

Summary
ניתוחי RNA-רצף וביואינפורמטיקה שמשו לזהות גורמי שעתוק הביעו באופן משמעותי ואופן דיפרנציאלי בתת אוכלוסיות לין-CD34 + ולין-CD34- של EMLcells עכבר. גורמי שעתוק אלו עשויים לשחק תפקיד חשוב בקביעה לעבור בין תאי לין-CD34- לין-CD34 + ומובחן באופן חלקי עצמי חידוש.
Abstract
תאי גזע hematopoietic (HSCs) משמשים קליניים לטיפול בהשתלה כדי לבנות מחדש את מערכת hematopoietic של מטופל במחלות רבות כגון לוקמיה ולימפומה. הבהרת מנגנוני שליטה חידוש עצמי HSCs ובידול חשוב ליישום של HSCs למחקר ושימושים קליניים. עם זאת, לא ניתן לקבל כמות גדולה של HSCs בשל חוסר היכולת שלהם להתרבות במבחנה. כדי להתגבר על מכשול זה, היינו קו עצם עכבר מח שמקורן בתאים, שורת תאי EML (erythroid, מיאלואידית, ולימפוציטית), כמערכת מודל למחקר זה.
RNA-רצף (RNA-Seq) כבר בשימוש יותר ויותר כדי להחליף microarray ללימודי ביטוי גנים. אנו מדווחים כאן שיטה מפורטת של שימוש בטכנולוגית RNA-Seq כדי לחקור את גורמי מפתח הפוטנציאל ברגולציה של תא EML התחדשות עצמית והבחנה. הפרוטוקול הניתן במאמר זה מחולק לשלושה חלקים. הנקוב הראשוןלא מסביר כיצד תרבות תאי EML ונפרד לין-CD34 + ותאי לין-CD34-. החלק השני של הפרוטוקול מציע נהלים מפורטים להכנת RNA מוחלטת ובניית הספרייה העוקבת ברצף תפוקה גבוהה. החלק האחרון מתאר את השיטה לניתוח נתונים RNA-Seq ומסביר כיצד להשתמש בנתונים כדי לזהות גורמי שעתוק הביעו באופן דיפרנציאלי בין לין-CD34 + ותאי לין-CD34-. גורמי השעתוק באופן משמעותי ביותר באו לידי ביטוי באופן דיפרנציאלי זוהו להיות הרגולטורים המרכזיים פוטנציאל השליטה תא EML התחדשות עצמית והבחנה. בחלקו המסכם של מאמר זה, אנו מדגישים את השלבים העיקריים לביצוע מוצלח של ניסוי זה.
לסיכום, מאמר זה מציע שיטה של שימוש בטכנולוגית RNA-Seq לזהות רגולטורי פוטנציאל של התחדשות עצמית והבחנה בתאי EML. הגורמים המרכזיים שזוהו נתונים לאנליזה פונקציונלית במורד הזרם במבחנה ואניvivo n.
Introduction
תאי גזע hematopoietic הם תאי דם נדירים שמתגוררים בעיקר בנישה של מח העצם הבוגר. הם אחראים לייצור של תאים נדרשים כדי לחדש את הדם והמערכת החיסונית 1. כסוג של תאי גזע, HSCs מסוגל גם התחדשות עצמית והבחנה. הבהרת מנגנונים ששולטים בהחלטת גורלו של HSCs, כלפי שתי התחדשות עצמית או בידול, יציע הדרכה רבת ערך על המניפולציה של HSCs למחקר מחלת דם ושימוש קליני 2. אחת בעיות שעומדות בפני החוקרים היא שאפשר לקיים HSCs והרחיב במבחנה למידה מוגבלת מאוד; רובם המכריע של צאצאיהם נבדלים באופן חלקי בתרבות 2.
על מנת לזהות רגולטורים מרכזיים השולטים על התהליכים של התחדשות עצמית והבחנה בקנה מידת הגנום, שנהגנו קו עכבר פרימיטיווי hematopoietic תא אב EML כמערכת מודל. ההוא שורת תאים נגזרה מ3,4 מח עצם העכברי. כאשר נמאס עם גורמי גדילה שונים, תאי EML יכולים להתמיין erythroid, מיאלואידית, ותאי הלימפה במבחנה 5. חשוב לציין, את שורות תאים יכולים להיות מופצים בכמות גדולה במדיום התרבות המכיל גורם תאי גזע (SCF) ועדיין שמירת multipotentiality. ניתן להפריד תאי EML לאוכלוסיות משנה של-SCA לין העצמי חידוש + CD34 + ומובחנים באופן חלקי תאי לין-SCA-CD34- המבוססים על סמני משטח CD34 וSCA 6. בדומה לטווח קצר HSCs, SCA + CD34 + תאים מסוגלים התחדשות עצמית. כאשר טופל בSCF, לין-SCA + תאי CD34 + במהירות יכול להתחדש אוכלוסייה מעורבת של לין-SCA + תאי CD34 + ולין-SCA-CD34- וממשיך להתרבות 6. שתי האוכלוסיות דומות במורפולוגיה ויש רמות דומות של mRNA c-kit וחלבון 6. תאי לין-SCA-CD34- מסוגלים הפצת באמצעי תקשורת המכילה IL-3 במקום SCF 3. Unveilinז הרגולטורים המרכזיים בהחלטת גורל תא EML תציע הבנה טובה יותר של מנגנונים תאיים ומולקולריים במעבר התפתחותי מוקדם במהלך hematopoiesis.
על מנת לחקור את ההבדלים מולקולריים שבבסיס בין-SCA לין העצמי חידוש + CD34 + ותאי לין-SCA-CD34- מובחנים באופן חלקי, היינו RNA-Seq לזיהוי גנים הביעו באופן דיפרנציאלי. בפרט, אנו מתמקדים בגורמי שעתוק, כגורמי שעתוק הם קריטיים בקביעת גורל תא. RNA-Seq הוא גישה שפותחה לאחרונה שמנצלת את היכולות של רצף הדור הבא טכנולוגיות (NGS) לפרופיל ולכמת RNAs עיבד מהגנום 7,8. בקיצור, רנ"א הכל הוא poly-נבחרים ומקוטע כתבנית template.The RNA הראשונית ולאחר מכן הוסבה לcDNA באמצעות טרנסקריפטאז ההפוך. על מנת למפות תעתיקי רנ"א באורך מלא, באמצעות RNA שלם, שאינו מושפל לבניית ספריית cDNA הוא חשוב. לPURפוזה של רצף, רצפי מתאם ספציפיים מתווספים לשני הקצוות של cDNA. ואז, ברוב המקרים, מולקולות cDNA הם מוגברים על ידי PCR והרצף באופן תפוקה גבוהה.
