

Summary
RNA测序和生物信息学分析被用来确定显著和差异表达的转录因子在小鼠EMLcells林-CD34 +和林-CD34-细胞亚群。这些转录因子可能在确定自我更新林的CD34 +和偏微分林CD34-细胞间的开关发挥重要作用。
Abstract
是造血干细胞(HSCs)用于临床移植治疗以重建病人的造血系统的许多疾病,如白血病和淋巴瘤。阐明控制造血干细胞的自我更新和分化的机制是用于研究和临床用途应用造血干细胞的重要。然而,这是不可能得到大量的造血干细胞,因为它们不能在体外增殖。为了克服这个障碍,我们使用了一个鼠骨髓衍生的细胞系,在EML(红系,髓系和淋巴细胞)细胞系,如用于这项研究的模型系统。
RNA的测序(RNA片段)已被越来越多地用于代替微阵列的基因表达研究。我们在这里报告使用RNA测序技术来调查潜在的关键因素在EML细胞的自我更新和分化的调节的具体方法。在本文提供的方法是分为三个部分。第一杆吨解释了如何培养EML细胞和独立的林CD34 +和林-CD34-细胞。该协议的第二部分提供了详细的程序总RNA制备和随后的图书馆建设高通量测序。最后一部分介绍的方法进行RNA测序数据的分析和解释了如何使用这些数据来确定林-CD34 +和林-CD34-细胞间差异表达的转录因子。最显著差异表达的转录因子被鉴定为潜在的关键调节控制EML的细胞的自我更新和分化的影响。在本文的讨论部分,我们强调这个实验的成功业绩的关键步骤。
综上所述,本文提供了使用RNA测序技术,以确定自我更新和分化中的EML细胞的潜在调节剂的方法。所确定的关键因素进行体外和我的下游功能分析Ñ 体内。
Introduction
造血干细胞是驻留主要在成人骨髓龛罕见的血细胞。他们负责生产补充血液所需的细胞和免疫系统1。作为一种干细胞,造血干细胞能自我更新和分化。阐明了控制造血干细胞的命运决定的机制,无论是对自我更新或分化,将造血干细胞上的操作对血液病的研究和临床应用2提供了宝贵的指导意见。面对的研究者的一个问题是造血干细胞可以维持并在体外扩增到非常有限的程度;他们的后代的绝大多数部分分化培养2。
为了确定控制自我更新和分化的过程中,在全基因组规模的关键调节剂,我们使用了一个鼠原始造血祖细胞系的EML作为模型系统。日是细胞系来自于小鼠骨髓3,4。当具有不同的生长因子供给,EML的细胞能够分化成红细胞,粒细胞,和淋巴细胞在体外 5。重要的是,该细胞系可以传播中大量含有培养基的干细胞因子(SCF)和仍保持其multipotentiality。 EML的细胞可以被分离成自我更新林的SCA + CD34 +亚群和偏微分根据表面标记物CD34和SCA的6林-SCA-CD34-细胞。类似的短期造血干细胞,SCA + CD34 +细胞能够自我更新。当与SCF,林SCA + CD34 +细胞治疗可迅速再生林SCA + CD34 +和林-SCA-CD34-细胞的混合群,并继续增殖6。两个种群在形态相似,并且具有的c-kit mRNA和蛋白6相似的水平。林SCA-CD34-细胞能够在传播媒体的含IL-3,而不是SCF 3。 Unveilin克在EML中的细胞命运决定,将造血过程中提供更好的理解的细胞和分子机制在早期发育过渡的关键调节剂。
为了研究的自我更新林的SCA + CD34 +和偏微分林SCA-CD34 - 细胞间的潜在的分子差异,我们使用RNA测序,以确定差异表达的基因。特别是,我们专注于转录因子,转录因子是决定细胞命运的关键。 RNA测序是最近发展起来的方法,利用新一代测序的能力(NGS)技术来分析和量化的RNA基因组,从7,8转录。简言之,总RNA是多聚A选择并割裂作为初始template.The RNA模板,然后转换成cDNA使用逆转录酶。为了映射全长的RNA转录物,用完整的,未降解的RNA构建cDNA文库,是非常重要的。对于PUR姿势测序,具体接头序列被添加到cDNA的两端。然后,在大多数情况下,cDNA分子通过PCR扩增和测序的高通量方式进行。
测序后,由此产生的读取可以对齐到参考基因组和转录组数据库。的读取数映射到参考基因进行计数,并且这个信息可以被用来估计的基因表达水平。的读取,也可以组装从头无参考基因组,可实现转录的非生物模型9的研究。 RNA的以次技术也已用于检测剪接异构体10-12,新转录13和基因融合14。除了 对编码蛋 白质的基因的检测,RNA测序,也可用于检测新的和分析的非编码RNA,如转录水平长的非编码RNA 15,16,微小RNA 17的siRNA 等 18。由于的t这种方法的他准确性,它已被用于检测的单核苷酸变异19,20。
