

Summary
استخدمت RNA-التسلسل والتحليلات المعلوماتية الحيوية لتحديد عوامل النسخ أعرب كبير وبشكل مختلف في لين-CD34 + ولين CD34- القطعان من EMLcells الماوس. هذه عوامل النسخ قد تلعب أدوارا مهمة في تحديد التبديل بين الخلايا لين لين CD34--CD34 + ومتباينة جزئيا تجديد الذات.
Abstract
وتستخدم الخلايا الجذعية المكونة للدم (HSCs) سريريا للعلاج زرع لإعادة بناء نظام المكونة للدم المريض في العديد من الأمراض مثل سرطان الدم وسرطان الغدد الليمفاوية. توضيح آليات السيطرة HSCs تجديد الذات والتمايز المهم للتطبيق HSCs للبحوث واستخدامات السريرية. ومع ذلك، فإنه من غير الممكن الحصول على كمية كبيرة من HSCs بسبب عدم قدرتها على التكاثر في المختبر. للتغلب على هذه العقبة، استخدمنا خط خلية نخاع المستمدة الماوس العظام، وقائمة الأدوية الأساسية (محمر، متعلق بالنخاع الشوكي، والليمفاوي) خط الخلية، كنظام نموذج لهذه الدراسة.
RNA-التسلسل (RNA تسلسل) قد استخدمت على نحو متزايد ليحل محل ميكروأري للدراسات التعبير الجيني. نحن هنا تقرير مفصل لطريقة استخدام التكنولوجيا-RNA بعدها للتحقيق في العوامل الرئيسية المحتملة في تنظيم خلية EML تجديد الذات والتمايز. وينقسم البروتوكول المقدمة في هذه الورقة إلى ثلاثة أجزاء. على قدم المساواة الأولر يشرح كيفية الثقافة خلايا EML، وفصل لين-CD34 + والخلايا لين CD34-. الجزء الثاني من البروتوكول يوفر إجراءات مفصلة لمجموع RNA إعداد وبناء مكتبة لاحق الإنتاجية العالية التسلسل. ويصف الجزء الأخير طريقة لتحليل البيانات RNA وبعدها يشرح كيفية استخدام البيانات لتحديد عوامل النسخ أعرب تفاضلي بين لين-CD34 + والخلايا لين CD34-. تم تحديد عوامل النسخ الأهم أعرب تفاضلي أن المنظمين الرئيسيين المحتملين السيطرة على خلية EML تجديد الذات والتمايز. في قسم مناقشة هذه الورقة، ونحن تسليط الضوء على الخطوات الرئيسية للأداء الناجح لهذه التجربة.
وباختصار، فإن هذه الورقة تقدم طريقة استخدام التكنولوجيا لتحديد تسلسل الحمض النووي الريبي التنظيمية المحتملة لتجديد الذات والتمايز في الخلايا EML. تتعرض العوامل الرئيسية المحددة لتحليل وظيفي المصب في المختبر وأنان الجسم الحي.
Introduction
الخلايا الجذعية المكونة للدم هي خلايا الدم النادرة التي يقيمون بصورة رئيسية في نخاع عظم مكانة الكبار. أنها هي المسؤولة عن إنتاج الخلايا المطلوبة لتجديد الدم والجهاز المناعي 1. كنوع من الخلايا الجذعية، HSCs قادرون على حد سواء تجديد الذات والتمايز. توضيح الآليات التي تتحكم في قرار مصير HSCs، إما نحو التجديد الذاتي أو التمايز، سوف نقدم توجيهات قيمة بشأن التلاعب HSCs لأبحاث أمراض الدم والاستخدام السريري 2. واحد المشكلة التي يواجهها الباحثون هي أن HSCs يمكن الحفاظ على وتوسعت في المختبر على نطاق محدود جدا. ويفرق الغالبية العظمى من ذريتها جزئيا في الثقافة 2.
من أجل تحديد الجهات التنظيمية الرئيسية التي تتحكم في عمليات تجديد الذات والتمايز على نطاق الجينوم واسعة، وكنا ماوس البدائية المكونة للدم خط خلية سلفية EML كنظام نموذج. عشروتم اشتقاق خط خلية من الفئران نخاع العظم 3،4. عندما تتغذى على عوامل النمو المختلفة، يمكن أن تفرق في خلايا EML محمر، النخاعي، والخلايا اللمفاوية في المختبر 5. الأهم من ذلك، يمكن نشر هذا خط الخلية بكميات كبيرة في الثقافة المتوسطة التي تحتوي على الخلايا الجذعية عامل (SCF) والإبقاء على تعدد الإمكانيات التي لا يزال. يمكن فصل الخلايا EML إلى مجموعات سكانية فرعية من تجديد الذات لين-SCA + CD34 + وتختلف جزئيا خلايا لين-SCA-CD34- على أساس علامات السطحية CD34 وSCA 6. على غرار المدى القصير HSCs، SCA + CD34 + الخلايا قادرة على تجديد الذات. عندما تعامل مع SCF، لين-SCA خلايا CD34 + + يمكن أن تتجدد بسرعة اختلط فيها لين-SCA خلايا CD34 + + ولين-SCA-CD34- وتستمر في التكاثر 6. السكان متشابهان في التشكل ولها مستويات مماثلة من مرنا ج-طقم 6 والبروتين. خلايا لين-SCA-CD34- قادرة على نشر في وسائل الإعلام التي تحتوي على IL-3 بدلا من SCF 3. Unveilinز المنظمين الرئيسيين في قرار مصير خلية EML سوف نقدم فهم أفضل للآليات الخلوية والجزيئية في التحول التنموي في وقت مبكر خلال الدم.
من أجل التحقيق في الخلافات الجزيئية الكامنة بين تجديد الذات لين-SCA + CD34 + وخلايا متباينة جزئيا لين-SCA-CD34-، استخدمنا تسلسل الحمض النووي الريبي لتحديد الجينات وأعرب تفاضلي. على وجه الخصوص، ونحن نركز على عوامل النسخ، وعوامل النسخ حاسمة في تحديد مصير الخلية. تسلسل الحمض النووي الريبي هو نهج وضعت مؤخرا والتي تستخدم قدرات الجيل القادم التسلسل (خ ع) تقنيات للصفحة الشخصية وتحديد الرنا كتب من الجينوم 7،8. باختصار، هو الحمض النووي الريبي مجموع بولي-A المختارة ومجزأة كما هو ثم تحويل القالب template.The RNA الأولي في [كدنا] باستخدام الناسخ العكسي. من أجل تعيين النصوص الحمض النووي الريبي كامل طول، وذلك باستخدام سليمة، RNA غير المتدهورة لبناء مكتبة [كدنا] هو المهم. لبورتشكل التسلسل، يتم إضافة محول متواليات محددة لطرفي [كدنا]. بعد ذلك، في معظم الحالات، يتم تضخيم جزيئات [كدنا] بواسطة PCR والتسلسل بطريقة عالية الإنتاجية.
