGFP -ER-proteinet. Click här för att visa en större bild

Figur 3. FLIP experiment med COS-7 celler övergående transfekterade med d EGFP-ER. A) Den kontinuerliga blekning av en OSER (indikeras av den röda ROI) orsakar en progressiv minskning i fluorescens i resten av ER och i andra Oser strukturer inom samma cell (som den gula ROI). Den gula pilen visar ett parti av en oblekt cell i vilken fluorescenssignalen är konstant över tiden. (Skala bar 10 mikrometer). B) Kvantitativ analys av FLIP experimentet. Klicka här för att visa en större bild
Figur 4. Efter fixering och inbäddning ades celler som uttrycker höga nivåer av d EGFP-ER i vilken Oser strukturer kunde detekteras med hjälp av fluorescens-optiska mikroskopi observeras genom ett transmissionselektronmikroskop. A) låg förstoring vy av en del av cytoplasman i en cell som innehåller en OSER bestående av staplade cisternae och vågformade sinusformade membran. Mitokondrier (M) kan ses samlade kring de Oser strukturer, medan ribosomer dekorerar bara membranen i den yttersta cisternae (pilspetsar och infälld). Den 11 nm tjockt elektron-täta utrymmet mellan membranen är kontinuerlig med cytoplasma (pil och infälld) (L = lysosomer / (automatisk) phagosomes) (skal 1,5 um, infälld 0,25 mikrometer). B) En OSER kan bildas av lamellär ER: dvs högar av tillplattad ER cisternae som kan vara kontinuerlig eller fragmented i sitt utseende i tunna sektioner. Blåsor spirande från den yttersta cisternae av stapeln kan ibland observeras (asterisk) (PM, plasmamembran) (Skala bar 150 nm).
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Protokollen och avbildning som beskrivs i detta dokument har använts för att undersöka spridning och rörlighet för transfekterade TA fluorescerande proteiner bosatta i ER av levande celler. Vi har också analyserat effekten av överuttryck av dessa proteiner på arkitekturen av denna subcellulär avdelning med hjälp av ultrastrukturella analyser.
Kombinationen av live-cell konfokala imaging och elektronmikroskopi representerar är ett mycket kraftfullt medel för att undersöka de dynamiska egenskaperna hos proteiner, och kan ge viktig information om proteiners funktion. De beskrivna metoderna är inte tidskrävande (vanligtvis tre dagars arbete), samt utveckling av många användarvänliga program för bildtagning och analys gör fotoblekning-baserade, live-cell imaging relativt enkel.
Den största begränsningen av dessa tekniker är användningen av fluorescerande fusionsproteiner på grund av attfluorescerande markör kan påverka korrekt veckning och / eller sammansättning av proteinet av intresse. Dessutom överexpression kan ändra beteendet hos transfekterade, fluorescerande märkta proteiner, och kan därför inte återspegla de verkliga egenskaperna hos endogena proteiner, men kan detta övervinnas genom användning av inducerbara och stabilt transfekterade celler i vilka expressionsnivån kan vara exakt moduleras för att erhålla nivåer jämförbara med de av endogena protein 1,7. Tendensen av ramprogrammen till oligomerise är väldokumenterat och att kraftigt förändra beteendet (dvs. kinetik, oönskade protein-proteininteraktioner och bildning av aggregat) av chimära proteiner. Användningen av optimerade mono fluorescerande proteiner bör därför övervägas 17.
En annan viktig aspekt av dynamiska imaging studier med fluorescens och fotoblekning är den tid som behövs för att bleka fluorescens effektivt och mäta fluorescens reåterhämtning (och därmed protein rörlighet) exakt, vilket även beror på den del av avkastningen på investeringen och lokala celltjocklek. Om en viss GFP-märkta proteinet har en hög diffusionskoefficient, kan diffusion ske under blekning och därmed störa mätningarna återhämtningstid. För att få en snabb och effektiv blekning, rekommenderas det starkt att en "zoom in"-funktion (om tillgängligt) och mer än en laserlinje. Även om användningen av en snabb modul scan (dvs. en resonant skanner) i hög grad kan förbättra hastigheten på avbildning under återhämtningsfasen av ett experiment, i våra händer det också minskar avsevärt blekningseffektivitet. Däremot kan alternativa skanningssystem (till exempel en snurrande skiva försedd med en särskild fotoblekning enhet), och mer kraftfulla lasrar förbättra både blekningseffektiviteten och förvärvshastighet.