的RNA测序技术出现之前,微阵列被用来分析基因表达模式的主要方法。预先设计的探针被合成,随后附着于固相表面,以形成微阵列载玻 片21。 mRNA的提取并转化为cDNA。在反转录过程中,荧光标记的核苷酸被掺入到cDNA和该cDNA可以杂交到微阵列载玻片。从一个特定的点收集到的信号的强度依赖于cDNA的上点21结合的特异性探针的量。用RNA测序技术相比,芯片有几个限制。首先,微阵列依赖于基因注释的预先存在的知识,而RNA测序技术能够检测到新的转录物,在相对较高的背景水平,这限制了它的使用,当歌NE表达水平是低的。此外,RNA测序技术有高得多的动态范围检测(8000倍)7,而,由于背景和信号的饱和度,微阵列的准确性是有限的两个高和低表达基因7,22。最后,芯片探针有不同的杂交效率,一个样品23中比较不同转录本的相对表达水平时,这使结果不可靠。虽然RNA测序过微阵列具有许多优点,它的数据分析是复杂的。这是许多研究人员仍然使用RNA测序的微阵列代替的原因之一。各种生物信息学工具所需的RNA测序数据的处理和分析24。
在几种新一代测序(NGS)的平台上,454,Illumina公司,固体和离子洪流是最广泛使用的。 454是第一个商业化NGS平台。在对比的其它测序平台如Illumina公司和扎实,454平台生成更长的读长(平均700个碱基读取)25。较长的读取是transcriptiome的初始特性更好,由于其较高的装配效率25。 454平台的主要缺点是它的每个序列的兆碱基成本高。 Illumina公司和雄厚的平台生成读取增加的数量和长度短。每个序列的兆碱基的成本比454平台低得多。由于大量的短读取的Illumina的和固体的平台,数据分析是更计算密集的。仪器和试剂用于测序的离子洪流平台的价格比较便宜的测序时间短25。但是,错误率和每序列的兆碱基的成本都较高相比,Illumina的和固体的平台。不同的平台都有自己的优点和缺点,需要不同的方法对数据进行分析。解放军TForm的应基于测序的目的和资金的情况进行选择。
在本文中,我们采取的Illumina RNA测序平台为例。我们使用的EML细胞作为模型系统来调查在EML的细胞的自我更新和分化的关键调节剂,并提供RNA测序文库的构建和数据分析表达水平的计算和新的转录检测的详细方法。我们已经证明在我们以前的出版物,在EML的模型系统2的RNA及以下的研究中,加上功能测试( 例如,shRNA的击倒)提供一种有效的方法,在理解造血细胞分化的早期阶段的分子机制时,并且可以作为一个模型细胞自我更新和分化的一般分析。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. EML细胞培养和使用磁性细胞分选系统和Lin-CD34 +和林-CD34-细胞分离荧光激活细胞分选法
- 制备幼仓鼠肾(BHK)细胞培养基中对干细胞因子集合的:
- 培养BHK细胞中,在37℃,5%CO 2下在细胞培养孵育箱含有10%FBS中为25cm 2烧瓶中( 表1)的DMEM培养基中。
- 当细胞生长至80 - 90%汇合时,用10ml的PBS洗一次细胞。加入5毫升0.25%的胰蛋白酶-EDTA溶液,以单层和孵育的细胞在室温(RT)下1-5分钟,直到细胞分离。
- 吸取溶液,上下轻轻打散细胞团块。加入5 ml完全DMEM至烧瓶中以终止胰蛋白酶的活性。通过离心收集细胞,在200×g离心5分钟,在室温。
- 除去介质和重悬细胞沉淀在10ml新鲜的BHK细胞培养基。 传送2毫升细胞悬浮液从步骤1.1.4至一个新的75cm 2的烧瓶中,并加入加入48ml新鲜的BHK细胞培养基的烧瓶中。
- 培养的BHK细胞两天,收集培养基。通道通过0.45μm的过滤器中的介质。储存在-20℃下的介质,直到进一步使用。
- EML细胞培养:
- 培养EML细胞(悬浮)在EML的基本培养基中含有的BHK细胞培养基( 表1),在37℃,5%CO 2的细胞培养孵化器。
- 保持EML的细胞在低细胞密度(0.5-5×10 5个细胞/ ml)与峰密度小于6×10 5个细胞/ ml。分裂细胞每2-3天以1:5的比例。通过EML细胞传代10代后,轻轻地,丢弃文化。
- 枯竭谱系阳性细胞:
- 收获在EML的细胞通过离心分离,在200×g离心FOR 5分钟,并用PBS洗一次细胞。收集细胞,离心,在200×g离心5分钟。
- 重悬细胞,用PBS并用血细胞计数器计数细胞。确定抗体浓度在后续细胞分离步骤,根据所述单元的数目(请参阅由细胞分离系统的提供者所提供的说明)。
- 隔离谱系负(Lin-的)使用谱系抗体混合物(生物素标记的单克隆抗体的鸡尾酒细胞CD5,CD45R(B220),CD11b的,抗Gr-1(LY-6G / C),7-4和泰尔-119 ),并根据制造商的说明磁性活化细胞分选系统。
- 分离林CD34 +和林-CD34-细胞:
- 降速Lin-的细胞从步骤1.3.3在200×g离心5分钟。重悬细胞沉淀用PBS并用血细胞计数器计数细胞。
- 用FACS缓冲液洗涤细胞两次和颗粒细胞在200 XG5分钟。