بعد التسلسل، الناتجة يقرأ يمكن الانحياز إلى الجينوم مرجعية وقاعدة بيانات Transcriptome على. عدد يقرأ تلك الخريطة إلى الجين إشارة يحسب ويمكن استخدام هذه المعلومات لتقدير مستوى التعبير الجيني. ويمكن أيضا أن يتم تجميعها من جديد دون الجينوم إشارة، مما يتيح دراسة transcriptomes في الكائنات غير نموذج 9 يقرأ. كما تم استخدام تقنية الحمض النووي الريبي وما يليها لكشف الإسوية لصق 10-12، 13 رواية النصوص واندماج الجين 14. بالإضافة إلى الكشف عن الجينات المكودة للبروتين، يمكن أن تسلسل الحمض النووي الريبي أيضا أن تستخدم لكشف وتحليل مستوى الرواية نسخ من الرنا غير الترميز، مثل فترة طويلة غير الترميز الحمض النووي الريبي 15،16، 17 الرنا الميكروي، سيرنا الخ 18. بسبب رانه دقة هذه الطريقة، وقد استخدم ذلك للكشف عن الاختلافات النووية المنفردة 19،20.
قبل ظهور التكنولوجيا-RNA بعدها، كان ميكروأري الوسيلة الرئيسية المستخدمة لتحليل التعبير الجيني الشخصي. ويتم تصنيعه تحقيقات المصممة مسبقا وتعلق بعد ذلك إلى سطح صلب لتشكيل شريحة ميكروأري 21. يتم استخراج مرنا وتحويلها إلى [كدنا]. أثناء عملية النسخ العكسي، يتم دمج النيوكليوتيدات fluorescently المسمى في [كدنا] ويمكن تهجين [كدنا] على الشرائح ميكروأري. شدة الإشارة التي تم جمعها من بقعة معينة تعتمد على كمية [كدنا] ملزم لجنة التحقيق بشكل خاص على تلك البقعة 21. بالمقارنة مع التكنولوجيا RNA يليها، ميكروأري لديه العديد من القيود. أولا، يعتمد ميكروأري على المعرفة الموجودة مسبقا من الشرح الجينات، في حين تكنولوجيا RNA بعدها قادرة على كشف النصوص رواية النسبي في مستوى الخلفية عالية، الأمر الذي يحد من استخدامها عند جنرال الكتريكمستوى التعبير شمال شرق منخفض. الى جانب ذلك، تكنولوجيا RNA يليها ديها أعلى بكثير من المدى الديناميكي الكشف (8000 مرات) 7، في حين، بسبب الخلفية وإشارات تشبع، دقة ميكروأري محدودة لكل من الجينات وأعرب عالية ومتواضع 7،22. أخيرا، تحقيقات ميكروأري تختلف في الكفاءة التهجين، والتي جعل نتائج أقل موثوقية عند مقارنة مستويات التعبير النسبية للمحاضر مختلفة في عينة واحدة 23. على الرغم من تسلسل الحمض النووي الريبي لديه العديد من المزايا أكثر من ميكروأري، وتحليل بياناتها معقد. وهذا هو أحد الأسباب أن العديد من الباحثين لا يزالون يستخدمون ميكروأري بدلا من تسلسل الحمض النووي الريبي. ويلزم أدوات المعلوماتية الحيوية المختلفة لمعالجة البيانات وتحليل الحمض النووي الريبي يليها 24.
من بين العديد من الجيل المقبل من التسلسل (خ ع) المنصات، 454، البورشيد، الصلبة وايون سيل هم الأكثر استخداما على نطاق واسع. وكان 454 أول منصة تجارية NGS. خلافا لغيرها من المنصات التسلسلطول مثل البورشيد ومتين، ويولد 454 منصة تعد قراءة (متوسط 700 قاعدة يقرأ) 25. يعد يقرأ بشكل أفضل لتوصيف الأولي transcriptiome ترجع الزيادة إلى ارتفاع كفاءة تجميع 25. العيب الرئيسي للمنصة 454 هو التكلفة العالية في megabase التسلسل. البورشيد والمنصات الصلبة توليد يقرأ مع زيادة أعداد وأطوال قصيرة. التكلفة لكل megabase من تسلسل هو أقل بكثير من 454 منصة. بسبب الأعداد الكبيرة من يقرأ قصيرة لالبورشيد والمنصات الصلبة، تحليل بيانات أكثر كثافة حسابيا من ذلك بكثير. سعر الصك والكواشف لتسلسل لمنصة ايون سيل أرخص ووقت أقصر تسلسل 25. ومع ذلك، فإن نسبة الخطأ والتكلفة لكل megabase من تسلسل وأعلى بالمقارنة مع البورشيد والمنصات الصلبة. المنصات المختلفة لها مزاياها وعيوبها، وتتطلب أساليب مختلفة لتحليل البيانات. جيش التحرير الشعبى الصينىtform يجب أن يتم اختياره على أساس التسلسل الغرض وتوافر التمويل.
في هذه الورقة، ونحن نأخذ منصة البورشيد RNA تسلسل كمثال. كنا خلية EML كنظام نموذج للتحقيق المنظمين الرئيسيين في EML خلية تجديد الذات والتمايز، وفرت طرق مفصلة ل-RNA يليها بناء مكتبة وتحليل البيانات لحساب مستوى التعبير والكشف عن نسخة الرواية. أظهرنا في نشر رسائلنا السابقة أن الدراسة وما يليها-RNA في EML نظام نموذجي 2، عندما يقترن اختبار وظيفي (على سبيل المثال shRNA ضربة قاضية) توفير نهج قوي في فهم الآلية الجزيئية للمراحل المبكرة من التمايز للدم، ويمكن أن تكون بمثابة نموذج لتحليل خلية تجديد الذات والتمايز بشكل عام.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. EML الثقافة الخلية وفصل خلايا CD34 + لين ولين CD34- عن طريق خلية المغناطيسي نظام الفرز ومضان تنشيط الخلايا الفرز الطريقة
- إعداد الطفل الهامستر الكلى (BHK) مستنبت الخلايا الجذعية لجمع عامل الخلية:
- ثقافة خلايا BHK في DMEM المتوسط تحتوي على 10٪ FBS في 25 سم 2 قارورة (الجدول 1) عند 37 درجة مئوية و 5٪ CO 2 في حاضنة ثقافة الخلية.
- عندما تنمو الخلايا إلى 80-90٪ التقاء، وغسل خلايا مرة واحدة مع 10 مل من برنامج تلفزيوني. إضافة 5 مل من 0.25٪ التربسين-EDTA حل أحادي الطبقة واحتضان الخلايا لمدة 1-5 دقائق في درجة حرارة الغرفة (RT) حتى يتم فصل الخلايا.
- الماصة صعودا وهبوطا حل بلطف لتفريق كتل من الخلايا. إضافة 5 مل من DMEM الكامل إلى القارورة لوقف النشاط التربسين. جمع الخلايا بواسطة الطرد المركزي في 200 x ج لمدة 5 دقائق على RT.
