

Abstract
Es wurde zunehmend klar, daß die räumliche Verteilung und die Bewegung der Membran-Komponenten wie Lipide und Proteine sind Schlüsselfaktoren bei der Regulation vieler zellulärer Funktionen. Aufgrund der schnellen Dynamik und die winzigen Strukturen beteiligt sind, wird eine sehr hohe Raum-Zeit-Auflösung erforderlich ist, um das reale Verhalten von Molekülen zu fangen. Hier präsentieren wir die Versuchsprotokoll für die Untersuchung der Dynamik von fluoreszenzmarkierten Plasma-Membran-Proteine und Lipide in lebenden Zellen mit hoher örtlicher und zeitlicher Auflösung. Bemerkenswerterweise ist dieser Ansatz nicht jedes Molekül zu verfolgen, aber es Population Verhalten unter Verwendung aller Moleküle in einer bestimmten Region der Membran berechnet. Der Ausgangspunkt ist eine schnelle Bebilderung einer bestimmten Region auf der Membran. Danach wird eine komplette räumlich-zeitlichen Autokorrelationsfunktion korreliert erfassten Bilder mit zunehmender Zeitverzögerungen, beispielsweise alle 2, 3, n Wiederholungen berechnet. Es ist möglich zu zeigen, dass die Breiteder Peak der räumlichen Autokorrelationsfunktion nimmt bei zunehmender Zeitverzögerung in Abhängigkeit von der Partikelbewegung aufgrund von Diffusion. Daher, passend der Serie von Autokorrelationsfunktionen ermöglicht, extrahieren die tatsächliche Protein mittlere quadratische Verschiebung von Bildgebung (IMSD), hier in der Form des scheinbaren Diffusions vs mittlere Verschiebung vorgestellt. Dies ergibt eine quantitative Blick auf die durchschnittliche Dynamik einzelner Moleküle mit Nanometer-Genauigkeit. Durch die Verwendung eines GFP-markierten Variante des Transferrinrezeptors (TfR) und einer ATTO488 Bezeichnung 1-Palmitoyl-2-hydroxy-sn-glycero-3-phosphoethanolamin (PPE) ist es möglich, die Raumzeit-Regulierung von Protein-und Lipid-Diffusion beobachten um große Membranregionen der Mikro zu-Milli zweiten Zeitbereich.
Introduction
Ausgehend von der ursprünglichen "fluid mosaic"-Modell von Singer und Nicolson, das Bild der zellulären Plasmamembran kontinuierlich in den letzten Jahrzehnten um die Rolle der Schwellen Zytoskelett-und Lipid-Domänen enthalten 1,2 aktualisiert.
Die ersten Beobachtungen wurden durch Fluoreszenzerholung nach Photobleichen (FRAP) enthüllt, dass ein signifikanter Anteil von Membranproteinen unbeweglich 3-5 erhalten. Diese bahnbrechenden Studien, wenn auch sehr informativ, von der relativ schlechte Auflösung im Raum (Mikrometer) und die Zeit (in Sekunden) von FRAP-Setups gelitten. Auch als ein Ensemble Mittelung Messung, fehlt in FRAP mit Informationen über Einzelmolekülverhalten.
In diesem Zusammenhang ist die Möglichkeit, spezifisch zu markieren ein einzelnes Molekül mit sehr hellen Tags (die die Untersuchung des Diffusionsprozesses ein Molekül zu einem Zeitpunkt) ist sehr erfolgreich gewesen. Besonders, indem Sie dieZeitauflösung der Single Particle Tracking (SPT) Ansatz für die Mikrosekunden-Zeitskala, Kusumi et erhielten Zugang zu unbekannten Funktionen von Lipid-und Proteindynamik, die sich stark auf die Anerkennung der Rolle der Aktin-basierten Membranskelett in Membranphysiologie 6 beigetragen al. , 7. Diese Ergebnisse erzeugt die so genannte die "Lattenzaun und" Modell, in dem Lipid-und Proteindiffusion durch Aktin-Skelett-basierte geregelt. , Um den Zugriff auf die riesige Menge von Informationen, die von SPT viele experimentelle Fragen gestellt haben müssen jedoch angesprochen werden. Insbesondere wird das Markierungsverfahren typischerweise durch viele Schritte wie Herstellung, Reinigung und die Einführung der markierten Spezies in das System besteht. Weiterhin großen Labels, wie Quantenpunkte oder Metall-Nanopartikel, sind oft erforderlich, um die Sub-Millisekunden-Zeitskala und die Vernetzung der Zielmoleküle durch das Label nicht in vielen Fällen vermieden werden, zu erreichen. Schließlich sind viele Bahnenhaben aufzuzeichnenden statistischen Kriterien passen und damit eine niedrige Dichte des Etiketts erforderlich ist, um Tracking zu ermöglichen.
Im Vergleich zu SPT, Fluoreszenz-Korrelations-Spektroskopie (FCS), die Überwindung viele dieser Nachteile stellt einen vielversprechenden Ansatz, um Molekulardynamik zu studieren. Tatsächlich funktioniert FCS gut auch mit dim und dichte Etiketten, so dass die Dynamik des fluoreszierenden Protein-markierte Moleküle in transient transfizierten Zellen zu studieren. Außerdem ermöglicht es erreichte hohe Statistiken in einem begrenzten Zeitraum. Schließlich trotz der "hohen" Dichte von Etiketten FCS bietet einzelne Moleküle Informationen. Dank all dieser Eigenschaften stellt FCS einen sehr einfachen Ansatz und wurde ausgiebig angewendet, um Lipid und Proteindynamik sowohl in Modellmembranen und im Live-Zellen 8-10 studieren. Viele verschiedene Ansätze vorgeschlagen worden, um die Fähigkeit von FCS auf die Details der molekularen Diffusion zeigen erhöhen. Zum Beispiel war es sheigenen, die durch Ausführen FCS auf unterschiedlich großen Beobachtungsgebiete kann man ein "FCS Diffusionsgesetz" aufschlussreich versteckt Merkmale des Molekularbewegung 11,12 definieren. Neben der Verwendung in der Größe variiert wurde die Schwerpunktbereich auch dupliziert 13, bewegt im Raum entlang der Linien 14-20 bzw. konjugiert mit schnellen Kameras 21,22. Mit Hilfe dieser "Raum-Zeit-'Korrelation Ansätze wurden relevante biologische Parameter von mehreren Membrankomponenten quantitativ auf beiden Modellmembranen und der tatsächlichen biologischen diejenigen, wodurch man einen Einblick in die räumliche Organisation Membran beschrieben.
Jedoch in aller FRAP und FCS Anwendungen bisher beschriebenen die Größe der Fokusfläche eine Grenze der räumlichen Auflösung, die nicht überwunden werden können. Mehrere Super-Resolution-bildgebenden Verfahren wurden kürzlich entwickelt, um diese Grenze zu umgehen. Einige sind auf Lokalisierungsgenauigkeit basiert, wie stochastische optische Rekonstruktion Mikroskopie (STORM) <sup> 23,24, Photoaktivierung Lokalisationsmikroskopie (PALM) 25, Fluoreszenz PALM (FPALM) 26 und single-particle tracking PALM (sptPALM) 27: Die relativ große Menge an Photonen bei jeder Momentaufnahme erforderlich ist, begrenzt jedoch die Zeitauflösung Diese Verfahren, um mindestens einige Millisekunden, wodurch ihre Anwendbarkeit in vivo hemmt.
Im Gegensatz dazu haben eine vielversprechende Alternative zur Super-Resolution Imaging von räumlichen Modulation der Fluoreszenzemission mit Stimulated Emission Depletion-Methoden (STED oder reversible optische Sättigungsfluoreszenzübergänge (RESOLFT)) 28,29 geöffnet. Diese Ansätze kombinieren die Gestaltung der Beobachtungsvolumen deutlich unter der Beugungsgrenze mit der Möglichkeit, schnelle Scan-Mikroskope und Detektionssysteme zu verwenden. In Kombination mit Fluoreszenzfluktuationsanalyse, erlaubt der STED-Mikroskopie, um direkt Sonde die raumzeitliche Dynamik von nanoskaligen Lipide und proteins live Zellmembranen 30,31.