- 标签5的1.5 ml离心管中分别编号为1,2,3,4,5。重悬的细胞以每10 6个细胞(每管10 6个细胞)加入100μl的FACS缓冲液中。
- 加1微克抗小鼠CD34 FITC抗体对管1和管2和混合管轻轻。
- 孵育所有试管在4℃下1小时在黑暗中。
- 添加0.25 PE标记的抗SCA1抗体微克和20微升的APC标记的世系鸡尾酒抗体对管1中,0.25的PE缀合的抗SCA1抗体对管3微克和20μl的APC标记的世系鸡尾酒抗体来管4。
- 混合所有的管轻轻孵育的细胞中,在4℃下在黑暗中另外的30分钟。
- 添加300μl的FACS缓冲液至细胞和自旋向下的细胞在200×g离心5分钟。
- 用500μl的FACS缓冲液三次洗细胞。
- 重悬细胞沉淀在500μlFACS卜FFER。
- 使用该单元在管2,3,4和5,用于设置补偿。隔离林SCA + CD34 +和林-SCA-CD34-细胞在试管1使用流式细胞仪咏叹调。
2. RNA制备和图书馆建设的高通量测序
- 隔离,质量分析和RNA定量:
- 提取林CD34 +和林-CD34-细胞分别用Trizol按照制造商的协议总RNA。
- 除去受污染的使用DNA脱氧核糖核酸酶I(DNA酶I),按照生产商的方案。可选地,存储所述的RNA在-80℃下以供进一步使用此步骤。
- 评估根据由供应商提供的说明书使用生物分析总RNA的质量。利用RNA样品与RNA完整性(RIN)的啤酒比9。
- 图书馆建设与高通量测序:
注:本协议描述的RNA序列利用Illumina平台。为其他测序平台,不同的文库制备方法是必需的。- 使用0.1-4微克的高品质的总RNA,每个样品的文库制备。通常为2的总RNA微克可提取10 5 EML的细胞。
- 使用的RNA测序样品制备系统用于RNA纯化和片段化,第一和第二链cDNA合成,末端修复,3'端腺苷化,衔接子连接并进行PCR扩增,按照从提供者的说明,详细的标准程序。
- 积极选择聚腺苷酸mRNA的使用寡脱氧胸苷酸磁珠和片段的表达。
- 用随机引物来获得的cDNA,随后合成cDNA的第二链,以产生双股cDNA进行反转录。
- 除去3'突出,并填补了5'由DNA聚合酶出挑。腺苷酸3'末端,以防止cDNA片段从结扎到彼此。
- 添加多路分度适配器连接到dscDNA的两端。进行PCR对DNA片段的富集。
- 测量A260 / A280,得到约文库的使用分光光度计的浓度信息。
- 评估文库质量和测量用生物分析的DNA片段的大小范围。
3.数据分析
对于在这部分使用的软件参考,请参阅( 表2)。
- 数据文件的处理用于下游分析:
- 转换.bcl(基调用文件)文件中使用CASAVA软件(Illumina公司,1.8.2版本)来.fastq文件。
- 火起来的'终端'的Linux系统。转到包含从Illumina的HiSeq2000测序仪中的数据文件中的数据文件夹。假设结果文件夹是“NASboy1 / JiaqianLabData / HiSeq_RUN / 2013_07_11 / 130627_SN860_0309_A_2013-166_H0PW9ADXX /',类型在图S1A的命令,并输入数据的文件夹。
- 在Linux系统上安装CASAVA 1.8.2。假设outputfolder是“未对齐”,使用图S1B命令编写的配置文件转换。使用--fastq簇数0的选项,以确保只有一个.fastq文件是为每个样品创建。所生成的.fastq文件是在名为.gz格式。将它解压缩下游分析( 图S1B)。
- 已经产生了“不对齐”文件夹后,进入“未对齐”文件夹( 图S1C)。
- 图S1D命令开始转换过程。的'-j'参数提供将要使用的CPU的数量。
- 在系统完成转换过程,请在“未对齐”文件夹( 图S1E)结果的文件夹。
- 图S1F命令</ STRONG>解压缩.fastq.gz文件,将根据每个样品夹.fastq文件。
- 转换.bcl(基调用文件)文件中使用CASAVA软件(Illumina公司,1.8.2版本)来.fastq文件。
- 发现新的成绩单和利用Tuxedo的26套房评估的表达水平:
- 地图的配对末端的RNA序列读取到鼠标的参考基因组(UCSC版MM9,从获得的http://cufflinks.cbcb.umd.edu/igenomes.html采用高顶礼帽软件)(版本1.3.3)27,它使用该领结读取映射器(版本0.12.7)28。高顶礼帽提供与“-no-小说,juncs”选项,以提高表达水平的估计精度。
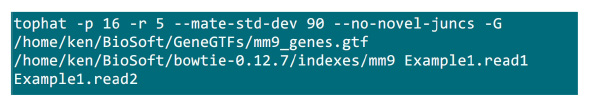
- 把.fastq文件所在的映射过程将执行一个文件夹中。假设有2 .fastq文件(重命名为Example1.read1,Example1.read2)用于配对末端测序的样品,使用命令, 如图S2中执行的映射(根据系统设定调整参数)。中的“-p”参数提供将要使用的CPU数目。在“-r”和“-mate-STD-dev的”参数可以从库QC索取或从排列的子集推断读取( 图S2)。