לאחר רצף, התוצאה כך קורא יכולה להיות מיושרת לגנום התייחסות ומסד נתוני transcriptome. מספר קורא את המפה לגן ההתייחסות נספרו, ומידע זה יכול לשמש כדי להעריך את רמת ביטוי הגן. קורא גם יכול להיות התאסף דה נובו ללא הגנום התייחסות, המאפשר המחקר של transcriptomes באורגניזמים שאינם מודל 9. טכנולוגית RNA-seq שימשה גם כדי לזהות isoforms אחוי 10-12, תמלילי רומן 13 ושילובי גן 14. בנוסף לזיהוי של גני המקודדים חלבונים, RNA-Seq יכול גם לשמש כדי לזהות רומן ולנתח ברמת שעתוק של RNA קידוד שאינו, כגון ארוך ללא קידוד 15,16 RNA, microRNA 17, siRNA וכו '18. בגלל tהוא הדיוק של שיטה זו, זה נוצל לצורך זיהוי של וריאציות נוקלאוטיד בודדות 19,20.
לפני הופעתו של טכנולוגיית RNA-Seq, microarray היה השיטה העיקרית המשמשת לניתוח פרופיל ביטוי גנים. בדיקות תוכנן מראש מסונתזות ולאחר מכן מחוברת למשטח מוצק ליצירת שקופיות microarray 21. mRNA מופק והמיר לcDNA. במהלך תהליך השעתוק לאחור, נוקלאוטידים שכותרתו fluorescently משולבים cDNA וcDNA יכול להיות הכלאה על שקופיות microarray. עוצמת האות שנאספו מנקודה מסוימת תלויה בכמות של cDNA מחייבת הבדיקה הספציפית באותו האזור 21. לעומת טכנולוגית RNA-Seq, יש microarray מספר מגבלות. ראשית, microarray מסתמך על הידע קיים מראש של ביאור גן, תוך טכנולוגית RNA-Seq היא מסוגלת לזהות תמלילי רומן ברמה היחסית גבוהה רקע, המגבילה את השימוש בו, כאשר GEרמת ביטוי ne היא נמוכה. חוץ מזה, יש לו את הטכנולוגיה RNA-Seq הרבה יותר גבוה טווח דינמי של זיהוי (8,000 לקפל) 7, ואילו, בשל רקע ורוויה של אותות, את הדיוק של microarray הוא מוגבל לשני הגנים הביעו מאוד ונח 7,22. לבסוף, בדיקות microarray שונות ביעילות ההכלאה שלהם, ההופכת את התוצאות פחות אמינות כאשר משווים רמות ביטוי היחסית של תעתיקים שונים בתוך דגימה אחת 23. למרות שיש RNA-Seq יתרונות רבים על פני microarray, ניתוח הנתונים שלה הוא מורכב. זה אחת הסיבות שחוקרים רבים עדיין משתמשים microarray במקום RNA-Seq. כלים ביואינפורמטיקה שונים הנדרשים לעיבוד נתונים RNA-Seq וניתוח 24.
בין כמה רצפים של הדור הבא פלטפורמות (NGS), 454, Illumina, Torrent מוצק והיון הם אלה בשימוש נרחב ביותר. 454 היו פלטפורמת NGS המסחרית הראשונה. בניגוד לפלטפורמות רצף האחריםאורך כגון Illumina ומוצק, הפלטפורמה 454 מייצרת לקרוא יותר (בסיס 700 ממוצע כניסות) 25. עוד קורא טוב יותר לאפיון ראשוני של transcriptiome בשלהם גבוהים יותר להרכיב את יעילות 25. החסרון העיקרי של הפלטפורמה 454 הוא העלות הגבוהה שלה לmegabase של רצף. Illumina והפלטפורמות מוצקים לייצר קוראים עם מספרים מוגברים ואורכים קצרים. העלות לmegabase של רצף היא נמוכה בהרבה מהפלטפורמה 454. בשל המספר הגדול של קצר קורא לIllumina ופלטפורמות מוצקים, ניתוח נתונים הוא הרבה יותר אינטנסיבי המחשוב. מחירו של המכשיר וריאגנטים עבור סידור עבור פלטפורמת Ion Torrent הוא זול יותר וזמן רצף קצר יותר 25. עם זאת, שיעור השגיאה ואת העלות לmegabase של רצף נמצאים גבוהה יותר בהשוואה לIllumina ופלטפורמות מוצקה. יש פלטפורמות שונות יתרונות וחסרונות משל ודורשות שיטות שונות לניתוח נתונים. PLAיש לבחור tform המבוסס על מטרת הרצף ואת הזמינות של מימון.
במאמר זה, אנחנו לוקחים את פלטפורמת Illumina RNA-Seq כדוגמא. אנחנו השתמשנו תא EML כמערכת מודל כדי לחקור את הרגולטורים המרכזיים בתא התחדשות עצמית EML ובידול, וסיפקנו שיטות מפורטות של בניית RNA-Seq ספרייה וניתוח נתונים לחישוב רמת ביטוי וגילוי תמליל רומן. שהראינו בפרסום הקודם שלנו שמחקר RNA-seq במערכת מודל EML 2, כאשר בשילוב עם בדיקה פונקציונלית (לדוגמא מציאה shRNA) מספק גישה חזקה בהבנת המנגנון המולקולרי של המוקדמים של בידול hematopoietic השלבים, ויכולים לשמש כ מודל לניתוח של תא התחדשות עצמית והבחנה באופן כללי.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
שיטה סלולארי מופעלת קרינת 1. EML תרבית תאים והפרדה של תאי לין-CD34 + ולין-CD34- באמצעות Cell מגנטי מיון מערכת ומיון
- הכנת כליות תינוק אוגר (BHK) תרבית תאים בינונית לאיסוף גורם תאי גזע:
- תאי BHK תרבות DMEM בינוניים מכילים 10% FBS ב 25 סנטימטר 2 בקבוק (טבלה 1) על 37 מעלות צלזיוס, 5% CO 2 באינקובטור תרבית תאים.
- כאשר תאים גדלים עד 80 - מפגש 90%, לשטוף תאים פעם אחת עם 10 מיליליטר של PBS. הוסף 5 מיליליטר של 0.25% פתרון טריפסין-EDTA לmonolayer דגירה התאים ל1-5 דקות בטמפרטורת חדר (RT) עד התאים מנותקים.
- Pipet הפתרון למעלה ולמטה בעדינות כדי לשבור את הגושים של תאים. הוסף 5 מיליליטר של DMEM המלא לבקבוק כדי להפסיק את פעילות טריפסין. איסוף תאים על ידי צנטריפוגה ב XG 200 במשך 5 דקות ב RT.