- إزالة المتوسطة و resuspend بيليه خلية في 10 مل من BHK جديدة خلية ثقافة المتوسط. نقل 2 مل من تعليق الخلية من الخطوة 1.1.4 جديدة إلى 75 سم 2 القارورة وإضافة 48 مل من الطازجة مستنبت خلايا BHK إلى القارورة.
- ثقافة الخلايا BHK لمدة يومين وجمع مستنبت. مرور المتوسطة من خلال مرشح 0.45 ميكرون. تخزين المتوسطة في -20 درجة مئوية حتى استخدامها مرة أخرى.
- زراعة الخلايا EML:
- خلايا EML الثقافة (في تعليق) في EML وسط قاعدي يحتوي BHK خلية ثقافة المتوسط (الجدول 1) عند 37 درجة مئوية و 5٪ CO 2 في حاضنة ثقافة الخلية.
- الحفاظ على خلايا EML في كثافة الخلية منخفضة (0،5-5 × 10 5 خلية / مل) مع ذروة الكثافة أقل من 6 × 10 5 خلية / مل. انقسام الخلايا كل 2-3 أيام في نسبة 1: 5. خلايا مرور EML بلطف والتخلص من الثقافة بعد الركض لمدة 10 أجيال.
- نضوب خلايا إيجابية النسب:
- حصاد الخلايا EML بواسطة الطرد المركزي في 200 x ج FOص 5 دقائق ويغسل خلايا مرة واحدة مع برنامج تلفزيوني. جمع الخلايا بواسطة الطرد المركزي في 200 x ج لمدة 5 دقائق.
- resuspend الخلايا مع برنامج تلفزيوني وعدد الخلايا مع عدادة الكريات. تحديد تركيز الأجسام المضادة في خطوة الانفصال خلية اللاحقة وفقا لعدد من الخلايا (يرجى الرجوع إلى الإرشادات المقدمة من قبل مزود للنظام العزلة الخلية).
- عزل سلبية النسب (Lin-) الخلايا باستخدام كوكتيل نسب الأجسام المضادة (مزيج من الأجسام المضادة وحيدة النسيلة البيوتين مترافق CD5، CD45R (B220)، CD11b، ومكافحة GR-1 (لي-6G / C)، 7-4 وتير-119 ) وتنشيط خلايا الجهاز المغناطيسي والفرز وفقا لتعليمات الشركة الصانعة.
- فصل خلايا CD34 + لين ولين CD34-:
- تدور باستمرار خلايا Lin- من الخطوة 1.3.3 في 200 x ج لمدة 5 دقائق. resuspend الكرية الخلية مع برنامج تلفزيوني وعدد الخلايا مع عدادة الكريات.
- غسل الخلايا مرتين مع العازلة FACS وبيليه الخلايا في 200 x جلمدة 5 دقائق.
- تسمية خمسة 1.5 مل أنابيب microcentrifuge مع رقم 1، 2، 3، 4، 5 على التوالي. resuspend الخلايا مع 100 ميكرولتر FACS العازلة في 10 6 خلايا (10 6 خلايا في أنبوب).
- إضافة 1 ميكروغرام المضادة للماوس CD34 FITC الأجسام المضادة لأنبوب 1 و 2 و أنبوب خلط أنابيب بلطف.
- احتضان جميع الأنابيب في 4 درجة مئوية لمدة 1 ساعة في الظلام.
- إضافة 0.25 ميكروغرام من-PE مترافق الأجسام المضادة لمكافحة Sca1 و 20 ميكرولتر من الأجسام المضادة سلالته كوكتيل APC-مترافق إلى أنبوب 1، 0.25 ميكروغرام من PE-مترافق الأجسام المضادة لمكافحة Sca1 لأنبوب 3، و 20 ميكرولتر من APC-مترافق الأجسام المضادة سلالته كوكتيل ل أنبوب 4.
- خلط جميع أنابيب بلطف واحتضان الخلايا عند 4 درجة مئوية لمدة 30 دقيقة إضافية في الظلام.
- إضافة 300 ميكرولتر من العازلة FACS إلى الخلايا وتدور أسفل الخلايا في 200 x ج لمدة 5 دقائق.
- غسل الخلايا مع 500 ميكرولتر من العازلة FACS لثلاث مرات.
- resuspend الكرية الخلية في 500 ميكرولتر من FACS بوffer.
- استخدام الخلايا في أنابيب 2، 3، 4، و 5 لإقامة التعويض. عزل الخلايا لين-SCA + CD34 + ولين-SCA-CD34- في أنبوب 1 باستخدام نظام مراقبة الأصول الميدانية الأغنية.
2. إعداد RNA وبناء مكتبة للإنتاجية عالية التسلسل
- العزلة، والتحليل النوعي والكمي لجودة RNA:
- استخراج الحمض النووي الريبي مجموع من لين-CD34 + والخلايا لين CD34- على التوالي باستخدام TRIzol التالية بروتوكول المصنوعات.
- إزالة الملوثة باستخدام الحمض النووي ديوكسي ريبونيوكلياز الأول (الدناز الأول) التالية بروتوكول للتصنيع. اختياريا، تخزين الحمض النووي الريبي في -80 درجة مئوية في هذه الخطوة لاستخدامها مرة أخرى.
- تقييم نوعية الحمض النووي الريبي مجموع باستخدام Bioanalyzer وفقا للتعليمات التي يقدمها المورد. استخدام عينة الحمض النووي الريبي RNA النزاهة مع عدد (رين) الجعة من 9.
- مكتبة البناء والإنتاجية العالية التسلسل:
ملاحظة: يصف هذا البروتوكول RNA تسلسل باستخدام منصة البورشيد. إلىالتسلسل منصات أخرى، يطلب من أساليب إعداد المكتبة المختلفة.- استخدام 0،1-4 ميكروغرام من الحمض النووي الريبي مجموع جودة عالية لكل عينة لإعداد مكتبة. عادة 2 ميكروغرام من الحمض النووي الريبي مجموع يمكن استخلاصها من 5 10 خلايا EML.
- استخدام نظام إعداد عينة الحمض النووي الريبي التسلسل لتنقية RNA والتشرذم، الأول والثاني التوليف حبلا [كدنا]، وإصلاح النهاية 3 'ينتهي adenylation، محول ربط PCR والتضخيم، واتباع الإجراءات القياسية مفصلة من التعليمات الموفر.
- إيجابي تحديد بوليا مرنا باستخدام بنسبة ضئيلة من DT حبات مغناطيسية وتفتيت مرنا.
- إجراء النسخ العكسي باستخدام بادئات عشوائية للحصول على [كدنا] وبعد ذلك تجميع الشق الثاني من [كدنا لتوليد المزدوج تقطعت بهم السبل [كدنا].
- إزالة "يتدلى وملء 5 '3 يتدلى من قبل البلمرة DNA. أدينيلات 3 'ينتهي لمنع شظايا [كدنا] من ligating لبعضها البعض.