- 组装映射读入使用袖扣软件RNA转录(版本1.3.0)29。使用已知的基因注释文件(使用高顶礼帽相同.gtf文件),并通过高顶礼帽产生.bam文件运行袖扣。
- 后高顶礼帽运行完毕后,在同一个文件夹中,使用图S3A要运行的命令袖扣构建转录和转录估计表达水平。在“mm9_repeatMasker.gtf'和'GenomeSeqMM9”文件夹中的基因组序列文件可以从UCSC基因组浏览器获得。
- 由此产生的genes.expr和transcripts.expr文件包含的基因和转录本(亚型)的表达式的值。复制和粘贴文件内容到一个Excel文件和处理与电子表格应用程序( 图S3B)。
- 如图S3C使用命令以确定新的转录所得'transcripts.gtf'文件的引用“mm9_genes.gtf'文件进行比较。
- 所得.tmap文件中包含的比较结果。复制文件内容粘贴到Excel文件和操作与电子表格应用程序。转录物与类代码“u”指可以被视为“新”相比.gtf文件的参考设置( 图S3D)。
注:对于下游分析的方便,设置FPKM值至0.1,如果值是低于0.1。
注: 步骤3.2.3 - 3.2.6是可选对于那些谁想要提高小说成绩单“表情估计精度。这将需要更长的时间,因为映射和转录施工需要为r未超过一次。
- 运行高顶礼帽使用默认参数,然后运行使用图S3E命令袖扣,以生成.gtf文件。
- 比较所得.gtf文件以使用图S3F命令中的参照基因组.gtf文件。
- 解析结果.tmap文件如在步骤3.2.2.4中描述。复制文件内容粘贴到Excel文件和操作与电子表格应用程序。转录物与类代码“u”指可以被视为“新”相比.gtf文件的参考设置。
- 在步骤3.2.5之后,有可被用作参考.gtf文件夹中的一个.combined.gtf文件。高顶礼帽和袖扣的第二次运行可以如在步骤3.2.1和3.2.2中所述,得到的新的转录本的更准确FPKM估计来进行。
- 地图的配对末端的RNA序列读取到鼠标的参考基因组(UCSC版MM9,从获得的http://cufflinks.cbcb.umd.edu/igenomes.html采用高顶礼帽软件)(版本1.3.3)27,它使用该领结读取映射器(版本0.12.7)28。高顶礼帽提供与“-no-小说,juncs”选项,以提高表达水平的估计精度。
- 检测differentiall使用DESeq包30岁表达的基因。
- DESeq的输入是原始读计数表。为了得到这样一个表,使用分布式与HTSeq Python包可从HTSeq网站下载的htseq计数脚本( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) 。
- 确保samtools,Python和htseq数programsare安装在系统中。通过使用图S4A命令获得高顶礼帽输出原始读取计数器值。
- 准备“Raw_Count_Table.txt”使用Excel,“ExperimentDesign.txt”文件。复制并保存为DESeq了R封装( 图S4B).txt格式的内容。
- 在系统中安装R程序。在终端,键入“R”,然后按ENTER.A屏幕信息将appearas显示在图S4C。
- 阅读“Raw_Count_Table.txt','用图S4D命令ExperimentDesign.txt“成R。
- 加载利用图S4E命令DESeq包。
- 比化条件R( 图S4F)。
- 使用图S4G命令就归伯爵表运行负二项分布的测试。
- 使用图S4H命令输出显著差异表达基因的.csv文件。
- DESeq的输入是原始读计数表。为了得到这样一个表,使用分布式与HTSeq Python包可从HTSeq网站下载的htseq计数脚本( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) 。
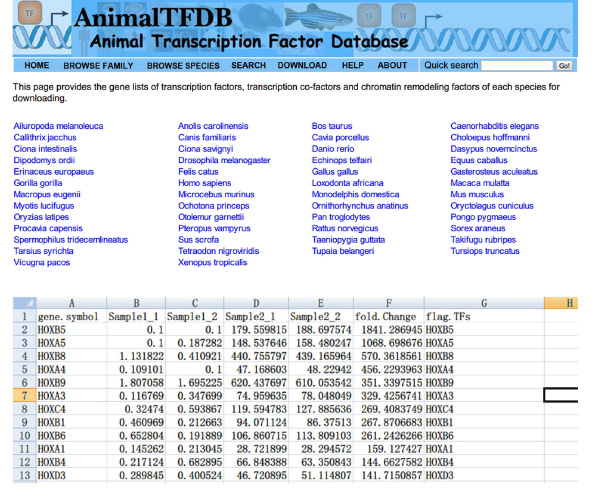
- 在整个样品查找转录因子'(TFS)FPKM值使用Excel。相交DE基因表和转录因子表。基因属于这两个表中差异表达的转录因子。
- 去网站http://www.bioguo.org/AnimalTFDB/download.php和下载的转录因子。那么查找的DE转录因子在Excel中(< STRONG>图S5)。
- 产生.bigwig文件UCSC基因组浏览器的可视化。
- 从网站上下载“bedtools”软件包https://github.com/arq5x/bedtools2并安装该软件系统中的31。从网站下载UCSC工具“bedGraphToBigWig” http://hgdownload.cse.ucsc.edu/admin/exe/并安装该软件在系统中。