Die gleichen physikalischen Größen STED-Mikroskopie anhand kann durch eine modifizierte raumzeitlichen Bildkorrelationsspektroskopie (Stics 32,33) Verfahren, die für die Untersuchung der Dynamik von fluoreszenzmarkierten Membranproteine und / oder Lipiden in lebenden Zellen erhalten werden und von einem kommerziellen Mikroskop. Das experimentelle Protokoll hier präsentiert wird nur von wenigen Schritten zusammen. Die erste erfordert eine schnelle Abbildung des Bereichs von Interesse auf der Membran. Dann wird der resultierende Stapel von Bildern verwendet, um die durchschnittlichen räumlichen-zeitlichen Korrelationsfunktionen zu berechnen. Vs -Mittlere Verschiebung Grundstück - von der Montage der Serie von Korrelationsfunktionen, die molekulare 'Diffusionsgesetz "kann direkt von der Bildgebung in Form einer scheinbaren Diffusionskoeffizienten (D app) erhalten werden. Dieses Grundstück kritisch hängt von der Umgebung durch die Moleküle erforscht und ermöglicht direkt die Anerkennung der tatsächlichen Diffusionsartender Lipid / Protein von Interesse.
In mehr Details, wie zuvor gezeigt, 34, die räumlich-zeitliche Autokorrelationsfunktion des erworbenen Bild-Serie hängt entscheidend von der Dynamik der Moleküle sich in den gesammelten Bild-Serie (bitte beachten Sie, dass die gleiche Argumentation kann in einer Linie Erwerb angewendet werden wo nur eine Dimension im Raum wird berücksichtigt). Insbesondere definieren wir die Korrelationsfunktion als:
 (1)
(1)
wo  die gemessene Fluoreszenzintensität an der Position x, y, und zum Zeitpunkt t,
die gemessene Fluoreszenzintensität an der Position x, y, und zum Zeitpunkt t, ![]() und
und ![]() stellt den Abstand in der x-undy-Richtung jeweils
stellt den Abstand in der x-undy-Richtung jeweils ![]() repräsentiert die Zeitverzögerung, und
repräsentiert die Zeitverzögerung, und ![]() stellt den Mittelwert. Diese Funktion kann wie folgt ausgedrückt werden:
stellt den Mittelwert. Diese Funktion kann wie folgt ausgedrückt werden:
 (2)
(2)
wobei 'n' die durchschnittliche Anzahl der Moleküle in dem Überwachungsbereich, ![]() stellt die Faltungsoperation im Raum, und
stellt die Faltungsoperation im Raum, und  stellt die Autokorrelation der Instrumental Taille. Diese letztere kann als Maß dafür, wie die Photonen eines einzelnen Emitter sind verstreut im Raum aufgrund der optischen / Recording Setup interpretiert werden (die so genannte Point Spread Function, PSF, Gen-Kundgebung gut durch eine Gauß-Funktion angenähert). Schließlich
stellt die Autokorrelation der Instrumental Taille. Diese letztere kann als Maß dafür, wie die Photonen eines einzelnen Emitter sind verstreut im Raum aufgrund der optischen / Recording Setup interpretiert werden (die so genannte Point Spread Function, PSF, Gen-Kundgebung gut durch eine Gauß-Funktion angenähert). Schließlich  die Wahrscheinlichkeit, ein Teilchen in einem Abstand zu finden
die Wahrscheinlichkeit, ein Teilchen in einem Abstand zu finden ![]() und
und ![]() nach einer Zeitverzögerung
nach einer Zeitverzögerung ![]() . Wenn wir ein Diffusionsdynamik, in denen Teilchen bewegen sich zufällig in alle Richtungen und Nettoflüsse vorhanden sind nicht der Ansicht, diese Funktion ist auch gut mit einer Gauß-Funktion, bei der die Varianz kann als der mittlere quadratische Verschiebung (MSD) des beweglichen Teilchen identifiziert werden angenähert . Somit ist die Taille der Korrelationsfunktion (auch bezeichnet als
. Wenn wir ein Diffusionsdynamik, in denen Teilchen bewegen sich zufällig in alle Richtungen und Nettoflüsse vorhanden sind nicht der Ansicht, diese Funktion ist auch gut mit einer Gauß-Funktion, bei der die Varianz kann als der mittlere quadratische Verschiebung (MSD) des beweglichen Teilchen identifiziert werden angenähert . Somit ist die Taille der Korrelationsfunktion (auch bezeichnet als ![]() ), Kann als die Summe der Partikel MSDS und Instrumental Taille festgelegt und kann durch eine Gaußsche Form gemessen werdenlung der Korrelationsfunktion für jede Zeitverzögerung. Die gemessene I MSD verwendet, um eine scheinbare Diffusivität der beweglichen Moleküle zu berechnen
), Kann als die Summe der Partikel MSDS und Instrumental Taille festgelegt und kann durch eine Gaußsche Form gemessen werdenlung der Korrelationsfunktion für jede Zeitverzögerung. Die gemessene I MSD verwendet, um eine scheinbare Diffusivität der beweglichen Moleküle zu berechnen ![]() und einen mittleren Verschiebungs
und einen mittleren Verschiebungs ![]() Beispiel:
Beispiel:
 (3)
(3)
 (4)
(4)
Paar Überlegungen über die verwendeten Versuchsaufbau können die Leser in den folgenden Abschnitten zu führen. Um die Fluorophore auf der Basalmembran von lebenden Zellen werden wir eine Totalreflexion (TIR) Beleuchtung zu verwenden, mit einem handelsüblichen TIR Fluoreszenz selektiv anzuregen (TIRF)-Mikroskop (Details finden Sie in der Materialabschnitt zu finden). Darüber hinaus, um zu sammeln the Fluoreszenz wir eine hohe Vergrößerung Ziel verwenden und ein EMCCD Kamera (physikalische Größe der Pixel auf dem Chip 16 um) (100X NA 1,47, wird eine hohe numerische Apertur für TIRF-Beleuchtung erforderlich). Um eine Pixelgröße von 100 nm zu erreichen wenden wir eine zusätzliche Vergrößerungslinse von 1.6X. Wie nachstehend erörtert, würde eine Zeitauflösung von weniger als 1 msec benötigt, um richtig beschreiben die Dynamik schneller Membranlipide unter 100 nm ist. Um diese zeitliche Auflösung zu erreichen müssen wir eine Region of Interest (ROI) wählen kleiner als der ganze Chip der Kamera (512 x 512). Auf diese Weise wird die Kamera eine verringerte Anzahl von Leitungen die Erhöhung der zeitlichen Auflösung zu lesen. Jedoch wird in diesem Auslese Regime die Rahmenzeit würde durch die erforderliche Zeit, um die Ladungen von dem Kontakt mit dem Auslesechip der Kamera zu verschieben und in der Regel in der Größenordnung von Millisekunden für 512 x 512 Pixel EMCCD- begrenzt. Um diese Grenze zu schlagen, ermöglicht eine neue Technologie Verschieben der den gesamten Rahmen ROI-Linien nur statt, wit einem praktischen effektive Reduktion des freigelegten Chipgröße (genannt Gestellte Sensor-Modus in unserem EM-CCD). Für diese Konfiguration wirksam zu sein, muss der Chip außerhalb der ROI von einem Paar von Schlitzen in dem optischen Weg angeordnet abgedeckt werden. Dank dieser Einrichtung eine Zeitauflösung von bis zu 10 -4 Sekunden erreicht werden kann. Bitte beachten Sie jedoch, dass dieser Ansatz mit vielen verschiedenen Versuchsanordnungen gekoppelt werden, wie im Abschnitt "Diskussion", erklärte.
Demonstration des Verfahrens wird in lebenden Zellen bereitgestellt werden, indem sowohl ein ATTO488 Bezeichnung 1-Palmitoyl-2-hydroxy-sn-glycero-3-phosphoethanolamin (ATTO488-PPE) und ein GFP-markierten Variante des Transferrinrezeptors (GFP TFR). Im Fall von ATTO488-PPE kann dieser Ansatz erfolgreich eine nahezu konstante D app als Funktion der durchschnittlichen Verschiebung angibt, dass sie eine freie Diffusion zu erholen, wie zuvor berichtet 30,35. Dagegen TfR-GFP zeigt eine abnehmende D
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Systemkalibrierung
- Point Spread Function (PSF) Kalibrierung
- Verdünne 10 ul 30 nm Fluoreszenz Wulst Lösung (etwa 5 um) in 90 ul destilliertem Wasser und dann beschallt die Lösung für 20 min. Schneiden Sie ein Quadrat (1 cm x 1 cm) Stück Agarose-Gel (3%) und beträgt 10 ul der Lösung auf die Oberseite des Gels. Drehen Sie das Stück Gel auf der unteren Glas einer 2 cm Petrischale und drücken Sie die Tropfen auf dem Glas.