- إضافة محولات الفهرسة متعددة لطرفي dscDNA. أداء PCR لإثراء شظايا الحمض النووي.
- قياس A260 / A280 للحصول على معلومات حول تركيز مكتبة باستخدام مقياس الطيف الضوئي.
- تقييم جودة مكتبة وقياس مدى حجم شظايا الحمض النووي باستخدام Bioanalyzer.
3. تحليل البيانات
للإشارة من البرمجيات المستخدمة في هذا الجزء، يرجى الاطلاع على الجدول (2).
- معالجة ملف البيانات لتحليل المصب:
- تحويل .bcl (ملف دعوة القاعدة) ملف إلى ملف .fastq باستخدام برنامج CASAVA (البورشيد، الإصدار 1.8.2).
- اطلاق النار حتى على "محطة" في نظام لينكس. انتقل إلى المجلد الذي يحتوي على بيانات ملف البيانات من جهاز التسلسل البورشيد HiSeq2000. لنفترض المجلد النتيجة هي "NASboy1 / JiaqianLabData / HiSeq_RUN / 2013_07_11 / 130627_SN860_0309_A_2013-166_H0PW9ADXX / '، نوعفي الأمر في الشكل S1A، وأدخل مجلد البيانات.
- تثبيت CASAVA 1.8.2 في نظام لينكس. افترض أن outputfolder هو "محاذاتها"، استخدم الأمر في الشكل S1B لإعداد ملف التكوين للتحويل. استخدام الخيار --fastq الكتلة العد 0 لضمان يتم إنشاء ملف .fastq واحد فقط لكل عينة. ملف .fastq ولدت في شكل .gz. فك الضغط لتحليل المصب (الشكل S1B).
- بعد أن تم إنشاء مجلد "محاذاتها"، انتقل إلى المجلد "محاذاتها" (الشكل S1C).
- استخدم الأمر في الشكل S1D لبدء عملية تحويل. المعلمة "-j" وازم عدد وحدة المعالجة المركزية التي سيتم استخدامها.
- بعد الانتهاء من عملية تحويل نظام، انتقل إلى المجلد النتيجة ضمن مجلد "محاذاتها" (الشكل S1E).
- استخدم الأمر في الشكل S1F </ strong> لضغط الملف .fastq.gz في .fastq الملف تحت كل مجلد العينة.
- تحويل .bcl (ملف دعوة القاعدة) ملف إلى ملف .fastq باستخدام برنامج CASAVA (البورشيد، الإصدار 1.8.2).
- كشف النصوص رواية وتقييم مستوى التعبير باستخدام سهرة جناح 26:
- خريطة نهاية تقرن RNA تسلسل لجينوم يقرأ اشارة الماوس (UCSC نسخة MM9، تم الحصول عليها من http://cufflinks.cbcb.umd.edu/igenomes.html ) باستخدام برنامج Tophat (الإصدار 1.3.3) 27، والذي يستخدم وبووتي قراءة مخطط (الإصدار 0.12.7) 28. يتم تزويد Tophat مع "-لا-الرواية-juncs" خيار لتحسين دقة تقدير مستوى التعبير.
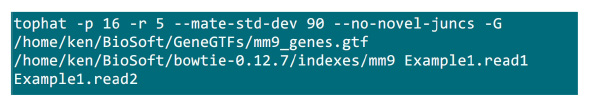
- وضع الملفات في مجلد .fastq حيث سيتم تنفيذ عملية رسم الخرائط. لنفترض أن هناك 2 ملفات .fastq (إعادة تسمية لExample1.read1، Example1.read2) لعينة التسلسل تقرن نهاية، استخدم الأمر في الشكل S2 للقيام تعيين (ضبط المعلمات وفقا لإعداد النظام).المعلمة "-p" وازم عدد وحدة المعالجة المركزية التي سيتم استخدامها. ويمكن الحصول على المعلمات "-R" و "-mate-STD-DEV" من مكتبة QC أو الاستدلال من مجموعة فرعية من محاذاة يقرأ (الشكل S2).
- تجميع معين يقرأ في النصوص RNA باستخدام برنامج أزرار أكمام (الإصدار 1.3.0) 29. أزرار تشغيل باستخدام ملف الشرح من الجينات المعروفة (نفس الملف .gtf يستخدمها Tophat) وملف .bam التي تنتجها Tophat.
- بعد الانتهاء Tophat على التوالي، في نفس المجلد، استخدم الأمر في الشكل S3A لتشغيل أزرار أكمام لبناء Transcriptome على النص وتقدير مستوى التعبير. و"mm9_repeatMasker.gtf" تسلسل الجينوم والملفات في المجلد "GenomeSeqMM9" يمكن الحصول عليها من UCSC متصفح الجينوم.
- وgenes.expr وtranscripts.expr الملفات الناتجة تحتوي على قيمة التعبير عن الجينات والنصوص (الإسوية). نسخ ولصقمحتويات الملف إلى ملف اكسل والتلاعب مع تطبيق جداول البيانات (الشكل S3B).
- استخدم الأمر في الشكل S3C لمقارنة الملف الناتج "transcripts.gtf" إلى مرجعية "mm9_genes.gtf" ملف من أجل تحديد النصوص الجديدة.
- يحتوي الملف .tmap الناتجة نتيجة المقارنة. نسخ ولصق محتويات الملف إلى ملف Excel والتعامل مع تطبيق جدول البيانات. النصوص مع رمز الطبقة "U" يمكن اعتبار رواية "مقارنة مرجعية .gtf ملف المقدمة (الشكل S3D).
ملاحظة: للحصول على الراحة تحليل المصب، تعيين قيم FPKM إلى 0.1 إذا كانت القيم هي تحت 0.1.
ملاحظة: الخطوة 3.2.3 - 3.2.6 اختيارية بالنسبة لأولئك الذين يرغبون في تحسين دقة التعبير تقدير النصوص الجديدة. هذا سوف يستغرق وقتا أطول بكثير، لأن رسم الخرائط والبناء Transcriptome على أن يكونوا صالأمم المتحدة أكثر من مرة.
- تشغيل Tophat استخدام المعلمات الافتراضية ثم قم بتشغيل أزرار أكمام لإنشاء ملف .gtf باستخدام الأمر في الشكل S3E.
- مقارنة الملف .gtf مما أدى إلى ملف .gtf الجينوم مرجعية باستخدام الأمر في الشكل S3F.
- تحليل الملف .tmap أدى كما هو موضح في الخطوة 3.2.2.4. نسخ ولصق محتويات الملف إلى ملف Excel والتعامل مع تطبيق جدول البيانات. النصوص مع رمز الطبقة "U" يمكن اعتبار رواية "مقارنة مرجعية .gtf ملف المقدمة.