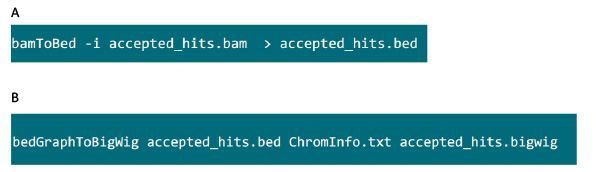
- 在包含.bam文件的文件夹,使用图S6A命令来转换通过高顶礼帽到.bed文件生成.bam文件。
- 该.bed文件产生后,使用图S6B命令来生成.bigwig文件。文件'ChromInfo.txt“可以从以下网址获得:ARGET =“_blank”> http://hgdownload.cse.ucsc.edu/goldenPath/mm9/database/chromInfo.txt.gz。
- 观察UCSC基因组浏览器自定义的轨道。请参阅网站http://genome.ucsc.edu/goldenPath/help/customTrack.html如何显示使用UCSC基因组浏览器自定义的轨道。

图S1:转换.bcl文件使用CASAVA软件.fastq文件。

图S2:读取映射使用高顶礼帽到参考基因组。
图S3:检测新转录和表达水平的估计。

图S4:使用DESeq包呼唤差异表达的基因。

图S5:差异表达的转录因子的鉴定。

图六:转换映射结果进行数据可视化。
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
为了分析差异表达基因的林CD34 +和林-CD34-细胞EML,我们使用了RNA测序技术。 如图1所示的程序流程。谱系阴性细胞的磁性细胞分选分离后,我们就分开了林SCA + CD34 +,并使用流式细胞仪咏叹调林SCA-CD34-细胞。林富EML的细胞用抗CD34,抗SCA1和谱系鸡尾酒抗体。只有Lin-被门控的SCA1和CD34的表达分析。两个群体(SCA + CD34 +和SCA-CD34- EML的细胞)可以通过FACS分析( 图2)6被观察到。
细胞分离后,我们提取的总RNA的CD34 +和CD34 - 细胞分别和分析RNA的质量。的RNA测序数据的准确度在很大程度上依赖于RNA测序文库的质量和总RNA的质量是制备高质量文库至关重要。高品质的RNA样品应具有1之间的OD 260/280值。8和2.0。除了 使用分光光度计,RNA质量进一步评估与由生物分析仪更高的精确度。 图3示出了具有RIN的高质量的RNA样品等于9.4的结果。只用RIN值大于9的高品质的总RNA样品用于提取mRNA和随后文库的构建程序。
核糖体RNA是最丰富的类型细胞的RNA。目前两种主要的策略,rRNA的枯竭或积极选择聚腺苷酸化的mRNA(聚-A的mRNA)的,用于靶RNA文库构建之前富集。非聚腺苷酸化RNA种类的聚-A基因的选择过程中丢失。与此相反,rRNA的耗竭的方法,如RIBOMINUS可以保持非聚腺苷酸化的RNA种类。我们的研究目的是寻找差异表达的编码基因在两种细胞类型,因此,我们所用的多聚A mRNA的选择方法对靶RNA的富集文库CONSTRU前ction。当库施工完成后,在文库DNA片段的大小使用生物分析仪测序之前进行检查。 图4示出了与该片段大小峰的质量好文库,在约300bp。
在随后的步骤中,该库进行高通量测序。原则上,较长的读长将是为读出的映射有帮助的。它可以减少读出被映射到多个位置,由于重复的基因或基因家族成员之间的相似性的概率。作为一对末端测序序列来自所述片段的两端,所选择的读出长度应小于一半的平均片段长度。如果实验的主要目的是测量的表达水平,而不是构建转录结构,单端读出(75或100碱基对)可以降低成本,而不会丢失太多的信息。配对末端测序是用于转录的结构构造和较短的更有益阅读长度可用于降低成本。当然,当有足够的资金可用,更长的读取长度是首选。
为差异表达分析,有很多比DESeq其他替代算法。也有一包括在名为cuffdiff 32袖扣包。 DESeq是基于最广泛使用的计数DE基因分析算法之一。 DESeq方法是基于一种充分表征的统计模型 - 负二项式分布。根据我们的经验,DESeq更稳定比较cuffdiff。早期版本的cuffdiff经常给显著不同数量DE基因。因此,我们采用DESeq为DE的分析在这里。
由于转录因子是细胞命运决定的关键,我们专注于显著差异表达的转录因子33。 TFS的改变林CD34 +和林-CD34-间> 1.5倍,发现并显示在热图( 造型玩具Ë5)2。值得注意的是,TCF7在林CD34 +细胞的相对表达水平比林CD34-细胞超过100倍以上。因此TCF7被选作进一步的ChIP测序(染色质免疫沉淀和测序)分析和功能测试,以确认TCF7的在EML细胞的自我更新和分化2的调节功能。

图1:的程序工作流林的CD34 +和Lin - CD34 -细胞中通过磁性细胞分离系统和荧光激活细胞分选法分离。提取总RNA,接着用mRNA纯化和文库构建。文库质量分析后,样品进行高通量测序。数据进行分析和差异表达的转录因子进行鉴定。

