

Summary
RNAシークエンシングおよびバイオインフォマティクス解析は、マウスEMLcellsのリン、CD34 +およびリン-CD34-亜集団において有意にかつ差次的に発現する転写因子を同定した。これらの転写因子は、自己再生リン-CD34 +を、部分的に分化したリン-CD34-細胞との間の切り替えを決定する際に重要な役割を果たしている可能性がある。
Abstract
造血幹細胞(HSC)は、白血病やリンパ腫などの多くの疾患において、患者の造血系を再構築するために、移植治療のために臨床的に使用されている。 HSCの自己再生および分化を制御するメカニズムを解明することは研究および臨床用途のためのHSCのアプリケーションのために重要である。しかし、によりインビトロで増殖することができないことにHSCを大量に得ることができない。このハードルを克服するために、我々は、この研究のためのモデル系として、マウス骨髄由来の細胞株、EML(赤血球、骨髄、およびリンパ球)細胞株を使用した。
RNAシークエンシング(RNA-配列)がますます遺伝子発現研究のためのマイクロアレイを置き換えるために使用されてきた。ここではEML細胞の自己再生および分化の調節において潜在的な主要因を調査するためにRNA-配列技術を使用する具体的な方法を報告する。このホワイトペーパーで提供されるプロトコルは、3つの部分に分かれています。最初のパーtはどのように文化のEML細胞と独立したLIN-CD34 +およびLIN-CD34-細胞に説明しています。プロトコルの第2の部分は、トータルRNA調製およびハイスループット配列決定のための後続のライブラリー構築のための詳細な手順を提供しています。最後の部分は、RNA配列データ解析のための方法を説明し、リン、CD34 +およびリン-CD34-細胞との間の差次的に発現する転写因子を同定するためにデータを使用する方法について説明します。最も有意に差次的に発現される転写因子はEML細胞の自己再生および分化を制御する潜在的な重要な調節因子であると同定された。本稿の議論のセクションでは、この実験の成功パフォーマンスのための主要なステップを強調表示します。
要約すると、本論文では、EML細胞中で自己複製と分化の潜在的な調節因子を同定するために、RNA-配列技術を使用する方法を提供しています。特定された主要な要因は、in vitroおよびiの下流の機能解析に供されているn個の生体内。
Introduction
造血幹細胞は、成体骨髄ニッチに主に存在する稀な血液細胞である。これらは、血液および免疫系1を補充するために必要な細胞の産生を担う。幹細胞の種類としては、HSCは、自己再生および分化の両方が可能である。 HSCの運命決定を制御するメカニズムの解明、自己複製または分化のどちらかに向かって、血液疾患の研究や臨床使用量2のためのHSCの操作に貴重なガイダンスを提供します。研究者が直面する問題の一つは、HSCが維持され、非常に限られた範囲でインビトロで増殖することができることである。その子孫の大部分は、部分的に文化2で分化されている。
ゲノムワイドスケールでの自己再生および分化の過程を制御する重要な調節因子を同定するために、我々は、モデル系としてマウス原始造血前駆細胞株EMLを使用した。目細胞株は、マウス骨髄3,4から導出された。異なる成長因子を供給すると、EML細胞は、in vitroで 5 において赤血球、骨髄、およびリンパ系細胞に分化することができる。重要なことに、この細胞株は、幹細胞因子(SCF)を含む培地中で大量に増殖させることができ、それでもそれらの多能性を維持する。 EML細胞は、自己再生リン-SCA + CD34 +表面マーカーCD34およびSCA 6に基づいて、部分的に分化したリン-SCA-CD34-細胞の亜集団に分けることができる。短期HSCのと同様に、SCA + CD34 +細胞は、自己複製が可能である。 SCFで処理した場合、LIN-SCA + CD34 +細胞は急速に林-SCA + CD34 +およびLIN-SCA-CD34-細胞の混合集団を再生成し、6を増殖し続けることができます。つの集団は、形態学的に類似しており、c-キットmRNAおよびタンパク質6の同様のレベルを有する。リン-SCA-CD34-細胞はIL-3の代わりに、SCF 3を含有する培地中で増殖させることが可能である。 UnveilingのEML細胞の運命決定に重要な調節因子は、造血時の初期発生の移行における細胞および分子メカニズムのより良い理解を提供します。
自己複製リン-SCA + CD34 +および部分的に分化リン-SCA-CD34-細胞との間の根本的な分子の違いを調べるために、我々は、差次的に発現される遺伝子を同定するために、RNA配列を使用した。転写因子は、細胞の運命を決定する上で重要であるように、特に、我々は、転写因子に焦点を当てる。 RNA-seqがゲノム7,8から転写されたRNAをプロファイリングし、定量化するために、次世代シーケンシング(NGS)技術の能力を利用して最近開発されたアプローチです。簡単に述べると、トータルRNAは、ポリ選択と初期template.The RNAテンプレートは次いで逆転写酵素を用いてcDNAに変換されるように、断片化されている。 cDNAライブラリーを構築するために、無傷の、非分解RNAを用いて、完全長のRNA転写物をマッピングするために重要である。 PURのためシークエンシングの姿勢は、特定のアダプター配列は、cDNAの両端に付加される。そして、ほとんどの場合、cDNA分子は、PCRによって増幅され、ハイスループット様式で配列決定した。
シーケンシング後、得られたは参照ゲノムとトランスクリプトームデータベースに整列させることができる読み取ります。数は、参照遺伝子のマップがカウントされ、この情報は、遺伝子発現のレベルを推定するために使用できることを読み取る。読み込みは、非モデル生物9にトランスクリプトームの研究を可能にする、参照ゲノムなしデノボ組み立てることができる。 RNA-seqの技術は、スプライスアイソフォーム10-12、新規な転写物13および14の遺伝子融合を検出するために使用されてきた。タンパク質をコードする遺伝子の検出に加えて、RNA-配列はまた、新規に検出し、そのような18の非コードRNA 15,16、マイクロRNA 17は、siRNA などの長い非コードRNAの転写レベルを分析するために使用され得る。なぜなら、tの彼は、この方法の精度は、単一ヌクレオチド変異19,20の検出に利用されている。