- بعد الخطوة 3.2.5، وهناك ملف .combined.gtf في المجلد الذي يمكن استخدامه مثل ملف .gtf المرجعية. والجولة الثانية من Tophat وأزرار يمكن أن يؤديها كما هو موضح في الخطوة 3.2.1 و 3.2.2 للحصول على تقدير FPKM أكثر دقة من النصوص الجديدة.
- خريطة نهاية تقرن RNA تسلسل لجينوم يقرأ اشارة الماوس (UCSC نسخة MM9، تم الحصول عليها من http://cufflinks.cbcb.umd.edu/igenomes.html ) باستخدام برنامج Tophat (الإصدار 1.3.3) 27، والذي يستخدم وبووتي قراءة مخطط (الإصدار 0.12.7) 28. يتم تزويد Tophat مع "-لا-الرواية-juncs" خيار لتحسين دقة تقدير مستوى التعبير.
- كشف differentiallأعرب ذ الجينات باستخدام حزمة DESeq 30.
- مدخلات DESeq هو جدول التهم قراءة الخام. للحصول على مثل هذا الجدول، استخدام البرنامج النصي htseq العد توزيعها مع حزمة HTSeq بيثون التي يمكن تحميلها من موقع HTSeq ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- ضمان samtools، الثعبان، وhtseq العد programsare المثبتة في النظام. الحصول على الخام أرقام العد قراءة من الناتج tophat باستخدام الأمر في الشكل S4A.
- إعداد "Raw_Count_Table.txt، ملفات" ExperimentDesign.txt "باستخدام Excel. نسخ وحفظ المحتوى في الشكل. TXT لحزمة DESeq R (الشكل S4B).
- تثبيت برنامج R في النظام. في المحطة، وأظهرت سوف appearas نوع 'R' وENTER.A الصحافة رسالة الشاشة في الشكل S4C.
- قراءة 'Raw_Count_Table.txt '،' ExperimentDesign.txt "إلى R باستخدام الأمر في الشكل S4D.
- تحميل حزمة DESeq باستخدام الأمر في الشكل S4E.
- حلل إلى عوامل الظروف في R (الشكل S4F).
- استخدم الأمر في الشكل S4G لتشغيل اختبار الثنائي سلبي على الطاولة العد تطبيع.
- استخدم الأمر في الشكل S4H لإخراج التفاضلية كبير من الجينات التي أعرب عنها في ملف .csv.
- مدخلات DESeq هو جدول التهم قراءة الخام. للحصول على مثل هذا الجدول، استخدام البرنامج النصي htseq العد توزيعها مع حزمة HTSeq بيثون التي يمكن تحميلها من موقع HTSeq ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
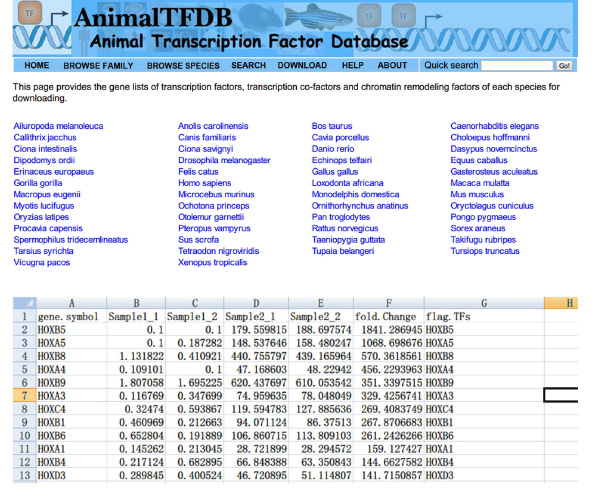
- بحث العوامل النسخ "(TFS) القيم FPKM عبر العينات باستخدام اكسل. تتقاطع DE الجدول الجينات والجدول TFS. الجينات تنتمي إلى وأعرب تفاضلي عوامل النسخ كلا الجدول.
- الذهاب إلى موقع http://www.bioguo.org/AnimalTFDB/download.php وتحميل عوامل النسخ. ثم المشاهده عوامل النسخ دي في اكسل (< قوي> الشكل S5).
- توليد ملف .bigwig عن التصور UCSC متصفح الجينوم.
- تحميل حزمة برامج "bedtools" من موقع https://github.com/arq5x/bedtools2 وتثبيت البرنامج في النظام 31. تحميل أدوات UCSC "bedGraphToBigWig" من موقع http://hgdownload.cse.ucsc.edu/admin/exe/ وتثبيت البرنامج في النظام.
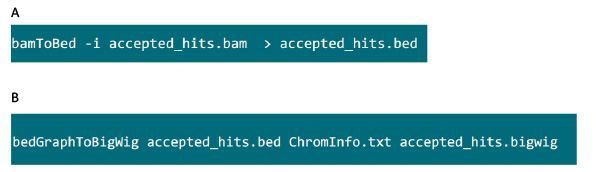
- في المجلد الذي يحتوي على الملف .bam، استخدم الأمر في الشكل S6A لتحويل ملف .bam الناتجة عن tophat في ملف .bed.
- بعد وينتج ملف .bed، استخدم الأمر في الشكل S6B لتوليد ملف .bigwig. الملف 'ChromInfo.txt "يمكن الحصول عليها من العنوان التالي:arget = "_blank"> http://hgdownload.cse.ucsc.edu/goldenPath/mm9/database/chromInfo.txt.gz.
- مراقبة مسار مخصص على UCSC متصفح الجينوم. الرجوع إلى الموقع http://genome.ucsc.edu/goldenPath/help/customTrack.html على كيفية عرض مسار مخصص باستخدام UCSC متصفح الجينوم.

الرقم S1: تحويل ملف .bcl ل.fastq ملف باستخدام برنامج CASAVA.

الرقم S2: رسم الخرائط يقرأ مرجع الجينوم باستخدام Tophat.
الرقم S3: الكشف عن النصوص رواية وتقدير مستوى التعبير.

الرقم S4: دعوة التفاضلية باستخدام الجينات أعرب DESeq الحزمة.

الرقم S5: تحديد عوامل النسخ أعرب تفاضلي.

الرقم S6: تحويل الخرائط نتيجة لتصور البيانات.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
من أجل تحليل الجينات وأعرب تفاضلي في لين-CD34 + والخلايا لين CD34- EML، استخدمنا التكنولوجيا-RNA يليها. ويظهر الشكل 1 سير الإجراءات. بعد عزل الخلايا النسب السلبية من قبل الفرز خلية المغناطيسي، وانفصلنا لين-SCA CD34 + + والخلايا لين-SCA-CD34- باستخدام نظام مراقبة الأصول الميدانية الأغنية. كانت ملطخة خلايا EML لين المخصب مع مكافحة CD34، ومكافحة Sca1 والأجسام المضادة كوكتيل النسب. تم بوابات خلايا Lin- فقط لتحليل Sca1 وCD34 التعبير. ويمكن ملاحظة اثنين من السكان (SCA CD34 + + والخلايا SCA-CD34- EML) من خلال تحليل FACS (الشكل 2) 6.