- הסר את המדיום וresuspend התא גלולה ב 10 מיליליטר של מדיום תרבות תא BHK הטרי. העברה 2 מיליליטר של ההשעיה התא מהצעד 1.1.4 ל75 סנטימטר 2 בקבוק חדש ולהוסיף 48 מיליליטר של מדיום תרבות תא BHK הטרי לבקבוק.
- תרבות תאי BHK במשך יומיים ולאסוף את מדיום התרבות. מעבר המדיום שדרך מסנן 0.45 מיקרומטר. אחסן הבינוני ב-20 מעלות צלזיוס עד לשימוש נוסף.
- תרבית תאי EML:
- תרבות תאי EML (בהשעיה) במדיום בסיסי EML המכילים תרבית תאים בינוני BHK (טבלה 1) על 37 מעלות צלזיוס, 5% CO 2 באינקובטור תרבית תאים.
- לשמור על תאי EML בצפיפות נמוכה תא (.5-5 x 10 5 תאים / מיליליטר) עם צפיפות השיא של פחות מ -6 x 10 5 תאים / מיליליטר. פיצול התאים כל 2-3 ימים ביחס של 1: 5. תאי המעבר EML בעדינות וזורקים את התרבות לאחר passaging במשך 10 דורות.
- דלדול של תאים חיוביים שושלת:
- קציר תאי EML ידי צנטריפוגה ב XG 200 FOדקות וr 5 לשטוף את התאים פעם עם PBS. לאסוף את התאים על ידי צנטריפוגה ב XG 200 במשך 5 דקות.
- Resuspend התאים עם PBS ולספור את התאים עם hemocytometer. קבע את ריכוז הנוגדנים בשלב הפרדת תאים שלאחר מכן לפי מספר התאים (נא עיין בהוראות המוצעות על ידי הספק של מערכת בידוד התא).
- לבודד את שלילי השושלת (Lin-) תאים באמצעות קוקטייל של נוגדני שושלת (קוקטייל של נוגדנים חד-שבטיים ביוטין מצומדות- CD5, CD45R (B220), CD11b, אנטי-Gr-1 (Ly-6G / C), 7-4 וטר-119 ) ומערכת מגנטית הופעלה תא מיון לפי הוראות יצרן.
- הפרדת תאי לין-CD34 + ולין-CD34-:
- ספין למטה תאי Lin- מהצעד 1.3.3 ב XG 200 במשך 5 דקות. Resuspend התא גלולה עם PBS ולספור את התאים עם hemocytometer.
- לשטוף את התאים פעמיים עם חיץ FACS וגלולת התאים ב XG 200למשך 5 דקות.
- לייבל חמישה צינורות 1.5 מיליליטר microcentrifuge עם המספר 1, 2, 3, 4, 5 בהתאמה. Resuspend התאים עם 100 μl חיץ FACS לכל 10 6 תאים (10 6 תאים לכל צינור).
- הוספת 1 מיקרוגרם של נוגדן אנטי העכבר CD34 FITC לצינור 1 וצינור 2 ומערבבים בעדינות את הצינורות.
- דגירה כל הצינורות על 4 מעלות צלזיוס במשך שעה 1 בחושך.
- להוסיף 0.25 מיקרוגרם של נוגדן Anti-Sca1 PE-מצומדות ו -20 μl של APC-מצומדות נוגדני קוקטייל Lineage לצינור 1, 0.25 מיקרוגרם של נוגדן Anti-Sca1 PE-מצומדות לצינור 3, ו -20 μl של נוגדני קוקטייל Lineage APC-מצומדות ל צינור 4.
- מערבבים את כל הצינורות בעדינות דגירה התאים ב 4 מעלות צלזיוס למשך 30 דקות נוספות בחושך.
- להוסיף 300 μl של חיץ FACS לתאים ולסובב את התאים ב XG 200 במשך 5 דקות.
- לשטוף את התאים עם 500 μl של חיץ FACS לשלוש פעמים.
- Resuspend התא גלולה ב 500 μl של bu FACSffer.
- השתמש בתאים בצינורות 2, 3, 4, ו -5 להקמת פיצוי. בודד תאי לין-SCA + CD34 + ולין-SCA-CD34- בצינור 1 באמצעות FACS Aria.
2. RNA הכנה וספריית בנייה לרצף תפוקה גבוהה
- בידוד, ניתוח איכות וכימות של RNA:
- לחלץ RNA הכולל מלין-CD34 + ותאי לין-CD34- בהתאמה באמצעות TRIzol הבאה הפרוטוקול מייצר ".
- הסר את deoxyribonuclease המזוהם ה- DNA באמצעותי (אני DNase) בעקבות הפרוטוקול של הייצור. לחלופין, לאחסן RNA ב -80 מעלות צלזיוס בשלב זה לשימוש נוסף.
- להעריך את האיכות של רנ"א הכל באמצעות Bioanalyzer על פי ההוראות המוצעות על ידי הספק. השתמש במדגם RNA עם RNA יושרה מספר (רין) לאגר מ -9.
- ספריית בנייה ורצף תפוקה גבוהה:
הערה: פרוטוקול זה מתאר RNA-Seq באמצעות פלטפורמת Illumina. לפלטפורמות רצף אחרות, שיטות הכנת ספרייה אחרות נדרשות.- השתמש .1-4 מיקרוגרם של RNA הכולל באיכות גבוהה לדגימה להכנת ספרייה. בדרך כלל 2 מיקרוגרם של רנ"א הכל ניתן לחלץ מן 10 5 תאי EML.
- השתמש במערכת הכנת מדגם RNA-רצף לטיהור RNA ופיצול, סינתזת cDNA הגדיל ראשונה ושנייה, תיקון הסוף, 3 'מסתיים adenylation, קשירת מתאם והגברת PCR, בעקבות הנהלים סטנדרטיים מפורטים מההוראות של הספק.
- חיובי לבחור הפולה mRNA באמצעות אוליגו-DT חרוזים מגנטיים וירסק את ה- mRNA.
- לבצע שעתוק לאחור באמצעות פריימרים אקראיים לקבל cDNA ולאחר מכן לסנתז הגדיל השני של cDNA ליצור גדילים כפולים cDNA.
- הסר את 'הסככות ולמלא 5' 3 סככות על ידי DNA פולימרז. Adenylate 3 'מסתיים כדי למנוע שברי cDNA מligating אחד לשני.
- להוסיף מתאמי אינדקס זמנית בשני הקצוות של dscDNA. לבצע PCR להעשרה של שברי DNA.
- מדוד את A260 / A280 כדי לקבל מידע על הריכוז של ספרייה באמצעות ספקטרופוטומטר.
- להעריך את איכות הספרייה ולמדוד את גודל הטווח של שברי DNA באמצעות Bioanalyzer.