RNA-配列技術の出現の前に、マイクロアレイは、遺伝子発現プロファイルを分析するために使用される主要な方法であった。予め設計されたプローブは、マイクロアレイスライド21を形成するように合成し、続いて固体表面に付着される。 mRNAを抽出し、cDNAに変換される。逆転写プロセスの間に、蛍光標識されたヌクレオチドは、cDNAに組み込まれ、cDNAはマイクロアレイスライドにハイブリダイズさせることができる。特定の場所から収集された信号の強度は、そのスポット21上の特異的プローブに結合したcDNAの量に依存する。 RNA-配列技術と比較して、マイクロアレイはいくつかの限界がある。 RNA-配列技術は、その使用が制限されるとき、比較的高いバックグラウンドレベルで新規な転写物を検出することが可能であるが、まず、マイクロアレイは、遺伝子アノテーションの既存の知識に依存している葛ね発現レベルが低い。による背景信号が飽和し、マイクロアレイの精度は両方の高度に発現される遺伝子と低い7,22のため制限され、一方でまた、RNA-配列技術は、検出(8000倍)7の非常に高いダイナミックレンジを有する。最後に、マイクロアレイプローブは、一つのサンプル23内の異なる転写物の相対発現レベルを比較した場合、結果はあまり信頼性を高めるそれらのハイブリダイゼーションの効率、が異なる。 RNA-配列がマイクロアレイに比べて多くの利点を有するが、そのデータ分析は複雑である。これは、多くの研究者がまだRNA-配列の代わりに、マイクロアレイを使用する理由の一つです。様々なバイオインフォマティクスツールは、RNA配列のデータ処理および分析24のために必要とされる。
いくつかの次世代シーケンサー(NGS)のプラットフォームのうち、454、イルミナ、SOLIDとイオントレントは、最も広く使用されているものである。 454は最初の商用NGSプラットフォームだった。他のシーケンシングプラットフォームとは対照的にそのようなイルミナ固体として、454プラットフォームは、より長い読みの長さ(平均700ベースの読み取り)25を生成します。長くは、その高い効率25を組み立てるためにtranscriptiomeの初期特性評価のための優れている読み取ります。 454プラットフォームの主な欠点は、シーケンスのメガベース当たりのコストが高いことである。イルミナとSOLIDプラットフォームは増大した数と短い長さで読み込み、生成する。シーケンスのメガベース当たりのコストは、454プラットフォームよりもはるかに低い。が短いため、大量のデータ分析がはるかに計算集約され、イルミナとSOLIDプラットフォーム用に読み取ります。イオントレントプラットフォーム用のシーケンシングするための機器および試薬の価格は安いですし、シーケンシング時間は25短い。しかし、エラーレートとシーケンスのメガベース当たりのコストは、イルミナとSOLIDプラットフォームに比べて高くなっている。異なるプラットフォームは、独自の長所と短所を持っており、データ解析のために異なる方法を必要とする。ナンプラーTFORMは、配列決定の目的や資金の利用可能性に基づいて選択する必要があります。
本稿では、例として、イルミナRNA-のSeqプラットフォームを取る。我々は、EMLの細胞の自己再生および分化において重要な調節因子を調査するためのモデル系としてEML細胞を使用し、発現量の計算および新規な転写産物を検出するためのRNA-配列ライブラリーの構築およびデータ分析の詳細な方法を提供した。我々は、機能テスト( 例えば shRNAのノックダウン)と結合EMLモデルシステム2のRNA-seqの研究は、造血分化の初期段階の分子機構を理解する上での強力なアプローチを提供し、として機能することができるという本発明者らの以前の刊行物に示されている一般に、細胞の自己再生および分化の分析のためのモデル。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. EML細胞培養およびシステムおよび磁気細胞ソーティングの使用LIN-CD34 +およびLIN-CD34-細胞の分離蛍光を活性化細胞選別法
- 幹細胞因子コレクションのベビーハムスター腎臓(BHK)細胞培養培地の調製:
- 37℃の細胞培養インキュベーター中、5%CO 2で25cm 2のフラスコ( 表1)、10%FBSを含むDMEM培地中で培養BHK細胞。
- 細胞が80まで成長する場合 - 90%コンフルエンス、10mlのPBSで細胞を1回洗浄します。単層に0.25%トリプシンEDTA溶液5ミリリットルを加え、細胞が剥離されるまで、室温(RT)で1-5分間細胞をインキュベート。
- ピペットで上下ソリューション優しく細胞の塊を分割する。トリプシン活性を停止するために、フラスコに完全DMEMを5ml追加します。 RTで5分間200×gでの遠心分離によって細胞を収集します。
- 培地を除去し、新鮮BHK細胞培養培地10ml中に細胞ペレットを再懸濁する。 転送2新しい75cm 2のフラスコにステップ1.1.4からの細胞懸濁液mlのフラスコに新鮮なBHK細胞培養培地の48ミリリットルを追加します。
- 培地を培養2日BHK細胞を収集。 0.45μmのフィルターを通過する媒体。さらに使用するまで-20℃でのメディアを保管してください。
- EML細胞培養:
- 細胞培養インキュベーター中で37℃、5%CO 2で、BHK細胞培養培地( 表1)を含むEML塩基性媒体中で培養EML細胞(懸濁液中)。
- ピーク未満の密度6×10 5細胞/ mlの低細胞密度(0.5〜5×10 5細胞/ ml)をEMLで細胞を維持する。 5:1の割合で2〜3日おきにセルを分割します。継代EML細胞を静かにし、10世代にわたって継代後の培養を捨てる。
- 系統陽性細胞の枯渇:
- 200×gでのFOでの遠心分離によってEML細胞を収穫R 5分間、PBSで細胞を1回洗浄します。 5分間200×gでの遠心分離によって細胞を収集します。
- PBSで細胞を再懸濁し、血球計算板を用いて細胞を数える。 (細胞単離システムのプロバイダによって提供される説明書を参照してください)は、細胞の数に応じてその後の細胞分離工程で抗体濃度を決定します。
- 系統陰性(リニア)を単離系統抗体カクテル(ビオチン結合モノクローナル抗体のカクテルを用いて細胞、CD5、CD45R(B220)、のCD11b、抗GR-1(LY-6G / C)、7-4とテル-119 )で製造業者の指示に従って、磁気活性化細胞選別システム。