بعد فصل الخلايا، فإننا استخراج الحمض النووي الريبي مجموع من CD34 + والخلايا CD34- على التوالي وتحليل نوعية الحمض النووي الريبي. دقة البيانات RNA يليها يعتمد إلى حد كبير على نوعية مكتبة RNA يليها ونوعية الحمض النووي الريبي مجموع أمرا حيويا لإعداد مكتبة جودة عالية. وينبغي أن عينة الحمض النووي الريبي جودة عالية وقيمة 260/280 OD بين 1.8 و 2.0. بالإضافة إلى استخدام مقياس الطيف الضوئي، وتم تقييم نوعية RNA كذلك مع مزيد من الدقة بواسطة Bioanalyzer الشكل 3 يظهر نتيجة عينة الحمض النووي الريبي جودة عالية مع رين تساوي 9.4. واستخدمت جودة عالية عينة الحمض النووي الريبي مجموع القيمة فقط مع رين أكبر من 9 لاستخراج مرنا وإجراءات بناء مكتبة اللاحقة.
الريباسي RNA هو النوع الأكثر وفرة من الحمض النووي الريبي في الخلايا. حاليا اثنين من الاستراتيجيات الرئيسية، ونضوب الريباسي أو إيجابا اختيار مذيل بعديد الأدينيلات مرنا (بولي-A مرنا)، وتستخدم لتخصيب اليورانيوم من الحمض النووي الريبي الهدف قبل بناء المكتبة. فقدت الأنواع RNA غير مذيل بعديد الأدينيلات خلال اختيار بولي-A مرنا. في المقابل، يمكن أن أساليب استنزاف الريباسي مثل RiboMinus الحفاظ على الأنواع غير مذيل بعديد الأدينيلات RNA. الغرض من دراستنا هو البحث عن الجينات وأعرب تفاضلي الترميز في أنواع الخلايا اثنين، وبالتالي استخدمنا-A بولي طريقة اختيار مرنا لإثراء الرنا الهدف قبل constru المكتبةction. حين انتهى بناء المكتبة، تم فحص حجم شظايا الحمض النووي في المكتبة قبل تسلسل باستخدام Bioanalyzer الشكل 4 يوضح مكتبة نوعية جيدة مع قمم حجم شظية في حوالي 300 شركة بريتيش بتروليوم.
في خطوة لاحقة، تعرضت المكتبة للالإنتاجية العالية التسلسل. من حيث المبدأ، فإن طول أطول قراءة تكون مفيدة لرسم الخرائط للقراءة. يمكن أن يقلل احتمال أن يتم تعيين لقراءة مواقع متعددة بسبب التشابه بين الجينات مكررة أو أفراد الأسرة الجينات. كما تسلسل تسلسل نهاية الزوج هي من طرفي شظايا، يجب أن يكون طول القراءة المختارة أقل من نصف متوسط طول شظايا. إذا كان الهدف الرئيسي من التجربة هو قياس مستوى التعبير بدلا من بناء بنية النص، نهاية واحدة قراءة (75 أو 100 بي بي) يمكن أن تقلل من التكاليف دون أن تفقد الكثير من المعلومات. إقران نهاية التسلسل هو أكثر فائدة لبناء بنية النص وأقصرقراءة طول يمكن استخدامها للحد من التكاليف. بالتأكيد، عندما التمويل الكافي هو متاح، ويفضل طول تعد القراءة.
لتحليل التعبير التفاضلي، هناك العديد من الخوارزميات بديلة أخرى من DESeq. وهناك أيضا واحد تضمينها في حزمة أزرار أكمام اسمه cuffdiff 32. DESeq هو واحد من أكثر تستخدم على نطاق واسع العد مقرها DE خوارزميات تحليل الجينات. ويستند أسلوب DESeq على نموذج إحصاءات تتميز أيضا - توزيع ذي الحدين السلبي. في تجربتنا، DESeq أكثر استقرارا مقارنة cuffdiff. الإصدارات القديمة من cuffdiff غالبا ما تعطي أرقام مختلفة بشكل كبير من الجينات دي. لذا كنا DESeq لتحليل DE هنا.
بسبب عوامل النسخ حاسمة لتحديد مصير الخلية، وركزنا على النسخ بشكل ملحوظ أعرب تفاضلي العوامل 33. غيرت TFS> 1.5 أضعاف بين لين-CD34 + ولين CD34- عثر وترد على heatmap (شملت رقمه 5) (2). والجدير بالذكر أن مستوى التعبير النسبي Tcf7 في لين CD34 + الخلايا أكثر من 100 أضعاف أعلى من ذلك في الخلايا لين CD34-. وهكذا تم اختيار Tcf7 لمزيد الشذرة التسلسل (مناعي لونين وتسلسلها) تحليل واختبار وظيفي لتأكيد وظيفة Tcf7 'S في تنظيم EML خلية تجديد الذات والتمايز 2.

تم فصل سير العمل في إجراءات لين-CD34 + ولين CD34- الخلايا عن طريق نظام الفصل المغناطيسي الخلية والخلية مضان تنشيط طريقة الفرز: الرقم 1. تم استخراج الحمض النووي الريبي مجموع تليها تنقية مرنا وبناء المكتبة. بعد تحليل نوعية المكتبة، وتعرضوا لعينات عالية الإنتاجية التسلسل. وقد تم تحليل البيانات وأعرب تفاضلي عوامل النسخ تم تحديدها.

الشكل 2: فصل لين-CD34 + ولين CD34- EML خلايا تم إثراء 6 خلايا Lin- EML قبل الفرز خلية المغناطيسي. كانت ملطخة خلايا Lin- مع مكافحة CD34، ومكافحة Sca1 والأجسام المضادة خليط النسب. تم بوابات خلايا Lin- للتعبير عن CD34 وSca1. تم فرز لين-CD34 + + SCA والسكان خلية لين-CD34-SCA- EML.

الرقم 3: ممثل ذات جودة عالية عينة الحمض النووي الريبي مجموع وتم تقييم نوعية الحمض النووي الريبي مجموع من قبل Bioanalyzer. الحمض النووي الريبي عدد النزاهة هي 9.4 (FU، الإسفار وحدات).

الشكل 4: نطاق حجم أجزاء من مكتبة المقترنة-إنهاء تم تحليل الحمض النووي توزيع حجم المكتبة باستخدام Bioanalyzer. معظم الشظايا هي ضمن نطاق حجم 250-500 مضت.

الرقم 5: أعرب تفاضلي عوامل النسخ (> 1.5 أضعاف) بين لين-CD34 + الخلايا وخلايا لين CD34- 2 لكل نوع من الخلايا، وأجريت تجربتين مستقلة. يشار يصل ينظم الجينات كما يشار إلى اللون الأحمر والجينات تنظم أسفل مثل اللون الأخضر.