De flesta fluorescerande proteiner som används i FRAP och FLIP experiment visar en viss grad av reversibel fotoblekning och BLInking som måste beaktas vid utförande av kvantitativa analyser. Svängningarna mellan fluorescerande och mörka stater förekommer i det andra för minut tidsskala. För EGFP, har det visat sig att under blekningsexperiment, kan fluorescens variationer innebär mindre än 10% av molekylerna, vilket i det nuvarande protokollet detta fenomen är försumbar. Om alla förhållanden hålls konstanta, kommer detta att införa en konstant bias i resultaten. Om andra fluorescerande proteiner används, där den reversibla fraktionen är betydligt högre (dvs. YFP), eller för att upptäcka och bedöma fotoblekning reversibilitet, kan detta göras genom att mäta fluorescens återhämtning efter fotoblekning i hela levande cellen, om återhämtning observeras detta kan endast vara resultatet av fotoblekning reversibilitet 18.
Den potentiella toxiciteten av ljus under experimenten är en annan viktig faktor, framför allt därför att fotoblekning kräver stark belysning. Det är well känt att glada fluoroforer tenderar att reagera med syre för att producera fria radikaler som kan påverka olika intracellulära processer och även cellviabilitet 19, och därför är det nödvändigt att skapa en balans mellan effektiv blekning och minimal fototoxicitet, och dessutom bör cellviabiliteten alltid kontrolleras efter live-cell imaging experiment. Med tanke på den korta inspelningstiden, har vi inte betrakta den genotoxiska effekten av kortvågigt ljus (405 nm) i exemplet som beskrivs i detta dokument, men om det krävs en längre experiment, en 405 nm laserlinjen bör inte användas.
Vi valde att inte använda en motsvarande metod för transmissionselektronmikroskop på grund av den heterogena karaktären av OSER arkitektur och det faktum att vi ville observera så många celler (och strukturer) som möjligt. Mångfalden av den fina strukturen av proteinaggregat i celler kan vara en viktig del av olika sjukdomar och vi var intresserade av att få ett brett spektrum av SAmples, medan en motsvarande tillvägagångssätt tillåter observation av färre händelser under samma period tid. Dock bör korrelat ljus-elektronmikroskopi (CLEM) vara förstahandsvalet vid utredning av händelser i strukturer som inte lätt kan identifieras (t.ex. mindre framträdande ER domäner) eller i ett begränsat antal celler (såsom mikro-injicerade celler). Det är värt att notera att våra experiment som kännetecknas av en hög grad av transfektion effektivitet (minst 30% av cellerna transfekterades), annars möjligheten att observera Oser strukturer noncorrelatively är ganska begränsad.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Författarna har ingenting att lämna ut.
Acknowledgments
Författarna är tacksamma till Fondazione Filarete för dess hjälp och stöd i publiceringen av denna artikel. Vi vill också tacka Centro Europeo di Nanomedicina för användning av Tecnai G2 transmissionselektronmikroskop.
Materials
| Name | Company | Catalog Number | Comments |
| Dulbecco’s Modified Eagle Medium (DMEM) | Invitrogen | 41966029 | |
| Dulbecco’s Modified Eagle Medium (DMEM) w/o phenol red | Invitrogen | 31053028 | |
| Fetal Bovine Serum (FBS) | Invitrogen | 10270106 | |
| Pen/Strep | Invitrogen | 15140-122 | |
| L-Glutamine 200 mM solution | Invitrogen | 25030-024 | |
| jetPEI | Polyplus Transfection | PP10110 | |
| OxyFluor | Oxyrase Inc. | OF-0005 | |
| Glutaraldehyde Grade I | Sigma Aldrich | G5882 | |
| Sodium Cacodylate Trihydrate | Sigma Aldrich | C0250 | |
| Osmium Tetroxide 4% solution | Electron Microscopy Science | 19150 | |
| Uranyl Acetate Dihydrate | Sigma Aldrich | 73943 | slightly radioactive |
| Propylene Oxide | Sigma-Aldrich | 82320 | |
| EPON embedding medium kit | Sigma-Aldrich | 45359-1EA-F | |
| Lead Citrate | Electron Microscopy Science | 17800 | |
| Bench top centrifuge | Eppendorf | 5415 D | |
| Spectral Confocal Microscope | Leica Microsystems | TCS SP5 | |
| CO2 Microscope Cage Incubation System | OkoLab | ||
| Ultramicrotome | Leica Microsystems | UC6 | |
| Diamond knife | Diatome | Ultra 45 ° | |
| Transmission Electron Microscope | FEI | Tecnai G2 | |
| GraphPad Prism Software | GraphPad Software, Inc | ||
| Steel culture cell chamber for 24 mm coverslip | Bioscience Tools | CSC-25 | |
| Electron Microscopy grids | Electron Microscopy Science | G300Cu |
References
- Fasana, E., et al. A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 24, 1419-1430 (2010).
