

Summary
RNA-sequenziamento e bioinformatica analisi sono stati utilizzati per identificare i fattori di trascrizione espressi in maniera significativa e differenziale in Lin-CD34 + e CD34- Lin-sottopopolazioni di EMLcells mouse. Questi fattori di trascrizione potrebbero svolgere un ruolo importante nel determinare lo switch tra le cellule Lin-CD34- auto-rinnovamento Lin-CD34 + e parzialmente differenziato.
Abstract
Le cellule staminali ematopoietiche (CSE) sono utilizzati per il trattamento clinico del trapianto di ricostruire il sistema ematopoietico di un paziente in molte malattie come la leucemia e linfoma. Chiarire i meccanismi che controllano HSC self-renewal e la differenziazione è importante per l'applicazione di cellule staminali emopoietiche per la ricerca e usi clinici. Tuttavia, non è possibile ottenere grandi quantità di CSE causa della loro incapacità a proliferare in vitro. Per superare questo ostacolo, abbiamo utilizzato una linea cellulare derivata midollo osseo di topo, la linea cellulare EML (eritroide, mieloide, e linfocitaria), come sistema modello per questo studio.
RNA-sequencing (RNA-Seq) è stata sempre più utilizzata per sostituire microarray per studi di espressione genica. Riportiamo qui un metodo dettagliato di utilizzare la tecnologia RNA-Seq per indagare i potenziali fattori chiave nella regolazione delle cellule EML self-renewal e la differenziazione. Il protocollo fornito in questo documento è diviso in tre parti. La prima part spiega come la cultura cellule EML e separato Lin-CD34 + e le cellule Lin-CD34-. La seconda parte del protocollo offre procedure dettagliate per la preparazione di RNA totale e la costruzione della libreria successiva per alta sequenziamento. L'ultima parte descrive il metodo per l'analisi dei dati di RNA-Seq e spiega come utilizzare i dati per identificare i fattori di trascrizione espressi in modo differenziale tra Lin-CD34 + e le cellule Lin-CD34-. I fattori di trascrizione più significativamente differenzialmente espressi sono stati identificati come potenziali regolatori chiave di controllo delle cellule EML self-renewal e la differenziazione. Nella sezione di discussione di questo documento, si segnalano i passaggi chiave per performance di successo di questo esperimento.
In sintesi, questo lavoro offre un metodo di utilizzare la tecnologia RNA-Seq per identificare i potenziali regolatori di self-renewal e la differenziazione in cellule EML. I fattori chiave individuati sono sottoposti ad analisi funzionale a valle in vitro ed in vivo.
Introduction
Le cellule staminali ematopoietiche sono globuli rari che si trovano soprattutto nel midollo osseo degli adulti nicchia. Essi sono responsabili per la produzione di cellule necessarie per ricostituire il sangue e il sistema immunitario 1. Come una sorta di cellule staminali, cellule staminali emopoietiche sono in grado sia di auto-rinnovamento e differenziazione. Chiarire i meccanismi che controllano la decisione destino delle cellule staminali emopoietiche, nei confronti sia self-renewal o differenziazione, offrirà preziose indicazioni sulla manipolazione di cellule staminali emopoietiche per ricerche di malattie del sangue e l'utilizzo clinico 2. Un problema affrontato dai ricercatori è che CSE possono essere mantenuti e espanse in vitro in misura molto limitata; la maggior parte della loro progenie sono parzialmente differenziati in coltura 2.
Al fine di identificare regolatori chiave che controllano i processi di auto-rinnovamento e differenziazione in scala tutto il genoma, abbiamo usato un mouse primitiva linea di cellule progenitrici ematopoietiche EML come sistema modello. Thè linea cellulare è stata derivata da midollo osseo murino 3,4. Quando alimentato con diversi fattori di crescita, le cellule EML possono differenziarsi in eritroide, mieloide e cellule linfoidi in vitro 5. È importante sottolineare che questa linea cellulare può essere propagato in grande quantità nel terreno di coltura contenente fattore delle cellule staminali (SCF) e mantenendo la loro multipotenzialità. Cellule EML possono essere separati in sottopopolazioni di auto-rinnovamento Lin-SCA + CD34 + e le cellule parzialmente differenziate Lin-SCA-CD34- basati su marcatori di superficie CD34 e SCA 6. Simile a breve termine CSE, SCA + CD34 + cellule sono in grado di auto-rinnovamento. Se trattati con cellule CD34 + SCF, Lin-SCA + possono rapidamente rigenerare una popolazione mista di cellule CD34 + e Lin-SCA-CD34- Lin-SCA + e continuano a proliferare 6. Le due popolazioni sono simili nella morfologia e hanno simili livelli di c-kit mRNA e proteina 6. Cellule Lin-SCA-CD34- sono in grado di propagare in mezzi contenenti IL-3, invece di SCF 3. Unveiling i regolatori chiave nella decisione EML destino delle cellule offrirà una migliore comprensione dei meccanismi cellulari e molecolari nei primi mesi di transizione dello sviluppo durante l'ematopoiesi.