- Schalten Sie den Erwerb Setup, legen Sie die Probe in den Halter, stellen Sie die Kamera Belichtung und EMgain (100 ms und 1000 sind gute Parameter, sondern optimieren nach dem System) und warten Sie die Kamera abkühlen.
- Stellen Sie die Kamera Belichtung bis 100 ms, Kamera EMgain bis 1000, Erfassungsmodus zu übertragen, Wiederholung und 100 Auto-Save-Einstellung Rahmen.
- Mit Hilfe des Okulars und Durchlicht Fokus auf der Grenze des Gels und bewegen Sie dann das Ziel in der Mitte des Gels, stellen Sie den Fokus und start der Laserausrichtung Verfahren (LAS AF, wählen Sie "TIRF-Setup" und folgen Sie den Ausrichtungsvorgang auto).
- Finden Sie ein Sichtfeld mit isolierten einzelnen Spots, genau konzentrieren sich auf die helleren Fleck (die in der Regel stellt Perlen Aggregat) als Referenz, zu erwerben 100 Bilder und wiederholen Sie den Schritt 5-6 mal um mehrere einzelne Spots zu erwerben.
- Importieren Sie die erworbenen Serie mit einem Datenverarbeitungsprogramm und im Durchschnitt den Stapel in der Zeit (Abbildung 1A) und wählen Sie einen einzelnen isolierten Perle. Achten Sie darauf, um die Kleinsten zu wählen Partikelaggregate zu vermeiden.
- Die gewählten Intensitätsverteilung (ein Beispiel für Einzelperlen Profil ist in 1B vorgelegt) mit einer Gauß-Funktion mit dem Befehl "gaussfit" (in den ICS-Matlab-Tools in den Materialien in Matlab). Überprüfen Sie, ob die Güte der Anpassung durch Einsicht in die erhaltenen Reste (ein Beispiel für Einbau Gauß-Profil mit den entsprechenden Residuen vorgestellt in Abbildung 1B).
- Kamerakalibrierung
- Schalten Sie die Kamera, und warten Sie die Kamera abkühlen. Stellen Sie die Kamera Erwerb Einstellung, (dh für die verwendete Kamera setzen wir die Exposition gegenüber 0,5 ms, Kamera EMgain bis 1000, Erfassungsmodus zu Gestellte Modus, der ROI Größe 32 x 128, 10.000 Wiederholungen) und starten Sie die Übernahme von der Kamera Hintergrund Signal.
- Import erworben Rahmen Serie mit einem Datenverarbeitungsprogramm. Berechnen und überprüfen Sie die durchschnittliche Intensität in jedem Pixel, um zu überprüfen, dass die Kamera Hintergrund ist ungefähr Wohnung in der ausgewählten Region des Chips. In Freigestellte Modus, entfernen Sie den ersten und den letzten horizontalen Linien (3 bis 10, je nach der Größe der ROI) für jeden Rahmen, weil die Kamera Hintergrund wird in der Regel in den Grenzlinien vorgespannt ist.
- Erstellen Sie ein Histogramm der Werte (auch definiert digitale Ebene, DL) in aufgenommenen Bildern Stack (mit dem Befehl "hist" in Matlab) und plotten den Logarithmusder resultierende Frequenz (unter Verwendung semilogy Befehl in Matlab). Ein Beispiel für DL Verteilung Kamera Hintergrund ist in Figur 2 dargestellt.
HINWEIS: Wenn die Kamera gut funktioniert, wird das Grundstück eine etwa Gauß-Peak (ein Parabelprofil in Log-Skala), die die Verteilung der Werte auf Null Photon, gefolgt von einem exponentiellen Abfall (eine Linie mit negativer Steigung in Log-Skala zugeordnet zeigen ), die die Verteilung der Werte 1 Photon (Figur 2) zugeordnet sind, stellen. Insbesondere das Zentrum und die Varianz der Gaußschen Funktion zeigen die Kamera-Offset-Fehler und jeweils während der Zerfallskonstante des exponentiellen Teil stellt eine Schätzung des DL von der Kamera auf jedes einzelne Photon zugeordnet. In Matlab verwenden Sie den Abschnitt "CalibrateCamera" der Script in Hilfsmaterialien. - Wiederholen Sie den Vorgang für alle ausgewählten Kamera EMGain und Verstärkung.
2. beschriftetZellpräparation
- Um die für die Lipid-Einbau 36 erforderlich Liposomen Man löst getrennt 1 mg DOPE (1,2-Dioleoyl-sn-glycero-3-phosphoethanolamin), 1 mg DOTAP (1,2-Dioleoyl-3-trimethylammonium-propan), und 1 mg PPE-ATTO488 in 1 ml Chloroform. Mischen Sie 0,5 ml DOPE-Lösung, 0,5 ml DOTAP-Lösung und 25 ul PPE-ATTO488 Lösung und trocken unter Vakuum für 24 Stunden. 0,5 ml HEPES-Puffer, 20 mM, Vortex für 15 min mit Ultraschall 15 Minuten bei 40 ° C aufweist.
- Um die Zelle zu bereiten, waschen Sie 3-mal mit PBS eine p100 Gericht konfluenter CHO-K1 (Chinese Hamster Ovar), 1 ml Trypsin und speichern in Inkubator für 5 min. Aussetzen abgelösten Zellen die Zugabe von 9 ml DMEM / F12-Medium mit 10% FBS und Saatgut 150 ul Zell-Lösung in eine Petrischale mit 800 ul des gleichen Mediums.
- Lager im Brutschrank 24 h bei 37 ° C und 5% CO 2. Für die Lipid-Gründung, ersetzen Zellmedium mit 500ul serumfreiem Medium; nach 30 min, fügen 2 ul Liposomen-Lösung; nach 15 min mit Wasch PSB und neue DMEM / F12-Medium für die Bildgebung.
- Für die Transfektion Transfektion Zellen nach Lipofectamin-Protokoll (Hersteller Anleitung) mit TfR-GFP-Plasmid und Speicher 24 h im Inkubator vor Bildgebung.
3. Datenerfassung
- Setup Vorbereitung
- Um das Mikroskop Thermostat 24 Stunden vor dem Experiment Einschalten des Inkubators.
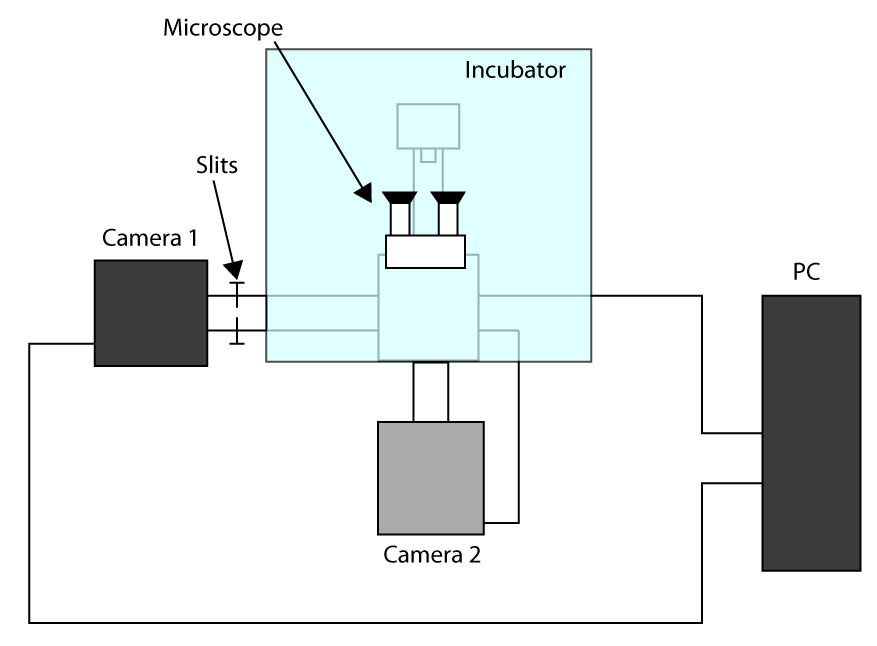
- Um die schnellsten erreichbaren Erfassungszeit gelten, die Arbeit in Freigestellte Sensor-Modus (siehe Einleitung) und verwenden Sie eine erste Kamera für die Bildgebung (Kamera 1) und zweiten Kamera, um die Zelle (Kamera 2) zu wählen. Ein Schema des Setup-Konfiguration ist in Abbildung S1 Ergänzende vorgestellt. Dann, um die zwei Kameras wiederum auf dem Mikroskop ausrichten und warten, bis die Kameras um sich abzukühlen.