图2:林-CD34 +和林-CD34-细胞EML 6 Lin-的EML细胞免疫磁珠富集分离 。线性细胞用抗CD34,抗SCA1和谱系混合物抗体。 Lin-被选通为CD34和SCA1的表达。林-CD34 + SCA +和林-CD34-的Sca-EML细胞群来分类的。

图3:高品质的总RNA样品的代表总RNA的质量由生物分析仪进行了评估。 RNA的完整性号是9.4(FU,荧光单位)。

图4:配对末端文库片段的大小范围的 用生物分析仪对文库的DNA大小分布进行了分析。多数片段的250-500碱基对大小范围内。

图5:差异表达的转录因子林CD34 +细胞和Lin - CD34 -细胞2之间(> 1.5倍),对于每种细胞类型的,两个独立的实验。上调的基因被表示为红色和下调的基因被表示为绿色。
表1:缓冲液和细胞培养基 。
| 软件 | 用法 | 参考 | |||
| 领结1.2.7 | 采用高顶礼帽映射 | [28] | |||
| 高顶礼帽1.3.3 | 读取映射回参考基因组 | [27] | |||
| 袖扣1.3.0 | 成绩单建设和表达水平估计 | [29] | |||
| DESeq 1.16.0 | 差异表达分析 | [30] | Bedtools 2.18 | 转换.bam文件到.bed文件 | [31] |
| bedGraphToBigWig | 转换.bed文件.bigwig文件 | http://genome.ucsc.edu/ |
表2:数据分析软件的列表 。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
哺乳动物转录是非常复杂的34-38。 RNA测序技术在转录组分析,新转录本的检测和单核苷酸变异的发现等的研究,越来越重要的作用,具有比其它方法进行基因表达分析的许多优点。如引言中所述,它克服了微阵列的杂交工件和可用于鉴定新的转录物从头 。 RNA的测序中的一个限制是比较Sanger测序相对短的读长。然而,随着测序技术的飞速进步,阅读长度不断增加。在本文中,我们提供了使用这种技术来识别鼠标EML细胞的自我更新和分化潜能的关键调节器的详细方法。
该协议的第一个关键步骤是EML细胞培养。虽然EML是造血前体细胞系,它可以是在传播大量与SCF。 EML的细胞的培养条件需要比通常的永生化细胞系的更多关注。细胞应供给和传代在用温和的操作定期;否则细胞可能会改变在自我更新和分化的它们的性质,并进行细胞死亡。作为收集足够的细胞后的第一个步骤中,我们使用磁激活细胞分选系统分离的谱系阴性细胞。然后我们使用荧光激活细胞分选分离的CD34 +和CD34 - 细胞。 EML的细胞使用用于RNA提取和CD34 +的数量和CD34-的细胞应该是分离后的类似前一般传代不超过10代。如果两个种群差别较大的细胞数,最好是放弃该培养和再解冻的细胞原液另一管中培养。
的CD34 +和CD34 - 细胞分离后,提取总RNA进行,对于本圣另一个重要步骤UDY。高质量的RNA是基础施工的高质量文库,其中承诺的测序数据的准确性。在这个关键步骤中,应避免用RNase任何接触。所有试剂应无RNA酶。戴上手套在所有时间,而处理RNA是重要的。高品质的RNA样品具有1.8和2.0之间的OD 260/280值。当收集含有RNA的水相,要小心,不要携带任何有机相中的RNA样品。任何残留的有机溶剂,如苯酚或氯仿中的RNA的结果将是OD260 / 280的值比1.65低。如果OD260 / 280值小于1.65时,再次沉淀RNA用乙醇。用75%乙醇洗涤后,没有干燥过度RNA沉淀。干燥RNA沉淀完全,会影响RNA的溶解度,导致RNA的产量低。
该协议的一个关键步骤就是库准备。总RNA提取,用DNA酶用于去除污染的DNA的工序后,我s极力推荐的,因为DNA污染可能导致总RNA的使用量的错误估计。建议之后RNA分离立即执行下行过程中,由于经过长期储存和冻融过程,核糖核酸会降低到一定程度。如果不能立即完成后分离RNA的后续步骤中,储存在-80℃下将RNA。前的总RNA用于mRNA纯化和cDNA合成,质量应始终检查。只有高品质的RNA可用于文库制备。使用低质量或降解的RNA可能导致过度的代表性的3'末端。测序之前,库质量进行评估,以确保最大的测序效率。
在数据分析中的一部分,没有一个参考转录组进行袖扣的运行后,我们结合了新颖的成绩单与已知的转录本形成.gtf文件并运行高顶礼帽和袖扣参考第二次。这两个程序运行,建议,因为这提供了更准确的FPKM估计比只运行一次。后的数据分析,差异表达的基因进行了鉴定。下游实验可以被执行以验证基因功能的体外和体内 。在我们以前的出版物2,我们选择了显著差异表达的转录因子,并通过进行染色质免疫沉淀法和测序(芯片起)确定这些因素的基因组中的结合位点。此外,我们采用的shRNA敲低实验来测试TCF7的功能效果。我们发现,在TCF7拦截细胞,上调的基因是高度富集CD34 -细胞中的基因,而被发现下调的基因中的CD34 +细胞可显著富集。因此,TCF7敲除细胞的基因表达谱移向一个部分分化CD34- state.Overall,使用EML细胞作为模型系统加上RNA的测序技术和功能分析,我们确定和证实TCF7作为EML的细胞的自我更新和分化的重要调节器。