Al fine di indagare le differenze molecolari alla base tra l'auto-rinnovamento Lin-SCA + CD34 + e le cellule Lin-SCA-CD34- parzialmente differenziati, abbiamo utilizzato l'RNA-Seq per identificare i geni differenzialmente espressi. In particolare, ci concentriamo su fattori di trascrizione, come fattori di trascrizione sono cruciali nel determinare il destino delle cellule. RNA-Seq è un approccio recentemente sviluppato che utilizza le capacità di sequenziamento di prossima generazione di tecnologie (NGS) per analizzare e quantificare RNA trascritto dal genoma 7,8. In breve, l'RNA totale è poli-A selezionato e frammentato come modello iniziale template.The RNA viene poi convertito in cDNA usando trascrittasi inversa. Al fine di mappare full-length trascritti di RNA, utilizzando intatto, RNA non degradato per la costruzione di libreria di cDNA è importante. Per la purposa di sequenziamento, specifiche sequenze adattatore vengono aggiunti a entrambe le estremità di cDNA. Poi, nella maggioranza dei casi, le molecole di cDNA vengono amplificati mediante PCR e sequenziati in un modo ad alta produttività.
Dopo la sequenza, la risultante si legge può essere allineato con un genoma di riferimento e un database del trascrittoma. Il numero di letture che mappa il gene di riferimento viene contato e questa informazione può essere utilizzata per stimare il livello di espressione del gene. La legge può anche essere assemblato de novo senza genoma di riferimento, consentendo lo studio di trascrittomi in organismi non modello 9. La tecnologia RNA-Seq è stato utilizzato anche per rilevare isoforme di splicing 10-12, nuove trascrizioni 13 e fusioni geniche 14. Oltre alla rilevazione dei geni codificanti proteine, RNA-Seq può anche essere utilizzato per rilevare romanzo e analizzare il livello di trascrizione di RNA non codificanti, come RNA non codificante lungo 15,16, 17 microRNA, siRNA 18 ecc. A causa di tegli accuratezza di questo metodo, è stata utilizzata per la rilevazione delle variazioni di singoli nucleotidi 19,20.
Prima dell'avvento della tecnologia RNA-Seq, microarray è stato il metodo principale utilizzato per l'analisi del profilo di espressione genica. Sonde pre-progettati sono sintetizzati e successivamente attaccati ad una superficie solida per formare una diapositiva microarray 21. mRNA viene estratto e convertito in cDNA. Durante il processo di trascrizione inversa, nucleotidi fluorescente sono incorporati nel cDNA e il cDNA possono essere ibridate sui vetrini microarray. L'intensità del segnale raccolto da un punto specifico dipende dalla quantità di cDNA legame specifico della sonda in quel luogo 21. Rispetto alla tecnologia RNA-Seq, microarray ha diversi limiti. In primo luogo, microarray basa sulla conoscenza preesistente gene annotazione, mentre la tecnologia RNA-Seq è in grado di rilevare nuovi trascritti a relativamente alto livello di fondo, che ne limita l'utilizzo quando gelivello di espressione ne è basso. Inoltre, la tecnologia RNA-Seq ha molto più alta gamma dinamica di rilevazione (8.000 volte) 7, considerando che, a causa di sfondo e la saturazione dei segnali, la precisione di microarray è limitata per entrambi i geni altamente espressi e umile 7,22. Infine, le sonde di microarray si differenziano per la loro efficienza di ibridazione, che rendono i risultati meno affidabili quando si confrontano i livelli di espressione relativa di diverse trascrizioni all'interno di un campione di 23. Sebbene RNA-Seq ha molti vantaggi rispetto microarray, la sua analisi dei dati è complessa. Questo è uno dei motivi che molti ricercatori utilizzano ancora microarray invece di RNA-Seq. Vari strumenti bioinformatici sono necessari per RNA-Seq l'elaborazione e analisi dei dati 24.