- Auf beiden Kameras die Parameter für Durchlicht-Bildgebung (dh
- Setzen Sie die Proben in der Halterung und konzentrieren sich mit Okular, senden Sie das Licht auf Kamera 1 und schieben Sie die Schlitze so dass das Licht nur auf der ROI für Cell Imaging (hier ein 32 x 32 Pixel ROI) verwendet.
- Bewegen Sie eine Zelle in der ausgewählten Region und senden Sie das Licht an der Kamera 2 ist, dann ziehen Sie eine ROI in der Software, Kamera 2, um eine Referenz zu haben steuern.
- Abbildungs (3A)
- Zunächst richten Sie den Laser TIRF nach dem Verfahren von Ihrem Setup. In unserem Setup, wählen Sie die "TIRF-Setup" und starten Sie die automatische Ausrichtungsverfahren. Wenn der Laser ausgerichtet Satz 70 nm Eindringtiefe (ca. 70 °).
- Stellen Sie die Belichtungszeit auf 70 ms und 100 EMGain sowohl auf Kamera 1 und Kamera 2; Dann wählen Sie eine Zelle mit Kamera 1, dann schicken Sie das Licht auf Kamera 2 und genau fokussieren die Zellmembran. Stellen Sie die minimale Exposition auf camera 2, 1000 EMGain, Bolero Sensor-Modus, 10 5 Wiederholungen und stellen Autosave als FITS-Dateien (Flexible Image Transport System, ein Format, das leicht sein können verwaltet werden).
- Starten Sie die Aufnahme, die Bildserien aufnehmen. Lassen Sie die Verstärkung und das zugeschnittene Mode auf Temperaturstabilisierung für den Erwerb eines neuen Zelle zu ermöglichen, dann wiederholen Sie die letzten beiden Schritte, um 8-10 Zellen zu erwerben.
4. Berechnung der mittleren quadratischen Verlagerung von Imaging (i MSD)
HINWEIS: Das folgende Protokoll kann direkt an Rohdaten angewendet werden. Zur gleichen Zeit ist die ganze Protokoll für Datenübernahmen simuliert sowohl in Matlab und in SimFCS. Der Link zu den entsprechenden Anleitungen finden Sie in der Rubrik "Materialien" gefunden werden.
- Berechnung von Matlab
- Importieren Sie die erworbenen Serie in Matlab mit ImportImageSeries Skript. Berechnen der durchschnittlichen Intensität von jedem Bild mit der Zeit zusammenmmand auf den ersten 2 Dimensionen bedeuten und benutzen Grundstück, um den resultierenden Vektor zu sehen.
- Wenn mehr als 10% der Photobleaching vorhanden ist, entsorgen Sie die Serie oder entfernen Sie den ersten Teil von ihnen. Wenn sie niedriger ist, versuchen, die Auswirkungen auf die Korrelationsfunktion durch Subtraktion für jedes Bild seine mittlere Intensität zu korrigieren, wie früher 37 gezeigt.
- Berechnen der durchschnittlichen Intensität von jedem Pixel unter Verwendung Schnitts auf die dritte Dimension und sehen sich ergebende Bild.
HINWEIS: Besondere Aufmerksamkeit ist erforderlich, um Artefakt Korrelationen zu vermeiden. In der Tat, wie zuvor für ähnliche Techniken 38, Zellgrenzen sowie unscharf Vesikel konnte gezeigt werden, eine starke Korrelation vor. Wenn die Kontrolle des Durchschnitts Bild deckt Zellgrenzen oder unscharf Vesikel, versuchen, die Region beteiligt ansonsten verwerfen die Übernahme auszuschließen. Um die Wirkung dieser unbeweglichen Strukturen korrigieren subtrahieren den durchschnittlichen zeitlichen Intensität von jedem Pixel 39. - Berechnen Sie ter räumlich-zeitliche Korrelation (G (ξ, χ, τ)) mit der Funktion CalculateSTICScorrfunc. Entfernen G (ξ, χ, 0), da die Korrelation aufgrund der Schrotrauschen bei schlechten Licht Regime dominiert G (0,0,0); die Korrelation durch den Detektor dominiert G (± 1,0,0) und Partikelbewegung während der Belichtungszeit kann G (ξ, χ, τ) für τ = 0 zu verformen, indem die gemessenen Taillen (dieser Effekt verschwindet τ > 0) 34.
- Durchschnittliche G (ξ, χ, τ> 0) unter Verwendung einer logarithmischen Zeit-Bin, um das Rauschen mittels der Funktion "LogBinStack" in Hintergrundmaterial zu reduzieren und dann passen die resultierende G (ξ, χ, τ) mit der Funktion "gaussfit" von die ICS-Matlab-Tools in den Materialien, die ich MSD (in der zweiten Spalte der resultierenden Array) erholen.
- Plotten der erhaltenen Taille σ (τ) 2 (i MSD) als Funktion der Zeit. Wenn die Daten zu laut, zu versuchen, die Anzahl der acqu erhöhenired Rahmen, erhöhen die Laserleistung, durchschnittlich mehr G (ξ, χ, τ) zusammen.
- Berechnung durch SimFCS
- Öffnen Sie die erworbenen Dateien mit ImageJ mit BioFormat Importeur Plugin und sparen erworben Serie als Tiff-Sequenz.
- Öffnen SimFCS und wählen RICS-Tool und wählen Sie Datei> Importieren mehrerer Bilder (Ergänzende Abbildung S2).
- Wählen Fit, legen Sie die richtigen Aufnahmeparameter und schließen Sie das Fenster passen (Ergänzende Abbildung S3).
- Wählen Sie Anzeige> durchschnittliche Intensität> CH1 und überprüfen Sie die Anwesenheit von Photobleaching (Ergänzende Abbildung S4).
- Wenn mehr als 10% der Photobleaching vorhanden Entsorgen Sie die Serie oder wenn es möglich ist, Last wieder die Bildfolge Entfernen des ersten Teils der Serie.
- Wenn Bleichen ist es weniger als 10% wählen Sie Extras> i MSD> Parameter einstellen, überprüfen 'Use gleitenden Durchschnitt', in der ROI-Panel auf der linken Seite ein gesetztumber des Rahmens für den gleitenden Durchschnitt zu achten, dass der Korrespondent Zeit höher ist als die charakteristische Diffusionszeit (für Teilchen, das sich in 1 um 2 s -1 eine Zeit von 10 s eine gute Bewegungsdurchschnitt)
- Wählen Sie Extras> IMSD> Berechnen i MSD (Ergänzende Abbildung S5) und Passform und exportieren Sie die i MSD aus dem Notizblock (Ergänzende Abbildung S6).
5. Berechnung des Diffusionsgesetz aus dem i MSD
- Setzen Sie die ersten paar Punkte, um den Achsenabschnitt (σ 0 2) extrapolieren (5 Punkte sind in der Regel genug, aber mehr Punkte eingepasst werden kann, wenn sie ein lineares Verhalten zeigen) und vergleichen Sie diesen Wert mit der zuvor gemessenen PSF 2. Wenn sie vergleichbar sind, wird die Dynamik des isolierten Fluorophore verfolgt. Im Gegensatz dazu, wenn σ 0 2 2 >> PSF versuchen schneller zu erwerben, um zu gewährleisten, dasskeine versteckten Dynamik vorhanden sind 34.
- Berechnen Sie den scheinbaren Diffusionskoeffizienten (D app) und die durchschnittliche Verschiebung (R) unter Verwendung der Gleichungen 3 und 4 (siehe Einleitung).