ניתוח 3. נתונים
להתייחסות של תוכנה המשמשת בחלק זה, ראה (טבלה 2).
- עיבוד קובץ נתונים לניתוח במורד הזרם:
- המרת .bcl (שיחת קובץ בסיס) להגיש ל.fastq קובץ באמצעות תוכנת CASAVA (Illumina, גרסת 1.8.2).
- לפטר את 'טרמינל' במערכת Linux. לכו לתיקיית שהנתונים המכילה את קובץ הנתונים ממכונת רצף Illumina HiSeq2000. נניח תיקיית התוצאה היא 'NASboy1 / JiaqianLabData / HiSeq_RUN / 2013_07_11 / 130627_SN860_0309_A_2013-166_H0PW9ADXX /', סוגבפקודה באיור S1A, והזן את תיקיית הנתונים.
- התקן CASAVA 1.8.2 במערכת Linux. נניח outputfolder הוא 'Unaligned', השתמש בפקודה באיור S1B להכין קובץ ההגדרות להמרה. השתמש באפשרות --fastq-אשכול-ספירת 0 כדי להבטיח רק קובץ אחד .fastq נוצר עבור כל דגימה. קובץ .fastq שנוצר הוא בפורמט .gz. לפתוח אותו לניתוח במורד הזרם (S1B איור).
- לאחר התיקייה 'Unaligned' כבר נוצרה, לכו לתיקייה 'Unaligned' (איור S1C).
- השתמש בפקודה באיור S1D כדי להתחיל בתהליך ההמרה. הפרמטר "-j 'מספק את כמות המעבד שישמש.
- לאחר שהמערכת סיימה את תהליך ההמרה, לכו לתיקיית שהתוצאה תחת תיקייה "Unaligned '(איור S1E).
- השתמש בפקודה באיור S1F </ Strong> כדי לבטל את דחיסת קובץ .fastq.gz ל.fastq קובץ תחת כל תיקיית מדגם.
- המרת .bcl (שיחת קובץ בסיס) להגיש ל.fastq קובץ באמצעות תוכנת CASAVA (Illumina, גרסת 1.8.2).
- זיהוי תמלילי רומן ולהעריך את רמת הביטוי באמצעות Suite טוקסידו 26:
- מפה לזווג סוף RNA-Seq קורא לגנום התייחסות עכבר (mm9 גרסת UCSC, המתקבל מhttp://cufflinks.cbcb.umd.edu/igenomes.html) באמצעות תוכנת Tophat (גרסה 1.3.3) 27, אשר עושה שימוש ב Bowtie לקרוא ממפה (גרסה 0.12.7) 28. Tophat מסופק עם אפשרות "-לא-הרומן-juncs" כדי לשפר את דיוק הערכה של רמת ביטוי.
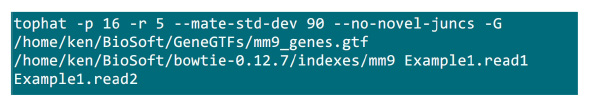
- לשים את קבצי .fastq בתיקייה שבה תהליך המיפוי יבוצע. נניח שיש 2 קבצי .fastq (לשנות את השם לExample1.read1, Example1.read2) למדגם רצף לזווג סוף, להשתמש בפקודה באיור S2 לעשות המיפוי (להתאים את הפרמטרים בהתאם להגדרת המערכת).הפרמטר "-p" מספק את כמות המעבד שישמש. ניתן להשיג את הפרמטרים "-r" ו- "-mate-STD-dev" מQC הספרייה או המשתמעות מקבוצת משנה של מיושר קורא (איור S2).
- הרכב את הממופה קורא לתעתיקי רנ"א באמצעות תוכנת חפתים (הגרסה 1.3.0) 29. חפתים להפעיל באמצעות קובץ ההסברים של גנים ידועים (אותו קובץ .gtf בשימוש על ידי Tophat) וקובץ .bam המיוצר על ידי Tophat.
- לאחר Tophat סיים ריצה, באותה התיקייה, השתמש בפקודה באיור S3A לרוץ חפתים לבנות רמת ביטוי transcriptome ותמליל הערכה. ניתן להשיג 'mm9_repeatMasker.gtf' וקבצי רצף הגנום בתיקייה 'GenomeSeqMM9' מהדפדפן הגנומי של האוניברסיטה קליפורניה.
- קבצי genes.expr וtranscripts.expr וכתוצאה מכך מכילים את ערך הביטוי של גנים ותמלילים (isoforms). העתק ודבקאת תוכן הקובץ לקובץ Excel ולטפל ביישום גיליון אלקטרוני (איור S3B).
- השתמש בפקודה באיור S3C כדי להשוות את הקובץ שנוצר "transcripts.gtf 'לקובץ' mm9_genes.gtf 'ההתייחסות על מנת לזהות תמלילי רומן.
- קובץ .tmap וכתוצאה מכך מכיל את תוצאת ההשוואה. להעתיק ולהדביק את תוכן הקובץ לקובץ Excel ולטפל ביישום גיליון אלקטרוני. תמלילים עם קוד בכיתה 'U' יכול להיחשב כ'רומן 'בהשוואה להתייחסות .gtf קובץ שסופקה (איור S3D).
הערה: לנוחיות ניתוח במורד הזרם, להגדיר את ערכי FPKM 0.1 אם הערכים נמצאים תחת 0.1.
הערה: שלב 3.2.3 - 3.2.6 היא אופציונלית למי שרוצה לשפר את רמת הדיוק של הערכת הביטוי 'תמלילי רומן. זה ייקח זמן רב יותר, משום שמיפוי ובניית transcriptome צריך להיות rהאו"ם יותר מפעם אחת.
- הפעל Tophat באמצעות פרמטרים ברירת מחדל ולאחר מכן להפעיל חפתים לקובץ .gtf שנוצר באמצעות הפקודה באיור S3E.
- השווה את קובץ .gtf וכתוצאה מכך לקובץ .gtf הגנום התייחסות באמצעות הפקודה באיור S3F.
- לנתח את קובץ .tmap הביא כמתואר בשלב 3.2.2.4. להעתיק ולהדביק את תוכן הקובץ לקובץ Excel ולטפל ביישום גיליון אלקטרוני. תמלילים עם קוד בכיתה 'U' יכול להיחשב כ'רומן 'בהשוואה להתייחסות .gtf קובץ שסופקה.
- לאחר השלב 3.2.5, יש קובץ .combined.gtf בתיקייה שבה ניתן להשתמש כקובץ .gtf ההתייחסות. ריצה שנייה של Tophat וחפת יכולה להתבצע כפי שמתוארת בשלב 3.2.1 ו3.2.2 לקבל הערכת FPKM מדויקת יותר של תמלילי רומן.