- LIN-CD34 +およびLIN-CD34-細胞の分離:
- 5分間200×gでのステップ1.3.3からのリニア細胞をスピンダウン。 PBSで細胞ペレットを再懸濁し、血球計算板を用いて細胞を数える。
- FACS緩衝液で2回細胞を洗浄し、200×gで細胞をペレット5分間。
- それぞれ番号1、2、3、4、5で5 1.5mlの微小管にラベルを付ける。 10 6個の細胞(試験管当たり10 6細胞)当たり100μlのFACS緩衝液で細胞を再懸濁。
- チューブ1とチューブ2に抗マウスCD34 FITC抗体1μgのを加え、穏やかにチューブを混ぜる。
- 暗闇の中で1時間、4℃で、全てのチューブをインキュベートします。
- にチューブ3にPE結合抗のSca1抗体のPE結合抗のSca1抗体及びチューブ1へのAPC結合リネージュカクテル抗体20μlの0.25μgの、0.25μgのを追加し、APC標識リネージュカクテル抗体20μlのチューブ4。
- 優しく全てのチューブを混合し、暗闇の中でさらに30分間、4℃で細胞をインキュベートする。
- 細胞にFACS緩衝液300μlのを追加し、5分間200×gで細胞をスピンダウン。
- 3回のFACS緩衝液500μlで細胞を洗浄。
- FACS BU500μlに細胞ペレットを再懸濁ffer。
- 補償を設定するために、チューブ2、3、4、および5の細胞を使用する。 FACSアリアを使用したチューブ1におけるLIN-SCA + CD34 +およびLIN-SCA-CD34-細胞を単離する。
2.高スループットシーケンシングのためのRNAの調製およびライブラリー構築
- 単離、RNAの品質分析および定量。
- 製造 'プロトコル以下のTRIzolを使用して、それぞれの林-CD34 +およびLIN-CD34-細胞から全RNAを抽出します。
- 製造業者のプロトコルに従って汚染されたDNAを用いて、デオキシリボヌクレアーゼI(DNaseI)を取り外します。必要に応じて、さらに使用するため、このステップで-80℃でRNAを保存する。
- 供給業者が提供する指示に従って、バイオアナライザーを用いて総RNAの品質を評価する。 RNA完全性番号(RIN)9よりラガーとRNAサンプルを使用してください。
- ライブラリーの構築およびハイスループットシークエンシング:
注:このプロトコルは、イルミナのプラットフォームを用いてRNA-配列について説明します。のために他のシーケンシングプラットフォームは、異なるライブラリの調製方法が必要とされる。- ライブラリ調製のためのサンプルあたりの高品質の全RNA 0.1-4μgのを使用してください。通常、2μgの全RNAは、10 5 EML細胞から抽出することができる。
- 第一および第二鎖cDNA合成、最後の修復、3 'プロバイダの指示の詳細な標準的手順に従って、アデニル化、アダプターライゲーションとPCR増幅を終了し、RNA精製および断片化RNA配列決定サンプル調製システムを使用する。
- 積極的にオリゴdT磁気ビーズを使用してポリA mRNAを選択して、mRNAを断片化する。
- cDNAを得るために、ランダムプライマーを用いて逆転写を行い、続いて二本鎖cDNAを生成するために、cDNAの第二鎖を合成する。
- 3 'オーバーハングを5埋める「取り外しDNAポリメラーゼによるオーバーハングを。アデニル3 'は、互いに連結することからcDNA断片を防止するために終了する。
- dscDNAの両端にマルチプレックスインデクシングアダプタを追加します。 DNA断片の濃縮のためのPCRを行います。
- 分光光度計を使用してライブラリの濃度に関する情報を得るためにA260 / A280を測定します。
- ライブラリーの質を評価し、バイオアナライザを用いてDNA断片のサイズ範囲を測定する。
3.データ解析
この部分で使用されているソフトウェアの参考のために、( 表2)を参照してください。
- 下流の分析のためのデータファイル処理:
- .bcl(ベースコールファイル)CASAVAソフトウェア(イルミナ、バージョン1.8.2)を使用してファイルを.fastqにファイル変換します。
- Linuxシステムの「ターミナル」を起動する。イルミナHiSeq2000シーケンシングマシンからデータファイルを含むデータのフォルダに移動します。結果フォルダである「NASboy1 / JiaqianLabData / HiSeq_RUN / 2013_07_11 / 130627_SN860_0309_A_2013-166_H0PW9ADXX / '、種類を仮定図S1A内のコマンドで、データのフォルダを入力してください。
- LinuxシステムでCASAVA 1.8.2をインストールします。 outputfolderが「アンアラインド」であるとし、変換するためのコンフィギュレーション·ファイルを準備するために、図S1Bでコマンドを使用します。一つだけ.fastqファイルは、各サンプルのために作成されていることを確認するために--fastqクラスタカウント0オプションを使用します。生成されたファイルは.gzの.fastq形式です。下流の分析( 図S1B)のためにそれを解凍します。
- 「アンアラインド'フォルダが生成された後、「アンアラインド'フォルダ( 図S1C)に行く。
- 変換プロセスを開始するために、図S1Dでコマンドを使用します。 「-j 'パラメータが使用され、CPUの数を提供しています。
- システムは変換処理を終えた後、「アンアラインド'フォルダ( 図S1E)の下で結果フォルダに移動します。
- <図S1Fでコマンドを使用します/ strong>の各サンプルフォルダの下.fastqファイルに.fastq.gzファイルを解凍します。
- .bcl(ベースコールファイル)CASAVAソフトウェア(イルミナ、バージョン1.8.2)を使用してファイルを.fastqにファイル変換します。
- 新規転写物を検出し、Tuxedoのスイート26を用いて発現レベルを評価します。
- RNA-seqが(から得UCSCのバージョンMM9、マウスの参照ゲノムに読み込み、ペアエンドにマップhttp://cufflinks.cbcb.umd.edu/igenomes.html使用しています)トップハットソフトウェアを使用します(バージョン1.3.3)27、ボウタイはマッパ(バージョン0.12.7)28をお読みください。トップハットは、発現量の推定精度を向上させる「-no-小説juncs」オプションが供給される。