Tra le diverse sequenziamento di prossima generazione di piattaforme (NGS), 454, Illumina, SOLID e Ion Torrent sono quelli più utilizzati. 454 è stata la prima piattaforma commerciale NGS. In contrasto con le altre piattaforme di sequenziamentolunghezza tale illumina e SOLID, la piattaforma 454 genera leggere più (in media 700 letture di base) 25. Legge più sono migliori per la caratterizzazione iniziale di transcriptiome a causa della loro maggiore efficienza montaggio 25. Lo svantaggio principale della piattaforma 454 è il suo alto costo per megabase di sequenza. Il Illumina e piattaforme SOLIDI generare legge con aumento del numero e lunghezze brevi. Il costo per megabase di sequenza è molto inferiore rispetto alla piattaforma 454. A causa del gran numero di breve recita per la Illumina e piattaforme SOLIDI, l'analisi dei dati è molto più computazionalmente intensive. Il prezzo dello strumento e dei reagenti per sequenziamento per la piattaforma Ion Torrent è più economico e il tempo di sequenza è più breve di 25. Tuttavia, il tasso di errore e il costo per megabase di sequenza sono più alti rispetto al Illumina e piattaforme SOLIDI. Piattaforme diverse hanno i loro vantaggi e svantaggi e richiedono diversi metodi per l'analisi dei dati. Il PLATForm devono essere scelti in base allo scopo di sequenziamento e la disponibilità di fondi.
In questo lavoro, prendiamo piattaforma Illumina RNA-Seq come esempio. Abbiamo utilizzato delle cellule EML come sistema modello per studiare i regolatori chiave di EML cellule auto-rinnovamento e differenziazione, e ha fornito una modalità dettagliate di costruzione della libreria di RNA-Seq e analisi dei dati per il calcolo del livello di espressione e di rilevamento romanzo trascrizione. Noi abbiamo indicato nel nostro precedente pubblicazione che lo studio RNA-Seq nel sistema modello EML 2, quando accoppiato con test funzionale (ad esempio shRNA atterramento) fornire un approccio efficace nella comprensione del meccanismo molecolare delle prime fasi di differenziazione ematopoietiche, e in grado di fungere da modello per l'analisi di cellule auto-rinnovamento e differenziazione in generale.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. EML coltura cellulare e la separazione delle cellule Lin-CD34 + e CD34- Lin-Utilizzo di cellule del sistema magnetico e ordinamento fluorescenza-attivato delle cellule metodo di ordinamento
- Preparazione di criceti neonati rene (BHK) terreno di coltura di cellule staminali per il fattore di cella di raccolta:
- Culture cellule BHK in DMEM contenente 10% FBS nel 25 cm 2 beuta (Tabella 1) a 37 ° C, 5% CO 2 in un incubatore di coltura cellulare.
- Quando le cellule crescono al 80 - 90% di confluenza, le cellule lavare una volta con 10 ml di PBS. Aggiungere 5 ml di 0,25% soluzione di tripsina-EDTA al monostrato e incubare le cellule per 1-5 minuti a temperatura ambiente (RT) fino a quando le cellule vengono staccate.
- Preparare la soluzione su e giù delicatamente per rompere grumi di cellule. Aggiungere 5 ml di DMEM completo al pallone per fermare le attività tripsina. Raccogliere le cellule per centrifugazione a 200 xg per 5 minuti a temperatura ambiente.
- Rimuovere il mezzo e risospendere il pellet cellulare in 10 ml di BHK fresco mezzo di coltura cellulare. Trasferire 2 ml della sospensione cellulare dal punto 1.1.4 a un nuovo pallone 75 centimetri 2 e aggiungere 48 ml di mezzo di coltura cellulare fresca BHK al pallone.
- Coltivare le cellule BHK per due giorni e raccolgono il terreno di coltura. Passaggio il mezzo attraverso un filtro da 0,45 micron. Conservare il mezzo di -20 ° C fino all'utilizzo.
- Coltura cellulare EML:
- Culture cellule EML (in sospensione) in ambiente basico EML contenenti BHK coltura cellulare (Tabella 1) a 37 ° C, 5% CO 2 in un incubatore di coltura cellulare.
- Mantenere le cellule EML a bassa densità cellulare (0,5-5 x 10 5 cellule / ml) con la densità di picco meno di 6 x 10 5 cellule / ml. Dividere le celle ogni 2-3 giorni nel rapporto di 1: 5. Cellule Passage EML delicatamente e scartare la cultura dopo passaging per 10 generazioni.
- L'esaurimento delle cellule positive lignaggio:
- Raccogliere le cellule EML per centrifugazione a 200 xg for 5 min e lavare le cellule una volta con PBS. Raccogliere le cellule per centrifugazione a 200 xg per 5 min.
- Risospendere le cellule con PBS e contare le cellule con un emocitometro. Determinare la concentrazione dell'anticorpo nella successiva fase di separazione cellulare secondo il numero delle celle (consultare le istruzioni offerte dal gestore del sistema di isolamento delle cellule).
- Isolare il negativo lineage (Lin-) cellule utilizzando un cocktail di anticorpi stirpe (cocktail di anticorpi monoclonali coniugati con biotina CD5, CD45R (B220), CD11b, Anti-Gr-1 (Ly-6G / C), 7-4 e Ter-119 ) ed un sistema magnetico attivato cell sorting secondo le istruzioni del produttore.