- מפה לזווג סוף RNA-Seq קורא לגנום התייחסות עכבר (mm9 גרסת UCSC, המתקבל מhttp://cufflinks.cbcb.umd.edu/igenomes.html) באמצעות תוכנת Tophat (גרסה 1.3.3) 27, אשר עושה שימוש ב Bowtie לקרוא ממפה (גרסה 0.12.7) 28. Tophat מסופק עם אפשרות "-לא-הרומן-juncs" כדי לשפר את דיוק הערכה של רמת ביטוי.
- זיהוי differentially ביטא גנים באמצעות חבילת DESeq 30.
- הקלט של DESeq טבלת ספירת קריאת גלם. כדי להשיג שולחן כזה, להשתמש בסקריפט htseq-הספירה מופץ עם חבילת HTSeq פייתון אשר ניתן להוריד מהאתר האינטרנט של HTSeq (http://www-huber.embl.de/users/anders/HTSeq/doc/count.html) .
- ודא שsamtools, פיתון, וprogramsare htseq-הספירה מותקנת במערכת. להשיג מספרי ספירת קריאת גלם מתפוקת tophat באמצעות הפקודה באיור S4A.
- הכן 'Raw_Count_Table.txt', הקבצים 'ExperimentDesign.txt' באמצעות Excel. העתק ולשמור את התוכן בתבנית txt לחבילת R DESeq (איור S4B).
- תכנית 'התקנת R במערכת. במסוף, הודעת מסך סוג 'R' וENTER.A עיתונאי appearas הראה בS4C איור.
- קראו 'Raw_Count_Table.txt ',' ExperimentDesign.txt 'לR באמצעות הפקודה באיור S4D.
- לטעון חבילת DESeq באמצעות הפקודה באיור S4E.
- תנאים לגורמים בR (איור S4F).
- השתמש בפקודה באיור S4G לרוץ מבחן binominal שלילי על שולחן הספירה המנורמל.
- השתמש בפקודה באיור S4H לגנים הביעו ההפרש משמעותי פלט בקובץ csv.
- הקלט של DESeq טבלת ספירת קריאת גלם. כדי להשיג שולחן כזה, להשתמש בסקריפט htseq-הספירה מופץ עם חבילת HTSeq פייתון אשר ניתן להוריד מהאתר האינטרנט של HTSeq (http://www-huber.embl.de/users/anders/HTSeq/doc/count.html) .
- ערכי גורמי שעתוק בדיקה '(TFS) FPKM פני דגימות באמצעות Excel. מצטלבים DE שולחן גן ושולחן TFS. גנים שייכים לשתי הטבלה הינן דיפרנציאלי הביעו גורמי שעתוק.
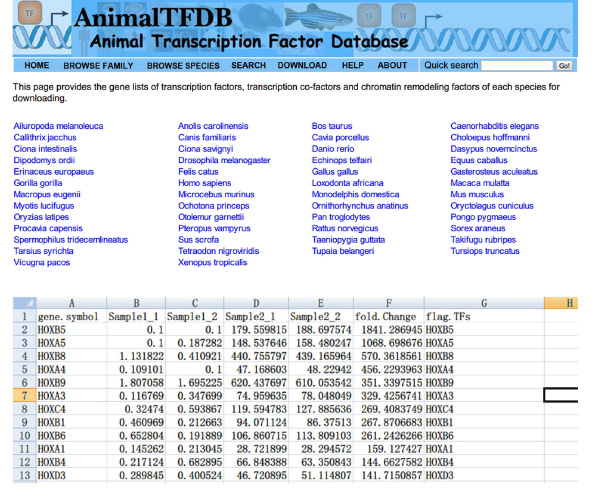
- עבור אל אתר האינטרנט http://www.bioguo.org/AnimalTFDB/download.php ולהוריד את גורמי השעתוק. ואז בדיקת מידע גורמי שעתוק DE ב- Excel (< strong> איור S5).
- יצירת קובץ .bigwig להדמיה דפדפן הגנום UCSC.
- להוריד את חבילת התוכנה של bedtools 'מאתר האינטרנט של https://github.com/arq5x/bedtools2 ולהתקין את התוכנה במערכת ביום 31 ב. להוריד את הכלים UCSC 'bedGraphToBigWig' מאתר האינטרנט של http://hgdownload.cse.ucsc.edu/admin/exe/ ולהתקין את התוכנה במערכת.
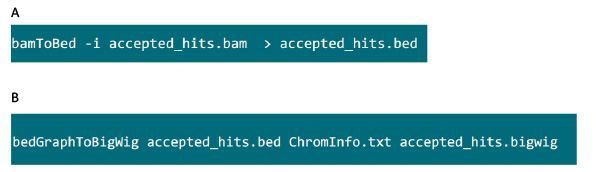
- בתיקייה המכילה את קובץ .bam, השתמש בפקודה באיור S6A להמיר קובץ .bam שנוצר על ידי tophat לתוך קובץ .bed.
- לאחר שקובץ .bed מיוצר, השתמש בפקודה באיור S6B ליצור קובץ .bigwig. ניתן להשיג את הקובץ 'ChromInfo.txt' מכתובת האתר הבא:arget = "_blank"> http://hgdownload.cse.ucsc.edu/goldenPath/mm9/database/chromInfo.txt.gz.
- שים לב מסלול מותאם אישית בדפדפן הגנומי של האוניברסיטה קליפורניה. עיין באתר האינטרנט של http://genome.ucsc.edu/goldenPath/help/customTrack.html על איך להציג מסלול מותאם אישית באמצעות דפדפן הגנום UCSC.

איור S1: המרת קובץ .bcl ל.fastq קובץ באמצעות תוכנת CASAVA.

איור S2: מיפוי קורא להתייחסות הגנום באמצעות Tophat.
איור S3: זיהוי של תמלילי רומן והערכת רמת ביטוי.

איור S4: קורא גן הביע ההפרש באמצעות DESeq חבילה.

איור S5: זיהוי של גורמי שעתוק הביעו באופן דיפרנציאלי.

איור S6: המרת תוצאת מיפוי ויזואליזציה נתונים.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
על מנת לנתח את גנים הביעו באופן דיפרנציאלי בלין-CD34 + ותאי לין-CD34- EML, אנו משתמשים בטכנולוגית RNA-Seq. איור 1 מציג את זרימת העבודה של הנהלים. לאחר הבידוד של תאים שליליים שושלת על ידי מיון תא מגנטי, נפרדנו לין-SCA + CD34 + ותאי לין-SCA-CD34- באמצעות FACS Aria. תאי EML מועשרים לין הוכתמו אנטי CD34, אנטי-Sca1 ונוגדני קוקטייל שושלת. רק תאי Lin- היו סגור לניתוח של ביטוי Sca1 וCD34. שתי אוכלוסיות (SCA + CD34 + ותאי SCA-CD34- EML) יכולות להיות שנצפו על ידי ניתוח FACS (איור 2) 6.