- Grundstück D App als Funktion der R, eine Diffusionsgesetz vergleichbar mit dem, was mit Spot-Variation auf der Basis FCS 12 (3D) gemessen zu erhalten.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Um die Taille Instrumental kalibrieren kann das Bild eines einzelnen fluoreszierenden Nano Wulst Maßnahme im Sinne des Protokolls Schritt 1.1 beschrieben. Ein typisches Fluoreszenzbild dieser Perlen ist in Abbildung 1 dargestellt. Die Montage Intensitätsverteilung durch eine 2D-Gauß-Funktion gibt zurück gute Residuen und ermöglicht die Messung der Taillen instrumental bei 270 nm auf. Dieser Wert ist in guter Übereinstimmung mit der von der Rayleigh-Gleichung geschätzt erwarteten Beugungsgrenze. Diese Kalibrierung ist nicht erforderlich, die Messung der Partikeldynamik, aber es ist erforderlich, um die scheinbare Teilchengrße zu messen.
Eine typische Frequenzverteilung des Hintergrundkamera ist in Figur 2 dargestellt. Wegen der Kamera ansprechend auf keine Photonen Der Peak bei etwa 180 DL ist, und den Beitrag der Analog Digital (AD)-Wandler darstellt. Dieser Beitrag kann als eine Gaußsche Verteilung zu schätzen, den Offset und die Varianz angenähert werdenvon der Signalaufzeichnung eingeführt. Oberhalb 200 DL der digitale Pegelverteilung wird exponentiell (linear im logarithmischen Maßstab), und stellt die durchschnittliche Kamera als Reaktion auf ein einzelnes Photon. Einbau dieses Teiles mit einer exponentiellen Verteilung ermöglicht die Messung der durchschnittlichen DL jedem Einzelphotonen zugeordnet. Je höher das Verhältnis zwischen der durchschnittlichen DL jedem Photon zugeordnet und den AD-Wandler-Fehler, wird die niedrigere der Lärm in der berechneten Korrelationsfunktion sein. Darüber hinaus ermöglicht die durchschnittliche Einzelphotonen Reaktion die Schätzung der Kamera Dynamikbereich.
Ein Diagramm des gesamten Versuchsdurchführung ist in Figur 3 zusammengefaßt und ein Bild von Atto488-PPE Insertion in die Membran ist in 4A dargestellt. Eine repräsentative TIRF der Basalmembran einer CHO-Zellen mit Atto488-PPE bezeichnet ist in 4B dargestellt. Mehrere helle Flecken außerhalb der Zelle aufgrund li vorhanden seinposomes auf dem Glas gestapelt. Sie können durch Auswählen eines ROI auf einem Membranabschnitt überwiegend einheitlichen Fluoreszenz verworfen (dh. Der zellulären Plasmamembran). Wie erwartet die gemessene Diffusionsgesetz (4C) für dieses Lipid ist flach, was auf eine meist freie Diffusion, wie zuvor von STED-FCS-Messungen 30,35 gezeigt. Es ist erwähnenswert, dass alle aufgeführten Verlagerungswerte unterhalb der Beugungsgrenze und offenbar zeigt die Fähigkeit von diesem Ansatz zu lösen wert super mittleren Molekular Verschiebungen auch unterhalb der Beugungsgrenze und bis zu einigen zehn Nanometern.
Eine Schematisierung TfR-GFP-Dimer Insertion in die Membran ist in 5A dargestellt. Viele Studien haben gezeigt, dass die cytoplasmatischen Schwanz des Rezeptors in Wechselwirkung mit dem Membranskelett, das wiederum wirkt als Zaun für den Rezeptor Mobilität 12,40. Ein Vertreter TIRF Bild einer CHO Zelle, die TfR-GFP ist Presein 5B nted. Niedrige Fluoreszenzintensität Zellen sollten bevorzugt werden, da die Membran ist näher an der nativen Zustand und die Wahrscheinlichkeit von Artefakten auf die Überexpression minimiert verwandt. Darüber hinaus sollte der zentrale Teil der Zelle vermieden werden, da die Auswirkungen der out-of-Fokus-Fluoreszenz (von Zytoplasma, zum Beispiel) können vorhanden sein. Wie erwartet ist die gemessene Diffusionsgesetz (5C) für TfR-GFP ist ein erstes Flachverhalten unterhalb von 100 nm, mit einem mittleren D app von etwa 0,7 um 2 s -1, gefolgt von der rasche Abnahme der scheinbaren Diffusions bis 0,2 um 2 s -1 (der Wert in der Regel durch gemessen beugungsbegrenzten FCS 12). Dieses Ergebnis zeigt, dass unser Ansatz kann leicht messen die durchschnittliche Verschiebung der GFP-markierten Proteine mit einer Auflösung von wenigen zehn Nanometern. Darüber hinaus ist die räumliche Maßstab, in dem D App startet, um Sätze verringern die charakteristischeräumlichen Skala von Protein teilweise Entbindung von der Membranskelett bei rund 120 nm, im Einklang mit früheren Schätzungen 6.

Abbildung 1. Kalibrierung von Point Spread Function. (A) Pseudo Bild eines isolierten Perle und Perlen Aggregate. (B) 3D-Plot der Intensitätsprofil eines isolierten Wulst zeigt eine gut definierte Gauß-Profil. (C) Fit der Intensitätsverteilung durch eine Gauß-Funktion (oben) mit den entsprechenden Residuen (unten). Die gute Übereinstimmung zwischen der angepassten Verteilung und der gemessenen Intensitätsprofil ist auch ein Beweis dafür, dass die instrumentale PSF kann durch eine Gauß-Funktion angenähert werden. Bitte klicken Sie hier, um vi ew eine größere Version dieser Figur.

Abbildung 2. Kalibrierung der Kamera Reaktion auf einzelne Photonen. Die Abbildung zeigt die Digital-Ebene (DL)-Verteilung für Kamera-Hintergrund in einem 32 x 128 ROI, Belichtung 0,5 ms, in Gestellte Sensor-Modus. Der Peak bei etwa 180 DL stellt die Kamera ansprechend auf keine Photonen. Insbesondere stellt sie den Beitrag des Analog-Digital (AD)-Wandler und kann durch eine Gauß-Funktion approximiert abzuschätzen Der Offset und die von der Signalaufzeichnungs eingeführt Varianz werden. Oberhalb 200 DL die Verteilung der digitalen Pegel wird exponentiellen und stellt die durchschnittliche Kamera als Reaktion auf ein einzelnes Photon. Die Messung dieser Parameter ermöglicht die Schätzung der Dichte der Photonen, die bei der Erfassung aufgezeichnet werden.s / ftp_upload / 51994 / 51994fig2highres.jpg "target =" _blank "> Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.

Abbildung 3. Schematisierung der Methode. (A) Weitfeld-Bildgebung von EMCCD Kamera wird angewendet, um Sub-Millisekunden-Auflösung zu erreichen, während TIRF Mikroskopie wird ausgenutzt, um eine genaue optische Schnitte von der Plasmamembran zu schaffen. (B) Die resultierende Stapel von Bildern, um die mittlere räumliche Autokorrelation berechnen -temporal Korrelationsfunktion. Diese Korrelationsfunktion ist gut durch eine Gauß-Funktion angenähert (siehe Einleitung), und es breitet sich in der Zeit nach Partikelverschiebungen. (C) So, um zu quantifizieren, die Ausbreitung der Korrelationsfunktion durch molekularen Verschiebung, passend mit einem Gauss Ian Funktion ausgeführt wird. Dies ermöglicht die Messung der Molekül 'Diffusionsgesetz' direkt von der Bildgebung in Form der scheinbaren Diffusions vs mittlere Verschiebung Grundstück. (D) Dank dieser Handlung, kann die molekulare Diffusion Modi direkt ohne die Notwendigkeit für ein Interpretationsmodell oder Annahmen über die identifiziert werden räumliche Organisation der Membran. In der Tat wird frei diffundierenden Moleküle eine konstante scheinbare Diffusivität anzuzeigen, wie die Mobilität nicht von der räumlichen Skala der Messung abhängen. Im Gegensatz dazu wird teilweise beschränkt Moleküle eine ganz konstante scheinbare Diffusionsvermögen für Verschiebungen kleiner als Haft Größe anzuzeigen, und eine abnehmende Diffusionsvermögen für Raumskalen größer als Haft Größe. Somit kann das Auftreten einer Verminderung der scheinbaren Diffusionskoeffizienten wie ein Fingerabdruck von transienten Haft interpretiert werden, während die damit verbundenen räumlichen Maßstab kann verwendet werden, um die räumliche Ausdehnung der Begrenzungs abzuschätzen. .jove.com / files / ftp_upload / 51994 / 51994fig3highres.jpg "target =" _blank "> Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.