الجدول 1: المخازن وخلية ثقافة وسائل.
| البرمجيات | استعمال | إشارة | |||
| ربطة 1.2.7 | يستخدمها Tophat لرسم الخرائط | [28] | |||
| 1.3.3 Tophat | يقرأ الخرائط إلى الجينوم مرجعية | [27] | |||
| أزرار أكمام 1.3.0 | بناء النصوص وتقدير مستوى التعبير | [29] | |||
| DESeq 1.16.0 | تحليل التعبير التفاضلي | [30] | Bedtools 2.18 | تحويل ملف .bam في ملف .bed | [31] |
| bedGraphToBigWig | تحويل ملف .bed ل.bigwig ملف | http://genome.ucsc.edu/ |
الجدول 2: قائمة من البرامج لتحليل البيانات.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Transcriptome على الثدييات معقد جدا 34-38. التكنولوجيا-RNA يليها تلعب دورا متزايد الأهمية في دراسات تحليل Transcriptome على، كشف النصوص رواية واحدة النوكليوتيدات اكتشاف الاختلاف وما إلى ذلك لديه العديد من المزايا أكثر من غيرها من الأساليب لتحليل التعبير الجيني. كما ذكر في المقدمة، فإنه يتغلب على التحف تهجين ميكروأري ويمكن استخدامها لتحديد النصوص الرواية من جديد. القيد واحدة من الحمض النووي الريبي التسلسل النسبي هو طول قصيرة قراءة مقارنة لسانجر التسلسل. ومع ذلك، مع التحسن السريع للتكنولوجيا التسلسل، وقراءة طول يتزايد باستمرار. في هذه الورقة، ونحن نقدم طرق تفصيلية لاستخدام هذه التكنولوجيا لتحديد المنظمين مفتاح المحتملة في الماوس خلية EML تجديد الذات والتمايز.
الخطوة الأساسية الأولى لهذا البروتوكول هي ثقافة الخلية EML. على الرغم من أن قائمة الأدوية الأساسية هو خط الخلية تمهيدا للدم ويمكن أن يكوننشر في كمية كبيرة مع SCF. حالة زراعة خلايا EML تتطلب المزيد من الاهتمام من خطوط الخلايا خلد المعتادة. ويجب أن تغذى الخلايا وpassaged على أساس منتظم مع عملية لطيف. وإلا فإن الخلايا قد يتغير في خصائصها على تجديد الذات والتمايز والخضوع لموت الخلايا. كخطوة أولى بعد جمع ما يكفي من خلايا، ونحن عزل الخلايا السلبية النسب باستخدام خلية تنشيط نظام الفرز المغناطيسي. ثم انفصلنا CD34 + والخلايا CD34- باستخدام مضان تنشيط الخلايا الفرز. و passaged الخلايا EML عادة أقل من 10 أجيال قبل استخدام لاستخراج الحمض النووي الريبي وأعداد CD34 + والخلايا CD34- يجب أن تكون مشابهة بعد الانفصال. إذا كان الشعبين تختلف اختلافا كبيرا في عدد الخلايا، فإنه من المستحسن أن تجاهل الثقافة وإعادة ذوبان الجليد أنبوب آخر من الأسهم خلية للثقافة.
بعد فصل CD34 + والخلايا CD34-، تم إجراء الكلي استخراج الحمض النووي الريبي، خطوة هامة أخرى لهذا الحادي وudy. عالية الجودة RNA هو الأساس لبناء مكتبة الجودة العالية، والتي تعد من دقة البيانات التسلسل. في هذه الخطوة حاسمة، وينبغي تجنب أي اتصال مع ريبونوكلياز. يجب ريبونوكلياز جميع الكواشف مجانا. من المهم أن ترتدي قفازات في جميع الأوقات أثناء التعامل مع الحمض النووي الريبي. عالية الجودة عينة الحمض النووي الريبي لديه OD 260/280 قيمة بين 1.8 و 2.0. عند جمع المرحلة المائية التي تحتوي على الحمض النووي الريبي، يجب الحرص على عدم تحمل أي مرحلة العضوية مع عينة الحمض النووي الريبي. ان أي المذيبات العضوية المتبقية مثل الفينول أو الكلوروفورم في الحمض النووي الريبي يؤدي إلى OD260 / 280 قيمة أقل من 1.65. إذا كان OD260 / 280 القيمة هي أقل من 1.65، يعجل RNA مرة أخرى مع الايثانول. بعد غسل مع 75٪ من الإيثانول، لا overdry بيليه RNA. سوف تجفيف بيليه RNA يؤثر تماما ذوبان من الحمض النووي الريبي ويؤدي إلى العائد المنخفض من الحمض النووي الريبي.
الخطوة الأساسية التالية لهذا البروتوكول هو إعداد مكتبة. بعد الكلي استخراج الحمض النووي الريبي، وهي خطوة من استخدام الدناز لإزالة الحمض النووي الملوث طS يوصي بشدة، لأن تلوث الحمض النووي قد يؤدي إلى التقدير الخاطئ من كمية الحمض النووي الريبي مجموع المستخدمة. فمن المستحسن لتنفيذ الإجراء المصب مباشرة بعد عزل الحمض النووي الريبي، لأن التخزين بعد فترة طويلة الأجل وتجميد ذوبان الإجراء، سوف تتحلل RNA إلى حد ما. إذا كانت الخطوات اللاحقة بعد عزل الحمض النووي الريبي لا يمكن أن يؤديها على الفور، وتخزين الحمض النووي الريبي في -80 درجة مئوية. قبل استخدام الحمض النووي الريبي مجموع لتنقية مرنا والتوليف [كدنا]، ينبغي دائما أن يتم التحقق من الجودة. فقط جودة عالية RNA يمكن استخدامها لإعداد مكتبة. استخدام منخفضة الجودة أو RNA المتدهورة قد تؤدي إلى الإفراط في تمثيل 3 'ينتهي. قبل التسلسل، وتم تقييم نوعية المكتبة لضمان أقصى قدر من الكفاءة التسلسل.
في الجزء تحليل البيانات، وبعد أداء سلسلة من أزرار أكمام دون Transcriptome على المرجعية، ونحن الجمع بين النصوص الجديدة مع النصوص المعروفة لتشكيل مرجعية .gtf ملف وتشغيل Tophat وأزرار القمصان للمرة الثانية.ويوصى هذا الإجراء تديرها اثنين، لأن هذا التقدير تقديم FPKM أكثر دقة من تشغيل مرة واحدة فقط. بعد تحليل البيانات، وقد تم تحديد الجينات وأعرب تفاضلي. تجارب المصب لا يمكن أن يؤديها للتحقق من صحة وظيفة الجينات في المختبر وفي الجسم الحي. لدينا في السابق نشر 2، اخترنا عوامل النسخ بشكل ملحوظ أعرب تفاضلي وحدد موقع ملزم الجينوم هذه العوامل عن طريق أداء مناعي لونين والتسلسل (رقاقة تسلسل). بالإضافة إلى ذلك، طبقنا فحص shRNA ضربة قاضية لاختبار تأثير وظيفي من Tcf7. وجدنا أن في الخلايا Tcf7 ضربة قاضية، كانت الجينات يصل ينظم الجينات التخصيب في الخلايا CD34-، في حين تم العثور على جينات أسفل التنظيم لتخصيبه بدرجة كبيرة في CD34 + الخلايا. وبالتالي، فإن الملف التعبير الجيني للخلايا ضربة قاضية Tcf7 تحول نحو متباينة جزئيا CD34- state.Overall، وذلك باستخدام الخلايا EML كنظام نموذجإلى جانب التكنولوجيا RNA-التسلسل والمقايسات الفنية، حددنا وأكد Tcf7 كمنظم مهم من خلية EML تجديد الذات والتمايز.