לאחר הפרדת תאים, אנו חילוץ RNA הכולל מCD34 + ותאי CD34- בהתאמה וניתחנו את האיכות של RNA. הדיוק של נתוני RNA-Seq מסתמך במידה רבה על האיכות של ספריית RNA-Seq והאיכות של רנ"א הכל היא חיונית להכנת ספרייה באיכות גבוהה. מדגם RNA באיכות גבוהה צריך להיות ערך 260/280 OD בין 1.8 ו 2.0. בנוסף לשימוש בספקטרופוטומטר, איכות RNA הוערכה נוסף עם דיוק רב יותר על ידי Bioanalyzer. תרשים 3 מראה תוצאה של מדגם RNA באיכות גבוהה עם רין שווה 9.4. רק מדגם RNA הכולל באיכות גבוהה עם ערך רין יותר מ -9 שימש להפקת mRNA ונהלי בניית ספרייה שלאחר מכן.
ריבוזומלי RNA הוא הסוג הנפוץ ביותר של RNA בתא. נכון לעכשיו שתי אסטרטגיות עיקריות, דלדול rRNA או חיובי בחירה של mRNA polyadenylated (פולי-mRNA), המשמשות להעשרה של RNA היעד לפני בניית ספרייה. מיני RNA polyadenylated ללא הולכים לאיבוד במהלך הבחירה של פולי-mRNA. בניגוד לכך, שיטות דלדול rRNA כגון RiboMinus יכולות לשמר מיני RNA polyadenylated שאינם. מטרת המחקר שלנו היא לחפש באופן דיפרנציאלי הביע גנים מקודדים בשני סוגי תאים, ובכך השתמשנו ב- פולי שיטת בחירת mRNA להעשרה של RNA היעד לפני constru הספרייהction. כאשר בניית ספרייה הייתה מוגמרת, בגודל של שברי DNA בספרייה נבדק לפני קביעת הרצף באמצעות Bioanalyzer. איור 4 מראה ספרייה באיכות טובה עם פסגות גודל שבר בכ -300 נ"ב.
בשלב שלאחר מכן, הספרייה הייתה נתון לרצף תפוקה גבוהה. באופן עקרוני, אורך לקרוא עוד יהיה מועיל למיפוי קריאה. זה יכול להפחית את ההסתברות שהקריאה ממופית ליעדים רבים בשל דמיון בין גנים כפולים או בני משפחת גן. כרצפי רצף זוג-הסוף הם משני קצותיו של הברים, אורך הקריאה הנבחר צריך להיות פחות ממחצית אורך ברים הממוצע. אם המטרה העיקרית של הניסוי היא למדוד את רמת הביטוי במקום בניית מבנה תמליל, חד-הסוף לקרוא (75 או 100 נ"ב) יכול להפחית את העלות מבלי לאבד יותר מדי מידע. רצף מותאם-הסוף הוא שימושי יותר לבניית מבנה תמליל וקצרה יותרלקרוא אורך יכול לשמש כדי להפחית את העלות. אין ספק, כאשר מימון מספיק נגיש, אורך לקרוא יותר עדיף.
לניתוח ביטוי ההפרש, יש הרבה אלגוריתמים חלופיים אחרים מאשר DESeq. יש גם אחד שנכלל בחבילת חפתים בשם cuffdiff 32. DESeq הוא אחד מאלגוריתמי הניתוח הגנטי DE נרחב ביותר ספירה משמשת בסיס. שיטת DESeq מבוססת על מודל סטטיסטי מאופיין היטב - התפלגות הבינומית שלילית. מניסיוננו, DESeq הוא יציב יותר בהשוואה לcuffdiff. גרסאות מוקדמות של cuffdiff לעתים קרובות לתת מספרים שונים באופן משמעותי מהגנים DE. לכן השתמשנו DESeq לניתוח DE כאן.
מכיוון שגורמי שעתוק הם חיוניים להגדרה גורל תא, התמקדנו בשעתוק באופן משמעותי הביע באופן דיפרנציאלי גורמי 33. TFS השתנה> 1.5 פי בין לין-CD34 + ולין-CD34- נמצאו ויוצג על המפה החמה (figurדואר 5) 2. יש לציין, את רמת הביטוי היחסית של Tcf7 בלין-CD34 + תאים היא יותר מזה בתאי לין-CD34- גבוהה יותר מ -100 לקפל. כך Tcf7 נבחר לשבב רצף נוסף (הכרומטין immunoprecipitation ורצף) ניתוח ובדיקה פונקציונלית כדי לאשר פונקציה של Tcf7 ברגולציה של תא התחדשות עצמית EML ובידול 2.

איור 1:. תאי זרימת עבודה של הנהלים לין-CD34 + ולין-CD34- הופרדו על ידי מערכת הפרדת תאים מגנטית ושיטת מיון תא הקרינה המופעל. RNA סה"כ הופק אחריו טיהור mRNA ובניית ספרייה. לאחר ניתוח איכות ספרייה, דגימות היו נתונים לקביעת רצף תפוקה גבוהה. הנתונים נותחו ואופן דיפרנציאלי הביעו גורמי שעתוק זוהו.

איור 2: הפרדה של לין-CD34 + ולין-CD34- EML תאים 6 תאי Lin- EML היו מועשרים על ידי מיון תא מגנטי.. תאי Lin- הוכתמו אנטי CD34, אנטי-Sca1 ונוגדני תערובת שושלת. תאי Lin- היו סגור לביטוי של CD34 וSca1. לין-CD34 + SCA + ואוכלוסיות תאי לין-CD34-SCA- EML מוינו.

איור 3:. נציג של מדגם RNA הכולל באיכות גבוהה באיכות של רנ"א הכל הוערכה על ידי Bioanalyzer. RNA היושרה מספר הוא 9.4 (FU, הקרינה יחידות).

איור 4:. טווח גודל שברים של ספרייה מותאם-End התפלגות גודל DNA של הספרייה נותחה באמצעות Bioanalyzer. רוב שברים נמצאים בטווח הגודל של 250-500 נ"ב.

איור 5:. גורמים הביעו באופן דיפרנציאלי שעתוק (> 1.5 פי) בין תאי לין-CD34 + ותאי לין-CD34- 2 לכל סוג תא, שני ניסויים בלתי תלויים שבוצעו. גנים מוסדרים עד מצוינים כצבע אדום והגנים למטה מוסדרים הם ציינו צבע ירוק.