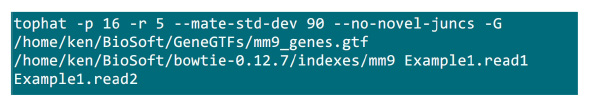
- マッピングプロセスが実装されたフォルダに.fastqファイルを置く。ペアエンドシーケンシングサンプルの2 .fastqファイル(Example1.read1、Example1.read2に名前変更)、(システム設定に応じてパラメータを調整する)マッピングを行うために、図S2でコマンドを使用があると仮定します。"-p"パラメータが使用され、CPUの数を提供しています。 「-r」と「-mate-STD-devの"パラメータは、ライブラリQCから得たり( 図S2は )整列のサブセットから読み取る推測することができる。
- カフスソフトウェアを使用してRNA転写物に読み込むマッピングされた(バージョン1.3.0)29を組み立てます。既知遺伝子(トップハットで使用されるのと同じ.gtfファイル)とトップハットによって生成.bamファイルの注釈ファイルを使用してファイル名を指定して実行カフス。
- トップハットは、実行が終了した後に、同じフォルダに、トランスクリプトームと推定転写産物の発現レベルを構築するためにカフスボタンを実行するために、図S3Aでコマンドを使用します。 「GenomeSeqMM9」フォルダ内の「mm9_repeatMasker.gtf 'とゲノム配列ファイルは、UCSCゲノムブラウザから得ることができる。
- その結果genes.exprとtranscripts.exprファイルが遺伝子や転写産物(アイソフォーム)の式の値が含まれています。コピーアンドペーストExcelへのファイルの内容をファイルやスプレッドシート·アプリケーション( 図S3B)で操作します。
- 小説転写物を同定するために、リファレンス「mm9_genes.gtf 'ファイルに生じた「transcripts.gtf'ファイルを比較するために、図S3Cでコマンドを使用します。
- 結果の.tmapファイルは、比較結果が含まれています。コピーしてExcelファイルにファイルの内容を貼り付け、スプレッドシートアプリケーションで操作します。クラスコードを持つ転写産物'U'が( 図S3D)提供されたファイル.gtfリファレンスに比べて「小説」として考えることができる。
注:値は0.1の下にある場合、下流の分析の利便性のために、0.1にFPKM値を設定します。
注: ステップ3.2.3 - 3.2.6は、新規転写産物'式の推定精度を向上させたい人のためのオプションです。マッピングおよびトランスクリプトームの構造をrにする必要があるため、これは非常に長い時間がかかる国連回以上。
- デフォルトのパラメータを使用して、トップハット実行し、 図S3Eでコマンドを使用して生成.gtfファイルへのカフスボタンを実行します。
- 図S3Fでコマンドを使用して、リファレンスゲノム.gtfファイルに結果の.gtfファイルを比較してください。
- ステップ3.2.2.4で説明したようになった.tmapファイルを解析します。コピーしてExcelファイルにファイルの内容を貼り付け、スプレッドシートアプリケーションで操作します。クラスコードを有する転写物uは提供されているファイル.gtf基準と比較して「新規」と考えることができる。
- ステップ3.2.5の後、ファイル.gtf基準として使用することができるフォルダ内.combined.gtfファイルが存在する。 ステップ3.2.1および3.2.2に記載したように、トップハットとカフスのセカンドランは、新規な転写産物のより正確なFPKM推定を得るために行うことができる。
- RNA-seqが(から得UCSCのバージョンMM9、マウスの参照ゲノムに読み込み、ペアエンドにマップhttp://cufflinks.cbcb.umd.edu/igenomes.html使用しています)トップハットソフトウェアを使用します(バージョン1.3.3)27、ボウタイはマッパ(バージョン0.12.7)28をお読みください。トップハットは、発現量の推定精度を向上させる「-no-小説juncs」オプションが供給される。
- differentiall検出DESeqパッケージ30を使用してyの発現された遺伝子。
- DESeqの入力は、生の読み取りカウントテーブルである。このような表を得るためには、HTSeqのウェブサイトからダウンロードすることができますHTSeq Pythonパッケージと一緒に配布htseqカウントスクリプトを使用( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) 。
- samtools、パイソン、およびhtseqカウントprogramsareがシステムにインストールされていることを確認してください。 図S4Aでコマンドを使用して、トップハット出力からの生のリードカウント数を取得します。
- 「Raw_Count_Table.txt「Excelを使用して、「ExperimentDesign.txt 'ファイルを準備します。 DESeq Rパッケージ( 図S4B)用の.txt形式でコンテンツをコピーして保存します。
- システム内のRプログラムをインストールします。ターミナルでは、タイプ「R」キーを押しENTER.A画面メッセージがappearasは、 図S4Cに示したでしょう。
- 「Raw_Cを読む図S4Dでコマンドを使用してRにount_Table.txt '、' ExperimentDesign.txt」。
- 図S4Eでコマンドを使用してDESeqパッケージをロードします。
- R( 図S4F)で因数分解条件。
- 正規化されたカウントテーブルの上に負の二項テストを実行するために、図S4Gでコマンドを使用します。
- .csvファイルに出力著しい差発現された遺伝子に、図S4Hでコマンドを使用します。
- DESeqの入力は、生の読み取りカウントテーブルである。このような表を得るためには、HTSeqのウェブサイトからダウンロードすることができますHTSeq Pythonパッケージと一緒に配布htseqカウントスクリプトを使用( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) 。