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| Antibiotic-Antimycotic | Invitrogen | 15240-062 | BHK cell culture |
| Anti-Mouse CD34 FITC | eBioscience | 11-0341-81 | FACS sorting |
| Anti-Mouse Ly-6A/E (Sca-1) PE | eBioscience | 12-5981-81 | FACS sorting |
| APC Mouse Lineage Antibody Cocktail | BD Biosciences | 558074 | FACS sorting |
| BD FACSAria Cell Sorter | BD Biosciences | Special offer sysmtem | FACS sorting |
| Corning™ Cell Culture Treated Flasks 75 cm2 | Corning incorporated | 430641 | Cell culture |
| Corning™ Cell Culture Treated Flasks 25 cm2 | Corning incorporated | 430639 | Cell culture |
| Deoxyribonuclease I, Amplification Grade | Invitrogen | 18068-015 | Library preparation |
| DMEM | Invitrogen | 11965-092 | BHK cell culture |
| DPBS | Gibco | 14190 | Cell culture |
| HI FBS | Invitrogen | 16140071 | BHK cell culture |
| Horse Serum | Invitrogen | 16050-122 | EML cell culture |
| IMDM | HyClone | SH30228.02 | EML cell culture |
| L-Glutamine | Invitrogen | 25030-081 | Cell culture |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | 130-090-858 | Isolation of lineage negative cells |
| NanoVue Plus spectrophotometer | GE Healthcare | 28-9569-62 | Quality control |
| Thermo Scientific™ Napco™ 8000 Water-Jacketed CO2 Incubators | Thermo Scientific | 15-497-002 | Cell culture |
| Penicillin-Streptomycin | Invitrogen | 15140-122 | EML cell culture |
| TRIzol® Reagent | Invitrogen | 15596-018 | RNA exraction |
| TruSeq™ RNA Sample Prep Kit v2 -Set B (48 rxn) | Illumina | RS-122-2002 | Library preparation |
| 2100 Electrophoresis Bioanalyzer Instrument | Agilent | G2939AA | Quality control |
| 0.25% Trypsin-EDTA | Gibco | 25200 | Cell culture |
| 0.45 µm Syringe Filters | Nalgene | 190-2545 | Cell culture |
References
- Chambers, S. M., Goodell, M. A. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 3, 201-211 (2007).
- Wu, J. Q., et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS genetics. 8, (2012).
- Ye, Z. J., et al. Complex interactions in EML cell stimulation by stem cell factor and IL-3. Proceedings of the National Academy of Sciences of the United States of America. 108, 4882-4887 (2011).
- Tsai, S., Bartelmez, S., Sitnicka, E., Collins, S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 8, 2831-2841 (1994).
- Weiler, S. R., et al. D3: a gene induced during myeloid cell differentiation of Linlo c-Kit+ Sca-1(+) progenitor cells. Blood. 93, 527-536 (1999).
- Ye, Z. J., Kluger, Y., Lian, Z., Weissman, S. M. Two types of precursor cells in a multipotential hematopoietic cell line. Proc Natl Acad Sci U S A. 102, 18461-18466 (2005).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10, 57-63 (2009).
- Chu, Y., Corey, D. R. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic acid therapeutics. 22, 271-274 (2012).
- Hornett, E. A., Wheat, C. W. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC genomics. 13, 361 (2012).
- Eswaran, J., et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific reports. 3, 1689 (2013).
- Wang, E. T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470-476 (2008).
- Wu, J. Q., et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proceedings of the National Academy of Sciences of the United States of America. 107, 5254-5259 (2010).
- Loraine, A. E., McCormick, S., Estrada, A., Patel, K., Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant physiology. 162, 1092-1109 (2013).
- Edgren, H., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome biology. 12, 6 (2011).
- Ilott, N. E., Ponting, C. P. Predicting long non-coding RNAs using RNA sequencing. Methods. 63, 50-59 (2013).
- Sun, L., et al. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC bioinformatics. 13, 331 (2012).
- Luo, S. MicroRNA expression analysis using the Illumina microRNA-Seq Platform. Methods in molecular biology. 822, 183-188 (2012).
- Bolduc, F., Hoareau, C., St-Pierre, P., Perreault, J. P. In-depth sequencing of the siRNAs associated with peach latent mosaic viroid infection. BMC molecular biology. 11, 16 (2010).
- Chepelev, I., Wei, G., Tang, Q., Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic acids research. 37, 106 (2009).
- Djari, A., et al. Gene-based single nucleotide polymorphism discovery in bovine muscle using next-generation transcriptomic sequencing. BMC genomics. 14, 307 (2013).
- Murphy, D. Gene expression studies using microarrays: principles, problems, and prospects. Advances in physiology education. 26, 256-270 (2002).
- Chen, K., et al. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS one. 8, 72567 (2013).
- Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 18, 1509-1517 (2008).
- Ramskold, D., Kavak, E., Sandberg, R. How to analyze gene expression using RNA-sequencing data. Methods in molecular biology. 802, 259-274 (2012).
- Glenn, T. C. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 11, 759-769 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 7, 562-578 (2012).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25, 1105-1111 (2009).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 10, 25 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature. 28, 511-515 (2010).
- Anders, S., Huber, W. Differential expression analysis for sequence count data. Genome biology. 11, 106 (2010).
- Quinlan, A. R., Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841-842 (2010).
- Cheranova, D., et al. RNA-seq analysis of transcriptomes in thrombin-treated and control human pulmonary microvascular endothelial cells. J Vis Exp. , (2013).
- Zhang, H. M., et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research. 40, 144-149 (2012).
- Wu, J. Q., et al. Systematic analysis of transcribed loci in ENCODE regions using RACE sequencing reveals extensive transcription in the human genome. Genome Biol. 9, 3 (2008).
- Wu, J. Q., et al. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques. 36, 690-696 (2004).
- Wu, J. Q., Shteynberg, D., Arumugam, M., Gibbs, R. A., Brent, M. R. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 14, 665-671 (2004).
- Dewey, C., et al. Accurate identification of novel human genes through simultaneous gene prediction in human, mouse, and rat. Genome Res. 14, 661-664 (2004).
- Wu, J. Characterize Mammalian Transcriptome Complexity. , LAP Lambert Academic Publishing. (2011).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}