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| Antibiotic-Antimycotic | Invitrogen | 15240-062 | BHK cell culture |
| Anti-Mouse CD34 FITC | eBioscience | 11-0341-81 | FACS sorting |
| Anti-Mouse Ly-6A/E (Sca-1) PE | eBioscience | 12-5981-81 | FACS sorting |
| APC Mouse Lineage Antibody Cocktail | BD Biosciences | 558074 | FACS sorting |
| BD FACSAria Cell Sorter | BD Biosciences | Special offer sysmtem | FACS sorting |
| Corning™ Cell Culture Treated Flasks 75 cm2 | Corning incorporated | 430641 | Cell culture |
| Corning™ Cell Culture Treated Flasks 25 cm2 | Corning incorporated | 430639 | Cell culture |
| Deoxyribonuclease I, Amplification Grade | Invitrogen | 18068-015 | Library preparation |
| DMEM | Invitrogen | 11965-092 | BHK cell culture |
| DPBS | Gibco | 14190 | Cell culture |
| HI FBS | Invitrogen | 16140071 | BHK cell culture |
| Horse Serum | Invitrogen | 16050-122 | EML cell culture |
| IMDM | HyClone | SH30228.02 | EML cell culture |
| L-Glutamine | Invitrogen | 25030-081 | Cell culture |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | 130-090-858 | Isolation of lineage negative cells |
| NanoVue Plus spectrophotometer | GE Healthcare | 28-9569-62 | Quality control |
| Thermo Scientific™ Napco™ 8000 Water-Jacketed CO2 Incubators | Thermo Scientific | 15-497-002 | Cell culture |
| Penicillin-Streptomycin | Invitrogen | 15140-122 | EML cell culture |
| TRIzol® Reagent | Invitrogen | 15596-018 | RNA exraction |
| TruSeq™ RNA Sample Prep Kit v2 -Set B (48 rxn) | Illumina | RS-122-2002 | Library preparation |
| 2100 Electrophoresis Bioanalyzer Instrument | Agilent | G2939AA | Quality control |
| 0.25% Trypsin-EDTA | Gibco | 25200 | Cell culture |
| 0.45 µm Syringe Filters | Nalgene | 190-2545 | Cell culture |
References
- Chambers, S. M., Goodell, M. A. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 3, 201-211 (2007).