- Excelを使用して試料にわたってルックアップ転写因子」(TFS)FPKM値。 DE遺伝子テーブルとのTFテーブルと交差する。遺伝子は、両方のテーブルに属し示差的転写因子を発現する。
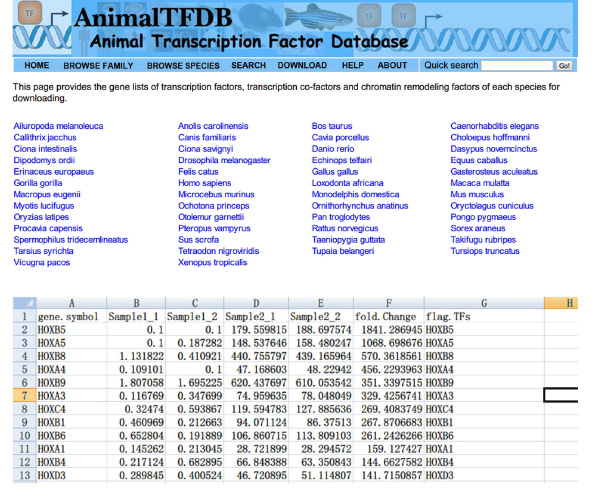
- ウェブサイトに行くhttp://www.bioguo.org/AnimalTFDB/download.phpおよび転写因子をダウンロードします。その後<(ExcelでDE転写因子を検索強い>図S5)。
- UCSCゲノムブラウザの可視化のための.bigwigファイルを生成する。
- ウェブサイトから「bedtools「ソフトウェアパッケージをダウンロードしhttps://github.com/arq5x/bedtools2し、システム31にソフトウェアをインストールします。 UCSCツール「bedGraphToBigWig」はウェブサイトからダウンロードしhttp://hgdownload.cse.ucsc.edu/admin/exe/およびシステム内のソフトウェアをインストールします。
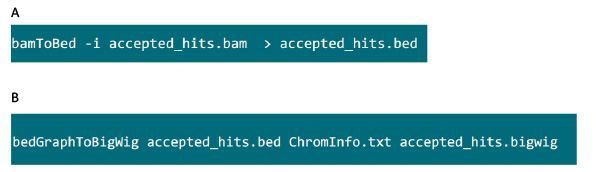
- .bamファイルを含むフォルダで、.bedファイルにトップハットによって生成された.bamファイルを変換するには、図S6Aでコマンドを使用します。
- .bedファイルが生成された後、.bigwigファイルを生成するために、図S6Bでコマンドを使用します。ファイル 'ChromInfo.txt'は、以下のURLから入手できます。ARGET = "_blank"> http://hgdownload.cse.ucsc.edu/goldenPath/mm9/database/chromInfo.txt.gz。
- UCSCゲノムブラウザ上のカスタムトラックを守ってください。 Webサイトを参照してくださいhttp://genome.ucsc.edu/goldenPath/help/customTrack.html UCSCゲノムブラウザを使用してカスタムトラックを表示する方法について。

図S1:CASAVAソフトウェアを使用してファイルを.fastqに変換する.bclファイル。

図S2:マッピングトップハットを使用してゲノムを参照するために読み取ります。
図S3:新規な転写産物の検出および発現量推定。

図S4:DESeqパッケージを使用して差分発現された遺伝子を呼び出す。

図S5:差次的に発現する転写因子の同定。

図S6:データ可視化のための変換マッピング結果。
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
リン- CD34 +およびCD34-リン-EML細胞において差次的に発現される遺伝子を分析するために、我々は、RNA配列の技術を使用した。 図1は、手順のワークフローを示している。磁気細胞選別による系統陰性細胞の単離後、我々は、LIN-SCA + CD34 +およびFACSアリアを使用してLIN-SCA-CD34-細胞を分離した。リン富化EML細胞を、抗CD34、抗のSca1および系統カクテル抗体で染色した。唯一のリニア細胞はのSca1及びCD34発現の分析のためにゲートした。二つの集団(SCA + CD34 +およびSCA-CD34- EML細胞)をFACS分析( 図2)6によって観察することができた。
細胞分離の後、我々は、それぞれCD34 +およびCD34-細胞からトータルRNAを抽出し、RNAの質を分析した。 RNA-配列データの精度は、主にRNA-配列ライブラリーの品質に依存して、総RNAの品質は、高品質のライブラリーを調製するために不可欠である。高品質のRNAサンプルは1〜OD 260/280の値を有するべきである。8および2.0。分光光度計を使用することに加えて、RNAの質は、さらにバイオアナライザーによって、より高い精度で評価した。 図3は、9.4に等しいRINを有する高品質RNA試料の結果を示す。 RIN値より大きい9のみ高品質の全RNAサンプルは、mRNA抽出およびその後のライブラリー構築の手順のために使用した。
リボソームRNAは、細胞中のRNAの最も豊富なタイプである。現在、二つの主な戦略は、rRNAの枯渇または正mRNAのポリアデニル化(ポリA mRNA)の選択は、ライブラリー構築の前に、標的RNAの濃縮のために使用される。非ポリアデニル化RNA種は、ポリA mRNAの選択中に失われます。対照的に、RiboMinusなどのrRNA枯渇法は、非ポリアデニル化RNA種を維持することができる。我々の研究の目的は、示差的に2つの細胞タイプで遺伝子をコードする発現さを探すためにこのようにして我々は、ライブラリconstru前に、標的RNAの濃縮のためにポリA mRNAの選択方法を使用しているction。ライブラリーの構築が終了した場合には、ライブラリー中のDNA断片の大きさは、バイオアナライザーを使用して配列決定する前に確認した。 図4は、約300bpでの断片サイズピークを有する良好な品質のライブラリーを示す。
続く工程では、ライブラリーは、ハイスループットシークエンシングに供した。原則的に、より長い読みの長さは、リードマッピングに役立つであろう。それは、リードにより重複遺伝子または遺伝子ファミリーメンバー間の類似度に複数の場所にマッピングされる確率を低減することができる。ペアエンドシーケンシング配列は、断片の両端からであるように、選択された読み取り長は、平均断片長の半分未満であるべきである。実験の主な目的ではなく転写物の構造を構築するの発現レベルを測定することである場合は、シングルエンドではあまりにも多くの情報を失うことなく、コストを低減することができる(75または100 bp)を読み取る。ペアエンド配列決定は、転写産物構造の構築と短いために、より便利です読み取り長は、コストを低減することができる。十分な資金が利用可能であるとき確かに、長く読ん長さが好ましい。
差次的発現分析のために、DESeq以外の多くの代替的なアルゴリズムが存在する。 32 cuffdiffという名前のカフスパッケージに含まれる1つもあります。 DESeqは、最も広く使用されているカウントベースのDE遺伝子解析アルゴリズムの一つである。負の二項分布 - DESeq法は十分に特徴付けられた統計モデルに基づいています。我々の経験では、DESeqはcuffdiffと比較し、より安定している。 cuffdiffの初期のバージョンは、多くの場合、DE遺伝子の有意に異なる数字を与える。そこで、ここではDE解析にDESeqを使用していました。