- Borgese, N., Francolini, M., Snapp, E. Endoplasmic reticulum architecture: structures in flux. Curr. Opin. Cell Biol. 18, 358-364 (2006).
- Ronchi, P., Colombo, S., Francolini, M., Borgese, N. Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J. Cell Biol. 181, 105-118 (2008).
- Lippincott-Schwartz, J., Snapp, E., Kenworthy, A. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2, 444-456 (2001).
- Lee, M. C., Miller, E. A., Goldberg, J., Orci, L., Schekman, R. Bi-directional protein transport between the ER and. 20, 87-123 (2004).
- Snapp, E. L., et al. Formation of stacked ER cisternae by low affinity protein interactions. J. Cell Biol. 163, 257-269 (2003).
- Papiani, G., et al. Restructured endoplasmic reticulum generated by mutant amyotrophic lateral sclerosis-linked VAPB is cleared by the proteasome. J. Cell Sci. 125, 3601-3611 (2012).
- Almsherqi, Z. A., Kohlwein, S. D., Deng, Y. Cubic membranes: a legend beyond the Flatland* of cell membrane organization. J. Cell Biol. 173, 839-844 (2006).
- Federovitch, C. M., Ron, D., Hampton, R. Y. The dynamic ER: experimental approaches and current questions. Curr. Opin. Cell Biol. 17, 409-414 (2005).
- Takei, K., Mignery, G. A., Mugnaini, E., Sudhof, T. C., De Camilli, P. Inositol 1,4,5-trisphosphate receptor causes formation of ER cisternal stacks in transfected fibroblasts and in cerebellar Purkinje cells. Neuron. 12, 327-342 (1994).
- Feldman, D., Swarm, R. L., Becker, J. Ultrastructural study of rat liver and liver neoplasms after long-term treatment with phenobarbital. Cancer Res. 41, 2151-2162 (1981).
- Sprocati, T., Ronchi, P., Raimondi, A., Francolini, M., Borgese, N. Dynamic and reversible restructuring of the endoplasmic reticulum induced by PDMP in cultured cells. J. Cell Sci. 119, 3249-3260 (2006).
- Costantini, L. M., Fossati, M., Francolini, M., Snapp, E. L. Assessing the tendency of fluorescent proteins to oligomerize under physiologic conditions. Traffic. 13, 643-649 (2012).
- Pyszniak, A. M., Welder, C. A., Takei, F. Cell surface distribution of high-avidity LFA-1 detected by soluble ICAM-1-coated microspheres. J. Immunol. 152, 5241-5249 (1994).
- Taylor, J. P., Hardy, J., Fischbeck, K. H. Toxic proteins in neurodegenerative disease. Science. 296, 1991-1995 (2002).
- Winklhofer, K. F., Tatzelt, J., Haass, C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 27, 336-349 (2008).
- Borgese, N., Gazzoni, I., Barberi, M., Colombo, S., Pedrazzini, E. Targeting of a tail-anchored protein to endoplasmic reticulum and mitochondrial outer membrane by independent but competing pathways. Mol. Biol. Cell. 12, 2482-2496 (2001).
- Chudakov, D. M., Matz, M. V., Lukyanov, S., Lukyanov, K. A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 90, 1103-1163 (2010).
- Bancaud, A., Huet, S., Rabut, G., Ellenberg, J. Fluorescence perturbation techniques to study mobility and molecular dynamics of proteins in live cells FRAP, photoactivation, photoconversion, and FLIP. Cold Spring Harb. Protoc. 2010, (2010).
- Michida, T., et al. Role of endothelin 1 in hemorrhagic shock-induced gastric mucosal injury in rats. Gastroenterology. 106, 988-993 (1994).

{kind=link}