Abbildung 4. ATTO488-PPE Diffusionsgesetz in Live-Zellmembranen (A) Schematische Darstellung der ATTO488-PPE Einsetzen in Zellmembran (B) TIRF Bild von CHO Basalmembran mit ATTO488-PPE beschriftet.:. Eine ROI (roter Kasten) ausgewählt ist in einer meist gleichmäßigen Teil der Zelle, die Vermeidung Zellenrand und stark fluoreszierenden Flecken. (C) Das Diffusionsgesetz in der ausgewählten ROI gemessen zeigt eine flache Verhalten bestätigt eine kostenlose Diffusionsmodell für diese Komponente. Bitte klicken Sie hier, um eine größere Version zu sehen diese Zahl.

Abbildung 5. TfR-GFP Diffusionsgesetz in Live-Zellmembranen (A) Schematische Darstellung der TfR-GFP Einsetzen in Zellmembran. Das Zytoplasmaschwanz des Rezeptors interagiert mit dem Membranskelett, das als Zaun für Mobilität Rezeptor wirkt (B. ) TIRF Bild von CHO-GFP exprimierenden TfR: ein ROI ausgewählt lieber niedrig exprimierenden Zellen zu Artefakten durch Überexpression zu vermeiden (C) die Diffusionsgesetz TfR (schwarze Punkte), im Gegensatz zu PSA (graue Linie, von der Abbildung 4 entnommen). zeigt das typische Verhalten von teilweise beschränkt Diffusion, wo eine erste flache Teil durch einen Rückgang der D App gefolgt. Bitte klicken Sie hier, um eine größere Version dieses fi sehengurieren.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Einzelpartikelverfolgung (SPT) ist eine der häufigsten Strategien zur Molekulardynamik zu studieren, und es hat den großen Vorteil der Messung Partikelbahnen. Dies wiederum ermöglicht Sondieren das Verhalten noch wenige markierten Partikel in einem komplexen System. Um jedoch diesen Vorteil SPT muss in der Regel eine geringe Dichte der Sonde und sehr hell Etiketten zu erreichen. In diesem Fall ein komplexes Verfahren der Produktion, die Kennzeichnung und das Einsetzen in das System notwendig ist: insbesondere auf hohe zeitliche Auflösung (us-Bereich) sind in der Regel anorganische Sonde erforderlich (zB Quantenpunkte oder Metall-Nanopartikel) zu gewinnen. Im Vergleich zu SPT das vorliegende Verfahren zeigt einige entscheidende Vorteile. Zunächst kann dieser Ansatz in Konjugation mit fluoreszierenden Proteinen verwendet werden. So wird im Vergleich zu SPT wird eine höhere zeitliche Auflösung erzielt (auf dem gleichen Etikett) durch die geringere Menge an Photonen benötigt 34. Mehr im Detail, ermöglicht diese Eigenschaft drückt die zeitliche Resolution unter 10-3 sec auch bei der Verwendung von fluoreszierenden Proteinen codierbar, und dieses Zeitskala gibt einen exklusiven Zugang zu der nanoskaligen Dynamik von Membranbestandteile. Schließlich ist es erwähnenswert, dass die molekulare Diffusion Gesetze werden durch die Analyse der volle Raum-Zeit-Korrelationsfunktion, ohne die Notwendigkeit, jedes Molekül verfolgen beschrieben.
Der Vergleich mit der STED-FCS-basierte ist auch interessant. In einem STED-FCS-Messung die mittlere Transitzeit der Moleküle zur Verringerung Beobachtungsvolumen wird durch die zeitliche Korrelation der Fluoreszenzsignal gemessen. Dies ermöglicht den Erhalt einer lokalen Messung der Molekulardynamik auch unterhalb der Beugungsgrenze. In der vorgestellten Ansatz der Diffusionsgesetz wird als der Durchschnitt aller Teilchen, die sich in der ausgewählten ROI, mittels Standard-, beugungsbegrenzten, Beobachtungsvolumen beobachtet gemessen. Jedoch berichteten Ergebnisse zeigen, daß dieses Verfahren nicht durch die Beugung begrenzt, sondern nur durch the zeitliche Auflösung zur Verfügung. Tatsächlich kann, obwohl eine beugungsbegrenzte Erfassung wird verwendet, um Schwankungen (analog zu dem, was in anderen Superauflösungstechniken, wie beispielsweise PALM und STORM gemacht), Molekular Verschiebungen deutlich unterhalb der Beugungsgrenze zu erkennen (direkt) berechnet, wie bereits durch die Verwendung Stics zu messen molekularen Beweis fließt 32. Darüber hinaus, im Gegensatz zu STED-FCS, dieser Ansatz kann leicht zu einer Vielzahl von kommerziellen und bestehende Setups Mikroskopie, wie Raster-Scanning-Mikroskope oder Weitfeldkamera-basierte Mikroskope angewendet werden. Es ist erwähnenswert, die STED-FCS-Messungen der molekularen Diffusion Gesetze ein Fluorophor abhängige Kalibrierung von der Größe des Instrumental Taille zwingend erforderlich. Entgegengesetzt, die Messung hier vorge erfordert keine Systemkalibrierung (nur für die Schätzung der Partikelgröße erforderlich).
Die tatsächliche Auflösung bei der Messung der Partikel Verschiebungen durch das vorgestellte Verfahren dehängt ab, wie genau können wir die Korrelationsfunktion zu messen. Folglich ist es nicht von Natur aus durch die Beugung begrenzt ist, analog zu dem Fall, wo die SPT Auflösung hängt von der Genauigkeit der Partikel "Bild" gemessen. Um eine signifikante Korrelation in weniger als 1 min für das Experiment zu messen, wenige Photonen (in der Regel unterhalb von 10 Photonen) für jedes Teilchen in jedem Rahmen genug sind. In der Tat ist der Beitrag aller beobachteten Teilchen zusammen gemittelt, wenn die Korrelationsfunktion berechnet wird, auch wenn Partikel nicht isoliert. Diese Eigenschaft ist Eigenschwankungskorrelationsmethoden und ermöglicht mit dunkel und dicht Labels wie fluoreszierende Proteine in lebenden Zellen transfiziert.
Vor diesem Hintergrund wird deutlich, daß die kleinste messbare Verschiebung ist abhängig von der Diffusionsfähigkeit des Teilchens und auf die zeitliche Auflösung der Bilddarstellungsaufbau. Als ein Beispiel, betrachten Sie bitte Diffusion von Molekülen auf der Zellmembran,wo die maximal gemessenen Diffusionskoeffizienten für Proteine oder Lipide ist etwa 5 um 2 sec -1. Unter diesen Bedingungen müssen wir eine Zeitauflösung von etwa 10 -4 s, um eine durchschnittliche Verschiebung von 50 nm zu fangen. Diese Zeitauflösung kann durch schnelles Scanning Mikroskopen einzelne Linien entlang oder durch schnelle EMCCD Kamera, wo Zeitauflösung der Belichtungszeit zusammenfällt, erreicht werden, wie hier gezeigt.