転写因子は、細胞運命決定に重要であるので、我々は、有意差次的に発現する転写因子33に焦点を当てた。のTFは、LIN-CD34 +およびLIN-CD34-間の1.5倍が検出されたとヒートマップ(FIGUR上に示されている>に変更E 5)2。注目すべきことに、リン- CD34 +細胞におけるTcf7の相対的な発現レベルは、リン-CD34-細胞のそれより100倍高い。したがってTcf7は EML細胞の自己複製と分化の調節に2 Tcf7の機能を確認するためにさらなるたChIP-シーケンシング(クロマチン免疫沈降および配列決定)分析と機能テストのために選択した。

図1:手順のワークフローリン、CD34 +およびリン-CD34-細胞は、磁気細胞分離システム、および蛍光活性化細胞選別法によって分離した。全RNAは、mRNA精製およびライブラリーの構築に続いて抽出した。ライブラリーの品質分析の後、サンプルを、ハイスループットシークエンシングに供した。データを解析し、差次的転写因子を発現させ同定された。

図2:LIN-CD34 +およびLIN-CD34- EML 6リニアEML細胞は、磁気細胞選別により濃縮した細胞の分離 。 LIN-細胞を抗CD34、抗のSca1および系統混合抗体で染色した。 LIN-細胞がCD34およびのSca1の発現についてゲーティングした。 LIN-CD34 + SCA +およびLIN-CD34-SCA- EML細胞集団を選別した。

図3:高品質の全RNAサンプルの代表的なトータルRNAの質は、バイオアナライザーにより評価した。 RNA完全性ナンバーは9.4(FU、蛍光単位)です。

図4:ペアエンドライブラリの断片サイズの範囲ザ·。 ライブラリーのDNAのサイズ分布は、バイオアナライザーを用いて分析した。ほとんどの断片は、250〜500塩基対のサイズの範囲内である。

図5:差次的に発現する転写因子のリン-CD34 +細胞およびリン-CD34-細胞2との間(> 1.5倍)は、各細胞型については、二つの独立した実験を行った。上方制御される遺伝子は赤色で示され、ダウンレギュレートされた遺伝子は、緑色で示されている。
表1:緩衝液および細胞培養培地 。
| ソフトウェア | 用法 | リファレンス | |||
| ボウタイ1.2.7 | マッピングのためのトップハットが使用する | [28] | |||
| トップハット1.3.3 | マッピングは、参照ゲノムに戻って読み込む | [27] | |||
| カフス1.3.0 | 転写物の構築および発現レベル推定 | [29] | |||
| DESeq 1.16.0 | 示差発現解析 | [30] | Bedtools 2.18 | .bedファイルに.bamファイルを変換する | [31] |
| bedGraphToBigWig | ファイルを.bigwigする.bedファイルを変換 | http://genome.ucsc.edu/ |
表2:データ分析のためのソフトウェアのリスト 。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
哺乳動物のトランスクリプトームは、34〜38は非常に複雑です。 RNA-配列技術はまた、遺伝子発現解析のための他の方法に勝る多くの利点を有する新規な転写産物の検出および一塩基変化の検出など 、トランスクリプトーム解析の研究においてますます重要な役割を果たしている。冒頭で述べたように、マイクロアレイのハイブリダイゼーションのアーチファクトを克服する新規の転写物のde novoを識別するために使用することができる。 RNAシークエンスの1つの制限は、サンガー配列と比較し、相対短い読み取り長である。しかし、シークエンシング技術の急速な改善と、長さが絶えず増加してお読みください。本稿では、マウスのEML細胞の自己再生および分化に潜在的な重要な調節因子を特定するためにこの技術を使用する詳細な方法を提供する。
このプロトコルの最初の重要なステップは、EML細胞培養である。 EMLは、造血前駆細胞株であり、それはあり得るがSCFを大量に増殖させた。 EML細胞の培養条件は通常の不死化細胞株よりも多くの注意が必要です。細胞が供給され、穏やかな操作で、定期的で継代されるべきである。そうでなければ細胞は、自己再生および分化のその特性が変化し、細胞死を受ける可能性がある。十分な細胞を収集した後の最初のステップとして、我々は、磁気活性化細胞ソーティングシステムを使用して、系統陰性細胞を単離した。その後、我々は、蛍光活性化セルソーティングを使用して、CD34 +およびCD34 - 細胞を分離した。 EMLの細胞は、通常のRNA抽出のために使用する前に、10未満の世代継代すると、CD34 +およびCD34-細胞の数は、分離後に類似していなければならない。 2集団は、細胞数が大きく異なる場合は、それが文化を破棄し、培養のための細胞株の別のチューブを再解凍することをお勧めします。
CD34 +およびCD34-細胞を分離した後、全RNA抽出は、このSTのための別の重要な一歩を行ったUDY。高品質のRNAが配列決定データの精度を約束する高品質のライブラリーの構築のためのベースである。この重要なステップでは、RNアーゼとの接触は避けるべきである。すべての試薬はRNaseフリーする必要があります。 RNAの取り扱いながら、常に手袋を着用することが重要です。高品質のRNAサンプルは、1.8と2.0の間のOD 260/280の値を有する。 RNAを含む水相を収集する際、RNAサンプルを持つ任意の有機相を運ぶしないように注意してください。そのようなRNA中のフェノール、クロロホルム等の任意の残留有機溶媒が1.65より低いOD260 / 280の値をもたらすであろう。 OD260 / 280の値が1.65未満である場合には、エタノールで再びRNAを沈殿させる。 75%エタノールで洗浄した後、過乾燥ないRNAペレットを行う。乾燥RNAペレットを完全にRNAの溶解性に影響を与え、RNAの低い収率につながる。