מדיומים חוצצים ותרבות נייד: טבלה 1.
| תוכנה | שימוש | התייחסות | |||
| עניבת הפרפר 1.2.7 | בשימוש על ידי Tophat למיפוי | [28] | |||
| 1.3.3 Tophat | מיפוי קורא חזרה לגנום התייחסות | [27] | |||
| חפתים 1.3.0 | בניית תמלילים והערכת רמת הביטוי | [29] | |||
| DESeq 1.16.0 | ניתוח ביטוי ההפרש | [30] | Bedtools 2.18 | להמיר קובץ .bam לתוך קובץ .bed | [31] |
| bedGraphToBigWig | להמיר קובץ .bed ל.bigwig קובץ | http://genome.ucsc.edu/ |
טבלה 2: רשימה של תוכנות לניתוח נתונים.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
transcriptome יונקים היא מאוד מורכבת 34-38. טכנולוגית RNA-Seq משחקת תפקיד חשוב יותר ויותר במחקרים של ניתוח transcriptome, גילוי תמלילי רומן וגילוי הווריאציה נוקלאוטיד הבודד וכו 'יש לו יתרונות רבים על פני שיטות אחרות לניתוח ביטוי גנים. כפי שהוזכר במבוא, שהוא מתגבר על חפצי הכלאה של microarray ויכול לשמש כדי לזהות את תמלילי רומן דה נובו. מגבלה אחת של RNA-רצף היא אורך קריאה קצרה יחסית בהשוואה לסנגר רצף. עם זאת, עם השיפור המהיר של טכנולוגיית ריצוף, לקרוא אורך הולך וגדל. במאמר זה, אנו מספקים שיטות מפורטות של שימוש בטכנולוגיה זו כדי לזהות רגולטורים מרכזיים פוטנציאליים בעכבר תא EML התחדשות עצמית והבחנה.
צעד מפתח הראשון לפרוטוקול זה הוא תרבית תאי EML. למרות EML הוא שורת תאי מבשר hematopoietic וזה יכול להיותמופץ בכמות גדולה עם SCF. מצב culturing של תאי EML דורש יותר תשומת לב מאשר שורות תאים הונצחו הרגילות. צריכים להיות מוזנים בתאים וpassaged באופן קבוע עם פעולה עדינה; אחרת התאים יכולים להשתנות בתכונותיהם של התחדשות עצמית והבחנה ועוברים מוות תאי. כצעד הראשון לאחר האיסוף מספיק תאים, אנו מבודדים תאים שליליים שושלת באמצעות מערכת תא מופעל מגנטית מיון. ואז נפרדנו + CD34 ותאי CD34- באמצעות מיון תא הקרינה המופעל. תאי EML בדרך כלל passaged פחות מ -10 דורות לפני השימוש להפקת RNA והמספרים של CD34 + ותאי CD34- צריך להיות דומה לאחר הפרדה. אם שתי האוכלוסיות להשתנות במידה רבה במספר תאים, מומלץ לבטל את התרבות מחדש להפשיר צינור נוסף של מניות תא לתרבות.
לאחר הפרדה + CD34 ותא CD34-, הכולל מיצוי RNA בוצע, צעד חשוב נוסף עבור st זהudy. RNA באיכות גבוהה הוא הבסיס לבנייה של ספרייה באיכות גבוהה, אשר מבטיחה את הדיוק של נתוני הרצף. בשלב קריטי זה, יש להימנע מכל מגע עם RNase. כל חומרים כימיים צריכים להיות RNase חינם. חשוב ללבוש כפפות בכל העת בזמן טיפול RNA. יש מדגם RNA באיכות גבוהה ערך OD 260/280 בין 1.8 ו -2.0. בעת האיסוף את השלב המימי המכיל RNA, להיזהר שלא לבצע כל שלב אורגני עם מדגם RNA. כל ממסים אורגניים שיורית כגון פנול או כלורופורם בRNA יביאו OD260 / 280 ערך נמוך מ1.65. אם OD260 הערך / 280 הוא נמוך יותר מאשר 1.65, לזרז RNA שוב עם אתנול. לאחר שטיפה עם אתנול 75%, לא גלולה RNA overdry. גלולה RNA ייבוש לחלוטין תשפיע על המסיסות של RNA ולהוביל לתשואה נמוכה של RNA.
צעד מפתח הבא לפרוטוקול זה הוא הכנת ספרייה. לאחר מיצוי RNA כולל, צעד של שימוש DNase להסרת DNA המזוהם אנישל מומלץ מאוד, שכן זיהום הדנ"א עלול לגרום להערכה שגויה של כמות הרנ"א מוחלט בשימוש. מומלץ לבצע את ההליך במורד הזרם מייד לאחר בידוד RNA, מאז אחסון לאחר לטווח ארוך ולהקפיא הפשרת הליך, RNA יהיה לבזות במידה מסוימת. אם לא ניתן לבצע את הפעולות הבאות לאחר בידוד RNA מייד, לאחסן RNA ב-80 מעלות צלזיוס. לפני רנ"א הכל משמש לטיהור mRNA וסינתזת cDNA, תמיד צריכה להיות בדקה את האיכות. רק RNA באיכות גבוהה יכול לשמש להכנת ספרייה. שימוש באיכות נמוכה או RNA המושפל עלולה להוביל ל'ייצוג יתר של 3 מסתיים. לפני הרצף, איכות ספרייה הוערכה על מנת להבטיח יעילות רצף מקסימלי.
בחלק ניתוח נתונים, לאחר ביצוע ריצה של חפתים ללא transcriptome התייחסות, אנחנו בשילוב תמלילי רומן עם תמלילים ידועים כדי ליצור התייחסות .gtf Tophat קובץ ולהפעיל וחפת, בפעם השנייה.הליך דו-טווח זה מומלץ, שכן זה מספק הערכת FPKM מדויקת יותר מאשר לרוץ רק פעם אחת. לאחר ניתוח הנתונים, גנים הביעו באופן דיפרנציאלי זוהו. ניתן לבצע ניסויים במורד הזרם כדי לאמת את הפונקציה של גנים במבחנת in vivo. בפרסום הקודם שלנו 2, בחרנו גורמי השעתוק באופן משמעותי הביעו באופן דיפרנציאלי וזיהינו את אתר קישור הגנום של גורמים אלה על ידי ביצוע immunoprecipitation הכרומטין וריצוף (שבב Seq). בנוסף, אנחנו מוחלים assay מציאה shRNA כדי לבחון את ההשפעה התפקודית של Tcf7. מצאנו כי בתאי Tcf7 מציאה, ויסות הגנים עד-היו גנים מועשרים בתאי CD34-, ואילו גנים למטה מוסדר נמצאו להיות מועשר באופן משמעותי בCD34 + תאים. לכן, פרופיל ביטוי גנים של תאי מציאה Tcf7 נע לעבר CD34- state.Overall מובחן באופן חלקי, באמצעות תא EML כמערכת מודלבשילוב עם טכנולוגית RNA-רצף ומבחנים תפקודיים, זיהינו ואישרתי Tcf7 כרגולטור חשוב של תא EML התחדשות עצמית והבחנה.