- Wu, J. Q., et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS genetics. 8, (2012).
- Ye, Z. J., et al. Complex interactions in EML cell stimulation by stem cell factor and IL-3. Proceedings of the National Academy of Sciences of the United States of America. 108, 4882-4887 (2011).
- Tsai, S., Bartelmez, S., Sitnicka, E., Collins, S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 8, 2831-2841 (1994).
- Weiler, S. R., et al. D3: a gene induced during myeloid cell differentiation of Linlo c-Kit+ Sca-1(+) progenitor cells. Blood. 93, 527-536 (1999).
- Ye, Z. J., Kluger, Y., Lian, Z., Weissman, S. M. Two types of precursor cells in a multipotential hematopoietic cell line. Proc Natl Acad Sci U S A. 102, 18461-18466 (2005).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10, 57-63 (2009).
- Chu, Y., Corey, D. R. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic acid therapeutics. 22, 271-274 (2012).
- Hornett, E. A., Wheat, C. W. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC genomics. 13, 361 (2012).
- Eswaran, J., et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific reports. 3, 1689 (2013).
- Wang, E. T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470-476 (2008).
- Wu, J. Q., et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proceedings of the National Academy of Sciences of the United States of America. 107, 5254-5259 (2010).
- Loraine, A. E., McCormick, S., Estrada, A., Patel, K., Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant physiology. 162, 1092-1109 (2013).
- Edgren, H., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome biology. 12, 6 (2011).
- Ilott, N. E., Ponting, C. P. Predicting long non-coding RNAs using RNA sequencing. Methods. 63, 50-59 (2013).
- Sun, L., et al. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC bioinformatics. 13, 331 (2012).
- Luo, S. MicroRNA expression analysis using the Illumina microRNA-Seq Platform. Methods in molecular biology. 822, 183-188 (2012).
- Bolduc, F., Hoareau, C., St-Pierre, P., Perreault, J. P. In-depth sequencing of the siRNAs associated with peach latent mosaic viroid infection. BMC molecular biology. 11, 16 (2010).
- Chepelev, I., Wei, G., Tang, Q., Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic acids research. 37, 106 (2009).
- Djari, A., et al. Gene-based single nucleotide polymorphism discovery in bovine muscle using next-generation transcriptomic sequencing. BMC genomics. 14, 307 (2013).
- Murphy, D. Gene expression studies using microarrays: principles, problems, and prospects. Advances in physiology education. 26, 256-270 (2002).
- Chen, K., et al. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS one. 8, 72567 (2013).
- Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 18, 1509-1517 (2008).
- Ramskold, D., Kavak, E., Sandberg, R. How to analyze gene expression using RNA-sequencing data. Methods in molecular biology. 802, 259-274 (2012).
- Glenn, T. C. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 11, 759-769 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 7, 562-578 (2012).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25, 1105-1111 (2009).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 10, 25 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature. 28, 511-515 (2010).
- Anders, S., Huber, W. Differential expression analysis for sequence count data. Genome biology. 11, 106 (2010).
- Quinlan, A. R., Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841-842 (2010).
- Cheranova, D., et al. RNA-seq analysis of transcriptomes in thrombin-treated and control human pulmonary microvascular endothelial cells. J Vis Exp. , (2013).
- Zhang, H. M., et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research. 40, 144-149 (2012).
- Wu, J. Q., et al. Systematic analysis of transcribed loci in ENCODE regions using RACE sequencing reveals extensive transcription in the human genome. Genome Biol. 9, 3 (2008).
- Wu, J. Q., et al. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques. 36, 690-696 (2004).
- Wu, J. Q., Shteynberg, D., Arumugam, M., Gibbs, R. A., Brent, M. R. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 14, 665-671 (2004).
- Dewey, C., et al. Accurate identification of novel human genes through simultaneous gene prediction in human, mouse, and rat. Genome Res. 14, 661-664 (2004).
- Wu, J. Characterize Mammalian Transcriptome Complexity. , LAP Lambert Academic Publishing. (2011).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}