このプロトコルのための次の重要なステップは、ライブラリの準備である。トータルRNA抽出、汚染DNAの除去のためにDNアーゼを使用するステップの後のiDNAの混入が使用された全RNA量の誤った推定につながる可能性があるため、sが強く推奨。それはすぐにRNAを単離した後、下流の手順を実行することが推奨され、長期保存後、凍結融解手順ので、RNAは、ある程度劣化する。 RNA単離の後、後続のステップが直ちに実行できない場合は、-80℃でRNAを保存する。総RNAは、mRNA精製及びcDNA合成のために使用される前に、品質が常にチェックしなければならない。唯一の高品質のRNAは、ライブラリーの調製のために使用することができる。低品質または分解されたRNAを使用することで過剰表現3 '末端のにつながるかもしれない。シークエンシングの前に、ライブラリーの品質を最大シーケンシング効率を確保するために評価した。
データ解析部では、参照トランスクリプトなしカフリンクスの実行を実行した後、我々は二度目のファイルと実行トップハットとカフス.gtfリファレンスを形成することが知られて転写産物を有する新規転写物を組み合わせる。これは一度だけ実行されているよりも正確FPKM推定を提供するため、この2ラン手順は、推奨されます。データ分析の後、示差的に発現された遺伝子を同定した。下流の実験は、インビトロおよびインビボでの遺伝子の機能を検証するために実行することができる。我々の以前の出版物2では、有意に差次的に発現する転写因子を選択し、クロマチン免疫沈降法および配列決定法(ChIP-配列)を行うことにより、これらの因子のゲノム結合部位を同定した。さらに、我々はTcf7の機能的効果を試験するためのshRNAノックダウンアッセイを適用した。我々は、ダウンレギュレートされた遺伝子が有意にCD34 +細胞が濃縮されることが見出されたままTcf7ノックダウン細胞ではアップレギュレートされた遺伝子は、高度にCD34-細胞が濃縮された遺伝子であったことを見出した。したがって、Tcf7ノックダウン細胞の遺伝子発現プロファイルは、モデル系としてEMLセルを用いて、部分的に分化したCD34- state.Overall側にシフトRNA-配列決定技術および機能アッセイと相まって、我々は同定し、EML細胞の自己再生および分化の重要な調節因子としてTcf7を確認した。
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| Antibiotic-Antimycotic | Invitrogen | 15240-062 | BHK cell culture |
| Anti-Mouse CD34 FITC | eBioscience | 11-0341-81 | FACS sorting |
| Anti-Mouse Ly-6A/E (Sca-1) PE | eBioscience | 12-5981-81 | FACS sorting |
| APC Mouse Lineage Antibody Cocktail | BD Biosciences | 558074 | FACS sorting |
| BD FACSAria Cell Sorter | BD Biosciences | Special offer sysmtem | FACS sorting |
| Corning™ Cell Culture Treated Flasks 75 cm2 | Corning incorporated | 430641 | Cell culture |
| Corning™ Cell Culture Treated Flasks 25 cm2 | Corning incorporated | 430639 | Cell culture |
| Deoxyribonuclease I, Amplification Grade | Invitrogen | 18068-015 | Library preparation |
| DMEM | Invitrogen | 11965-092 | BHK cell culture |
| DPBS | Gibco | 14190 | Cell culture |
| HI FBS | Invitrogen | 16140071 | BHK cell culture |
| Horse Serum | Invitrogen | 16050-122 | EML cell culture |
| IMDM | HyClone | SH30228.02 | EML cell culture |
| L-Glutamine | Invitrogen | 25030-081 | Cell culture |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | 130-090-858 | Isolation of lineage negative cells |
| NanoVue Plus spectrophotometer | GE Healthcare | 28-9569-62 | Quality control |
| Thermo Scientific™ Napco™ 8000 Water-Jacketed CO2 Incubators | Thermo Scientific | 15-497-002 | Cell culture |
| Penicillin-Streptomycin | Invitrogen | 15140-122 | EML cell culture |
| TRIzol® Reagent | Invitrogen | 15596-018 | RNA exraction |
| TruSeq™ RNA Sample Prep Kit v2 -Set B (48 rxn) | Illumina | RS-122-2002 | Library preparation |
| 2100 Electrophoresis Bioanalyzer Instrument | Agilent | G2939AA | Quality control |
| 0.25% Trypsin-EDTA | Gibco | 25200 | Cell culture |
| 0.45 µm Syringe Filters | Nalgene | 190-2545 | Cell culture |
References
- Chambers, S. M., Goodell, M. A. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 3, 201-211 (2007).