Eine weitere wesentliche Voraussetzung für diese Methode, um genau zu beschreiben, Molekulardynamik ist eine korrekte räumliche Abtastung. In der Tat, um die Korrelationsfunktion passen wir brauchen eine räumliche Abtastung (Pixelgröße) niedriger als die Taille des Instrumental PSF. In den meisten kommerziellen Mikroskope (konfokale oder weites Feld) umfasst der PSF Taille von 200 nm bis 500 nm (hauptsächlich in Abhängigkeit von der numerischen Apertur des ausgewählten Ziel und von der Wellenlänge verwendet werden) und kann leicht durch ein Experiment unter Verwendung von Nano Kalibrierung gemessen werden, große fluoreszierende Kügelchen. Thus kann eine Pixelgröße von 70-150 nm (3-mal niedriger als die Instrumental Taille) genug sein. Jedoch kann die Pixelgröße an das untersuchte System unter Berücksichtigung einer einfachen Regel anzupassen: Absenken der Pixelgröße, höhere Genauigkeit bei der Beschreibung der Korrelationsfunktion. Ferner weist die minimale Größe des Bildes, das erfaßt werden mindestens 3-mal größer als die maximale Verschiebung von Interesse (plus Instrumental Taille) sein. Dies ist erforderlich, um eine gute Konvergenz der Anpassungsalgorithmus und eine statistisch signifikante Abtastung des Molekular Verschiebungen erreichen erforderlich. Als ein Beispiel des durchschnittlichen Molekular Verschiebungen kleiner als einige hundert Nanometern (beispielsweise 200 nm) ist eine Bildgröße von wenigen Mikrometern reicht studieren. Darüber hinaus ist die Gesamtzahl der Pixel (unter Konstant die Pixelgröße) Auswirkungen auf die Qualität der Korrelationsfunktion. Tatsächlich erlaubt ein größeres Bild von durchschnittlich mehr Informationen in der Korrelationsfunktion, wenn auch auf Kosten der Zeit resolution. Über die Kamera-basierten System verwendet hier zu beachten, dass die physikalische Größe der Pixel auf dem Chip befestigt ist. Folglich Verringerung der Pixelgröße vermindert das Signal in jedem Pixel (das auf dem Quadrat der Pixelgröße abhängt), nimmt das Blickfeld, und erfordert höhere Vergrößerungsleistung. Andererseits wird in einem Scanning-System, wobei der Beobachtungsbereich befestigt ist, verringert die Pixelgröße führt gewöhnlich zu einer erhöhten Pixelzahl auf Kosten der Zeitauflösung.
Einige Details über den Detektor zu diskutieren sein. Im Gegensatz zu Einzelphotonendetektoren messen EMCCD Systeme eine durchschnittliche Intensität (digitaler Ebene, DL), die nicht direkt proportional zu dem gesammelten Licht durch die Anwesenheit einer Offset. Selbst wenn diese versetzt ist im Vergleich zu dem Dynamikbereich der Kamera (einige hundert Vergleich zu 2 16 in 16 Bit-Ausgabe) und vernachlässigbar in Experimenten, bei denen viele Photonen gesammelt werden, hat es in Betracht genommen werden,erhalten eine korrekte Normalisierung der Korrelationsfunktion. Auch kann der Offset als Referenz bei schlechten Lichtbedingungen, um die Menge des gesammelten Signals zu identifizieren. Darüber hinaus, um die durchschnittliche Menge von Photonen, die während der Erfassung erfasst werden schätzen, die durchschnittliche digitalen Ebene zu jeder gesammelten Photonen zugeordnet ist, zu messen. Diese Menge kann, indem die Kamera auf eine sehr niedrige Lichtintensität gefunden werden (beispielsweise das Hintergrundlicht im Raum); In der Tat, in diesem Fall können wir davon ausgehen, dass nur einzelne Photonen erreicht die Kamera, dh die gemessene Intensität auf nur Null oder Eins Photonen stehen.
Schließlich wollen wir, wie einige alternative Erfassungssysteme (dh verschiedene Mikroskopie-Setups) können verwendet werden, um die vorgestellten Messungen durchzuführen kommentieren. Zunächst kann die 'W'factor in Gleichung 2 (das die Autokorrelation des Instrumental PSF darstellt) angepasst werden zu ter insbesondere Erfassungssystem, um die experimentelle Korrelationsfunktion passen verwendet. Wie bereits gezeigt, 34, ist eine einfache Falle der Erwerb Whit ein Laser-Scanning-Mikroskop, wenn der Scan-Geschwindigkeit ist deutlich höher als Partikeldynamik. In einem solchen Fall wird in der Tat, die Bewegung von Partikeln während der Erfassungszeit (dh Zeilenzeit) kann als vernachlässigbar angesehen werden und die Korrelationsfunktion wird auch durch eine Gauß-Funktion approximiert. Im Rahmen der Schwellen Imaging-Technologien, ist ein interessanter Ansatz, über die Möglichkeit, sehr dünne Lichtbögen (1-2 um) durch die Probe 41 produzieren basiert. Der Lichtbogen ermöglicht die selektive Beleuchtung einer einzigen Ebene (Single Plane Illumination Mikroskopie, SPIM) in der Probe und in Kombination mit einem kamerabasierten Erfassungssystem, schnelle optische Schnitte in 3D 42. Aufgrund dieser Eigenschaften, SPIM wurde erfolgreich mit FCS 43 konjugiert und könnte eine Vali darstellend Werkzeug, um die vorgelegte Analyse von 3D-Umgebungen, wie beispielsweise das Cytoplasma oder den Zellkern von lebenden Zellen zu verlängern.
Zusammenfassend von einem experimentellen Standpunkt aus erfordert dieser Ansatz nur den Zugang zu einem Mikroskop mit einer schnellen Erfassungsmodul ausgestattet. Das Protein von Interesse kann mit einem beliebigen Fluoreszenzprotein oder organischen Fluorophor markiert werden, so dass auch für Mehrfarben-Bilderzeugung. In diesem Zusammenhang stellen wir uns die Möglichkeit, grenzüber verwenden i MSD-Analyse, um Sub-Populationen von Molekülen aus und zeigen Wechselwirkungen und Co-Diffusion auf Live-Zellmembranen. Schließlich glauben wir, dass dieser Ansatz ein leistungsfähiges Werkzeug, um Proteine und / oder Lipide unterziehen dynamische Partitionierung in Nanodomänen auf der Plasmamembran zu studieren darstellen. In diesem Fall wird die hoch variabler Größe und Lebensdauer der Nanodomänen vorstellen eine zusätzliche Ebene der Komplexität in der realen Daten, die weitere methodische Umsetzungen, darunter 2-Farbbild erfordern würde, lokaleAnalyse (zB 2D-Paarkorrelations) und / oder Fluoreszenz-Anisotropie.
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| iXon Ultra 897 | Andor | DU-897U-CS0 | |
| Solis | Andor | ||
| CHO-K1 | ATCC | CCL-61 | |
| ATTO 488 labeled PPE | ATTO-TEC GmbH | AD 488-151 | |
| DOPE | Avanti Polar Lipids, Inc. | 850725 | |
| DOTAP | Avanti Polar Lipids, Inc. | 890890 | |
| 100x Penicillin-Streptomycin-Glutamine | Gibco | 10378-016 | |
| DMEM/F-12 | Gibco | 21331 | |
| FBS | Gibco | 10082147 | |
| HEPES | Gibco | 15630-106 | |
| PBS | Gibco | 10010-023 | |
| SimFCS 3.0 | Globals Software | the software can be downloaded here: http://www.lfd.uci.edu/globals/ | |
| DMI6000 with TIRF modulus | Leica | ||
| LAS AF | Leica | ||
| Lipofectamine 2000 | Lipofectamine | 11668019 | |
| Matlab | MathWork | ||
| ImageJ | NIH |
| Name | Company | Catalog Number | Comments |
| C-terminal GFP tagged Tranferrin Receptor | OriGene | RG200980 | |
| Agar | Sigma Aldrich | A5306 | |
| Chloroform | Sigma Aldrich | 528730 | |
| Latex beads, fluorescent yellow-green, 30 nm | Sigma Aldrich | L5155 | |
| SONICA Ultrasonic Cleaners | SOLTEC | ETH S3 | |
| Petri Dishes | Willco | GWSt-3522 | |
| Bio-Format importer for Matlab | http://www.openmicroscopy.org/site/support/bio-formats5/users/matlab/ | ||
| ICS-MatLab Tools | https://www.cellmigration.org/resource/imaging/software/ICSMATLAB_28-02-06.zip | ||
| Simulation by Matlab Tutorial | https://www.cellmigration.org/resource/imaging/icsmatlab/ICSTutorial.html | ||
| Simulation by SimFCS Tutorial | https://www.cellmigration.org/resource/imaging/ppt-pdf/RICS%20Simulations.ppt |
References
- Engelman, D. M. Membranes are more mosaic than fluid. Nature. 438 (7068), 578-580 (2005).