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| Antibiotic-Antimycotic | Invitrogen | 15240-062 | BHK cell culture |
| Anti-Mouse CD34 FITC | eBioscience | 11-0341-81 | FACS sorting |
| Anti-Mouse Ly-6A/E (Sca-1) PE | eBioscience | 12-5981-81 | FACS sorting |
| APC Mouse Lineage Antibody Cocktail | BD Biosciences | 558074 | FACS sorting |
| BD FACSAria Cell Sorter | BD Biosciences | Special offer sysmtem | FACS sorting |
| Corning™ Cell Culture Treated Flasks 75 cm2 | Corning incorporated | 430641 | Cell culture |
| Corning™ Cell Culture Treated Flasks 25 cm2 | Corning incorporated | 430639 | Cell culture |
| Deoxyribonuclease I, Amplification Grade | Invitrogen | 18068-015 | Library preparation |
| DMEM | Invitrogen | 11965-092 | BHK cell culture |
| DPBS | Gibco | 14190 | Cell culture |
| HI FBS | Invitrogen | 16140071 | BHK cell culture |
| Horse Serum | Invitrogen | 16050-122 | EML cell culture |
| IMDM | HyClone | SH30228.02 | EML cell culture |
| L-Glutamine | Invitrogen | 25030-081 | Cell culture |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | 130-090-858 | Isolation of lineage negative cells |
| NanoVue Plus spectrophotometer | GE Healthcare | 28-9569-62 | Quality control |
| Thermo Scientific™ Napco™ 8000 Water-Jacketed CO2 Incubators | Thermo Scientific | 15-497-002 | Cell culture |
| Penicillin-Streptomycin | Invitrogen | 15140-122 | EML cell culture |
| TRIzol® Reagent | Invitrogen | 15596-018 | RNA exraction |
| TruSeq™ RNA Sample Prep Kit v2 -Set B (48 rxn) | Illumina | RS-122-2002 | Library preparation |
| 2100 Electrophoresis Bioanalyzer Instrument | Agilent | G2939AA | Quality control |
| 0.25% Trypsin-EDTA | Gibco | 25200 | Cell culture |
| 0.45 µm Syringe Filters | Nalgene | 190-2545 | Cell culture |
References
- Chambers, S. M., Goodell, M. A. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 3, 201-211 (2007).
- Wu, J. Q., et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS genetics. 8, (2012).
- Ye, Z. J., et al. Complex interactions in EML cell stimulation by stem cell factor and IL-3. Proceedings of the National Academy of Sciences of the United States of America. 108, 4882-4887 (2011).
- Tsai, S., Bartelmez, S., Sitnicka, E., Collins, S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 8, 2831-2841 (1994).
- Weiler, S. R., et al. D3: a gene induced during myeloid cell differentiation of Linlo c-Kit+ Sca-1(+) progenitor cells. Blood. 93, 527-536 (1999).
- Ye, Z. J., Kluger, Y., Lian, Z., Weissman, S. M. Two types of precursor cells in a multipotential hematopoietic cell line. Proc Natl Acad Sci U S A. 102, 18461-18466 (2005).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10, 57-63 (2009).
- Chu, Y., Corey, D. R. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic acid therapeutics. 22, 271-274 (2012).
- Hornett, E. A., Wheat, C. W. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC genomics. 13, 361 (2012).
- Eswaran, J., et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific reports. 3, 1689 (2013).
- Wang, E. T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470-476 (2008).
- Wu, J. Q., et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proceedings of the National Academy of Sciences of the United States of America. 107, 5254-5259 (2010).
- Loraine, A. E., McCormick, S., Estrada, A., Patel, K., Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant physiology. 162, 1092-1109 (2013).
- Edgren, H., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome biology. 12, 6 (2011).
- Ilott, N. E., Ponting, C. P. Predicting long non-coding RNAs using RNA sequencing. Methods. 63, 50-59 (2013).
- Sun, L., et al. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC bioinformatics. 13, 331 (2012).
- Luo, S. MicroRNA expression analysis using the Illumina microRNA-Seq Platform. Methods in molecular biology. 822, 183-188 (2012).
- Bolduc, F., Hoareau, C., St-Pierre, P., Perreault, J. P. In-depth sequencing of the siRNAs associated with peach latent mosaic viroid infection. BMC molecular biology. 11, 16 (2010).
- Chepelev, I., Wei, G., Tang, Q., Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic acids research. 37, 106 (2009).
- Djari, A., et al. Gene-based single nucleotide polymorphism discovery in bovine muscle using next-generation transcriptomic sequencing. BMC genomics. 14, 307 (2013).
- Murphy, D. Gene expression studies using microarrays: principles, problems, and prospects. Advances in physiology education. 26, 256-270 (2002).
- Chen, K., et al. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS one. 8, 72567 (2013).
- Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 18, 1509-1517 (2008).
- Ramskold, D., Kavak, E., Sandberg, R. How to analyze gene expression using RNA-sequencing data. Methods in molecular biology. 802, 259-274 (2012).
- Glenn, T. C. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 11, 759-769 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 7, 562-578 (2012).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25, 1105-1111 (2009).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 10, 25 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature. 28, 511-515 (2010).
- Anders, S., Huber, W. Differential expression analysis for sequence count data. Genome biology. 11, 106 (2010).
- Quinlan, A. R., Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841-842 (2010).
- Cheranova, D., et al. RNA-seq analysis of transcriptomes in thrombin-treated and control human pulmonary microvascular endothelial cells. J Vis Exp. , (2013).
- Zhang, H. M., et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research. 40, 144-149 (2012).
- Wu, J. Q., et al. Systematic analysis of transcribed loci in ENCODE regions using RACE sequencing reveals extensive transcription in the human genome. Genome Biol. 9, 3 (2008).
- Wu, J. Q., et al. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques. 36, 690-696 (2004).
- Wu, J. Q., Shteynberg, D., Arumugam, M., Gibbs, R. A., Brent, M. R. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 14, 665-671 (2004).
- Dewey, C., et al. Accurate identification of novel human genes through simultaneous gene prediction in human, mouse, and rat. Genome Res. 14, 661-664 (2004).
- Wu, J. Characterize Mammalian Transcriptome Complexity. , LAP Lambert Academic Publishing. (2011).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}