- Wu, J. Q., et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS genetics. 8, (2012).
- Ye, Z. J., et al. Complex interactions in EML cell stimulation by stem cell factor and IL-3. Proceedings of the National Academy of Sciences of the United States of America. 108, 4882-4887 (2011).
- Tsai, S., Bartelmez, S., Sitnicka, E., Collins, S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 8, 2831-2841 (1994).
- Weiler, S. R., et al. D3: a gene induced during myeloid cell differentiation of Linlo c-Kit+ Sca-1(+) progenitor cells. Blood. 93, 527-536 (1999).
- Ye, Z. J., Kluger, Y., Lian, Z., Weissman, S. M. Two types of precursor cells in a multipotential hematopoietic cell line. Proc Natl Acad Sci U S A. 102, 18461-18466 (2005).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10, 57-63 (2009).
- Chu, Y., Corey, D. R. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic acid therapeutics. 22, 271-274 (2012).
- Hornett, E. A., Wheat, C. W. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC genomics. 13, 361 (2012).
- Eswaran, J., et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific reports. 3, 1689 (2013).
- Wang, E. T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470-476 (2008).
- Wu, J. Q., et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proceedings of the National Academy of Sciences of the United States of America. 107, 5254-5259 (2010).
- Loraine, A. E., McCormick, S., Estrada, A., Patel, K., Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant physiology. 162, 1092-1109 (2013).
- Edgren, H., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome biology. 12, 6 (2011).
- Ilott, N. E., Ponting, C. P. Predicting long non-coding RNAs using RNA sequencing. Methods. 63, 50-59 (2013).
- Sun, L., et al. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC bioinformatics. 13, 331 (2012).
- Luo, S. MicroRNA expression analysis using the Illumina microRNA-Seq Platform. Methods in molecular biology. 822, 183-188 (2012).
- Bolduc, F., Hoareau, C., St-Pierre, P., Perreault, J. P. In-depth sequencing of the siRNAs associated with peach latent mosaic viroid infection. BMC molecular biology. 11, 16 (2010).
- Chepelev, I., Wei, G., Tang, Q., Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic acids research. 37, 106 (2009).
- Djari, A., et al. Gene-based single nucleotide polymorphism discovery in bovine muscle using next-generation transcriptomic sequencing. BMC genomics. 14, 307 (2013).
- Murphy, D. Gene expression studies using microarrays: principles, problems, and prospects. Advances in physiology education. 26, 256-270 (2002).
- Chen, K., et al. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS one. 8, 72567 (2013).
- Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 18, 1509-1517 (2008).
- Ramskold, D., Kavak, E., Sandberg, R. How to analyze gene expression using RNA-sequencing data. Methods in molecular biology. 802, 259-274 (2012).
- Glenn, T. C. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 11, 759-769 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 7, 562-578 (2012).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25, 1105-1111 (2009).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 10, 25 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature. 28, 511-515 (2010).
- Anders, S., Huber, W. Differential expression analysis for sequence count data. Genome biology. 11, 106 (2010).
- Quinlan, A. R., Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841-842 (2010).
- Cheranova, D., et al. RNA-seq analysis of transcriptomes in thrombin-treated and control human pulmonary microvascular endothelial cells. J Vis Exp. , (2013).
- Zhang, H. M., et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research. 40, 144-149 (2012).
- Wu, J. Q., et al. Systematic analysis of transcribed loci in ENCODE regions using RACE sequencing reveals extensive transcription in the human genome. Genome Biol. 9, 3 (2008).
- Wu, J. Q., et al. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques. 36, 690-696 (2004).
- Wu, J. Q., Shteynberg, D., Arumugam, M., Gibbs, R. A., Brent, M. R. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 14, 665-671 (2004).
- Dewey, C., et al. Accurate identification of novel human genes through simultaneous gene prediction in human, mouse, and rat. Genome Res. 14, 661-664 (2004).
- Wu, J. Characterize Mammalian Transcriptome Complexity. , LAP Lambert Academic Publishing. (2011).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}