- Vereb, G., et al. yet structured: The cell membrane three decades after the Singer-Nicolson model. Proc. Natl. Acad. Sci. U. S. A. 100 (14), 8053-8058 (1073).
- Ishihara, A., Hou, Y., Jacobson, K. The Thy-1 antigen exhibits rapid lateral diffusion in the plasma membrane of rodent lymphoid cells and fibroblasts. 84 (5), 1290-1293 (1987).
- Axelrod, D., et al. Lateral motion of fluorescently labeled acetylcholine receptors in membranes of developing muscle fibers. Proc. Natl. Acad. Sci. U. S. A. 73 (12), 4594-4598 (1976).
- Jacobson, K., Derzko, Z., Wu, E. S., Hou, Y., Poste, G. Measurement of the lateral mobility of cell surface components in single, living cells by fluorescence recovery after photobleaching. J. Supramol. Struct. 5 (4), 10-1002 (1976).
- Kusumi, A., et al. Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: high-speed single-molecule tracking of membrane molecules. Annu. Rev. Biophys. Biomol. Struct. 34, 351-378 (2005).
- Kusumi, A., Ike, H., Nakada, C., Murase, K., Fujiwara, T. Single-molecule tracking of membrane molecules: plasma membrane compartmentalization and dynamic assembly of raft-philic signaling molecules. Semin. Immunol. 17 (1), 3-21 (2005).
- Schwille, P., Korlach, J., Webb, W. W. Fluorescence correlation spectroscopy with single-molecule sensitivity on cell and model membranes. Cytometry. 36, 176-182 (1999).
- Gielen, E., et al. Diffusion of sphingomyelin and myelin oligodendrocyte glycoprotein in the membrane of OLN-93 oligodendroglial cells studied by fluorescence correlation spectroscopy. C. R. Biol. 328 (12), 1057-1064 (2005).
- Weiss, M., Hashimoto, H., Nilsson, T. Anomalous protein diffusion in living cells as seen by fluorescence correlation spectroscopy. Biophys. J. 84, 4043-4052 (2003).
- Wawrezinieck, L., Rigneault, H., Marguet, D., Lenne, P. F. Fluorescence correlation spectroscopy diffusion laws to probe the submicron cell membrane organization. Biophys. J. 89 (6), 4029-4042 (2005).
- Lenne, P. F., et al. Dynamic molecular confinement in the plasma membrane by microdomains and the cytoskeleton meshwork. EMBO J. 25 (14), 3245-3256 (2006).
- Ries, J., Schwille, P. Studying slow membrane dynamics with continuous wave scanning fluorescence correlation spectroscopy. Biophys. J. 91 (5), 1915-1924 (2006).
- Ruan, Q., Cheng, M. A., Levi, M., Gratton, E., Mantulin, W. W. Spatial-temporal studies of membrane dynamics: scanning fluorescence correlation spectroscopy (SFCS). Biophys. J. 87 (2), 1260-1267 (2004).
- Berland, K. M., So, P. T., Chen, Y., Mantulin, W. W., Gratton, E. Scanning two-photon fluctuation correlation spectroscopy: particle counting measurements for detection of molecular aggregation. Biophys. J. 71, 410-420 (1996).
- Heinemann, F., Betaneli, V., Thomas, F. A., Schwille, P. Quantifying lipid diffusion by fluorescence correlation spectroscopy: a critical treatise. Langmuir. 28 (37), 13395-13404 (2012).
- Cardarelli, F., Lanzano, L., Gratton, E. Capturing directed molecular motion in the nuclear pore complex of live cells. Proc. Natl. Acad. Sci. U. S. A. 109 (25), 9863-9868 (2012).
- Sanchez, S. A., Tricerri, M. A., Gratton, E. Laurdan generalized polarization fluctuations measures membrane packing micro-heterogeneity in vivo. Proc. Natl. Acad. Sci. U. S. A. 109 (19), 7314-7319 (2012).
- Cardarelli, F., Lanzano, L., Gratton, E. Fluorescence correlation spectroscopy of intact nuclear pore complexes. Biophys. J. 101 (4), 27-29 (2012).
- Di Rienzo, C., et al. Unveiling LOX-1 receptor interplay with nanotopography: mechanotransduction and atherosclerosis onset. Sci. Rep. 3, 10-1038 (2013).
- Unruh, J. R., Gratton, E. Analysis of molecular concentration and brightness from fluorescence fluctuation data with an electron multiplied CCD camera. Biophys. J. 95 (11), 5385-5398 (2008).
- Kannan, B., et al. Electron multiplying charge-coupled device camera based fluorescence correlation spectroscopy. Anal. Chem. 78 (10), 3444-3451 (2006).
- Jones, S. A., Shim, S. H., He, J., Fast Zhuang, X. three-dimensional super-resolution imaging of live cells. Nat. Methods. 8 (6), 499-508 (2011).
- Rust, M. J., Bates, M., Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy. 3 (10), 793-795 (2006).
- Betzig, E., et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 313 (5793), 1642-1645 (2006).
- Hess, S. T., Girirajan, T. P., Mason, M. D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 91 (11), 4258-4272 (2006).
- Manley, S., et al. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods. 5 (2), 155-157 (2008).
- Hell, S. W. Far-field optical nanoscopy. Science. 316 (5828), 1153-1158 (2007).
- Klar, T. A., Hell, S. W. Subdiffraction resolution in far-field fluorescence microscopy. Opt. Lett. 24 (14), 954-956 (1999).
- Eggeling, C., et al. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature. 457 (7233), 1159-1162 (2009).
- Hedde, P. N., et al. Stimulated emission depletion-based raster image correlation spectroscopy reveals biomolecular dynamics in live cells. Nat. Commun. 4, Forthcoming.
- Hebert, B., Costantino, S., Wiseman, P. W. Spatiotemporal image correlation spectroscopy (STICS) theory, verification, and application to protein velocity mapping in living CHO cells. Biophys. J. 88 (5), 3601-3614 (2005).
- Brown, C. M., et al. Probing the integrin-actin linkage using high-resolution protein velocity mapping. J. Cell Sci. 119, 5204-5214 (2006).
- Di Rienzo, C., Gratton, E., Beltram, F., Cardarelli, F. Fast spatiotemporal correlation spectroscopy to determine protein lateral diffusion laws in live cell membranes. Proc. Natl. Acad. Sci. U. S. A. 110 (30), 12307-12312 (2013).
- Mueller, V., et al. STED nanoscopy reveals molecular details of cholesterol- and cytoskeleton-modulated lipid interactions in living cells. Biophys. J. 101 (7), 1651-1660 (2011).
- Kleusch, C., Hersch, N., Hoffmann, B., Merkel, R., Csiszar, A. Fluorescent lipids: functional parts of fusogenic liposomes and tools for cell membrane labeling and visualization. Molecules. 17 (1), 1055-1073 (2012).
- Ries, J., Chiantia, S., Schwille, P. Accurate determination of membrane dynamics with line-scan FCS. Biophys. J. 96 (5), 1999-2008 (2009).
- Kolin, D. L., Wiseman, P. W. Advances in image correlation spectroscopy: measuring number densities, aggregation states, and dynamics of fluorescently labeled macromolecules in cells. Cell Biochem. Biophys. 49 (3), 141-164 (2007).
- Digman, M. A., et al. Measuring fast dynamics in solutions and cells with a laser scanning microscope. Biophys. J. 89 (2), 1317-1327 (2005).
- Ritchie, K., et al. Detection of non-Brownian diffusion in the cell membrane in single molecule tracking. Biophys. J. 88 (3), 2266-2277 (2005).
- Voie, A. H., Burns, D. H., Spelman, F. A. Orthogonal-plane fluorescence optical sectioning: three-dimensional imaging of macroscopic biological specimens. J. Microsc. 170, 229-236 (1993).
- Huisken, J., Swoger, J., Del Bene,, Wittbrodt, F., J,, Stelzer, E. H. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science. 305 (5686), 1007-1009 (2004).
- Wohland, T., Shi, X., Sankaran, J., Stelzer, E. H. Single plane illumination fluorescence correlation spectroscopy (SPIM-FCS) probes inhomogeneous three-dimensional environments. Opt. Express. 18 (10), 10627-10641 (2010).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}