- Separazione di cellule Lin-CD34 + e Lin-CD34-:
- Centrifugare le cellule lin- dal punto 1.3.3 a 200 xg per 5 min. Risospendere il pellet di cellule con PBS e contare le cellule con un emocitometro.
- Lavare le cellule due volte con tampone FACS e agglomerare le cellule a 200 xgper 5 min.
- Etichettare cinque provette da microcentrifuga da 1,5 ml con, rispettivamente, il numero 1, 2, 3, 4, 5. Risospendere le cellule con 100 microlitri tampone FACS per 10 6 cellule (10 6 cellule per provetta).
- Aggiungere 1 mg di anticorpo anti-topo CD34 FITC nella provetta 1 e 2 del tubo e mescolare delicatamente le provette.
- Incubare tutte le provette a 4 ° C per 1 ora al buio.
- Aggiungere 0,25 mg di anticorpi anti-Sca1 PE-coniugato e 20 l di APC-coniugato anticorpi Lineage Cocktail a tubo 1, 0,25 mg di PE-coniugato anticorpo anti-Sca1 a tubo 3, e 20 ml di anticorpi APC-coniugati Lineage cocktail a tubo 4.
- Mescolare tutti i tubi delicatamente e incubare le cellule a 4 ° C per altri 30 minuti al buio.
- Aggiungere 300 microlitri di tampone FACS di cellule e centrifugare le cellule a 200 xg per 5 min.
- Lavare le cellule con 500 microlitri di tampone FACS per tre volte.
- Risospendere il pellet di cellule in 500 ml di FACS buffer.
- Utilizzare le cellule in tubi 2, 3, 4 e 5 per l'impostazione di compensazione. Isolare le cellule Lin-SCA + CD34 + e Lin-SCA-CD34- in tubo 1 utilizzando FACS Aria.
2. RNA Preparazione e Biblioteca di costruzione di high-throughput Sequencing
- Isolamento, analisi della qualità e quantificazione di RNA:
- Estratto RNA totale da Lin-CD34 + e le cellule Lin-CD34- rispettivamente utilizzando TRIzol seguendo il protocollo produce '.
- Rimuovere il DNA utilizzando deossiribonucleasi contaminati I (DNasi I) seguendo il protocollo del produttore. Facoltativamente, conservare l'RNA a -80 ° C in questa fase per un ulteriore uso.
- Valutare la qualità di RNA totale usando Bioanalyzer secondo le indicazioni offerte dal fornitore. Utilizzare campione di RNA con RNA Integrity Number (RIN) lager di 9.
- Biblioteca Edilizia e high-throughput sequencing:
NOTA: Questo protocollo descrive RNA-Seq utilizzando la piattaforma Illumina. Peraltre piattaforme di sequenziamento, sono necessari diversi metodi di preparazione di libreria.- Utilizzare 0,1-4 mg di RNA totale di alta qualità a campione per la preparazione biblioteca. Normalmente 2 mg di RNA totale può essere estratto da 10 5 cellule EML.
- Utilizzare un sistema di preparazione del campione di RNA-sequenziamento per la purificazione dell'RNA e frammentazione, prima e seconda sintesi filamento di cDNA, riparazione fine, 3 'estremità adenylation, adattatore di legatura e amplificazione PCR, seguendo le procedure standard dettagliati di istruzioni del fornitore.
- Positivamente selezionare Polya mRNA utilizzando oligo-dT biglie magnetiche e frammentare l'mRNA.
- Eseguire la trascrizione inversa utilizzando primer casuali per ottenere il cDNA e successivamente sintetizzare il secondo filone di cDNA per generare doppia elica del DNA.
- Rimuovere le "sporgenze e riempire il 5 '3 sbalzi di DNA polimerasi. Adenylate 3 'estremità per evitare frammenti di cDNA dal legando tra loro.
- Aggiungi adattatori indicizzazione multiplex a entrambe le estremità del dscDNA. Eseguire PCR per l'arricchimento di frammenti di DNA.
- Misurare la A260 / A280 per ottenere informazioni sulla concentrazione di libreria utilizzando uno spettrofotometro.
- Valutare la qualità biblioteca e misurare la gamma di dimensioni dei frammenti di DNA utilizzando un Bioanalyzer.
Analisi 3. Dati
Per avere un riferimento del software utilizzato in questa parte, si veda (Tabella 2).
- L'elaborazione dei file di dati per l'analisi a valle:
- Convertire .bcl (file chiamata di base) file per .fastq di file utilizzando il software Casava (Illumina, versione 1.8.2).
- Accendi il 'Terminal' nel sistema Linux. Passare alla cartella di dati che contiene i file di dati da una macchina per il sequenziamento Illumina HiSeq2000. Supponiamo che la cartella risultato è 'NASboy1 / JiaqianLabData / HiSeq_RUN / 2013_07_11 / 130627_SN860_0309_A_2013-166_H0PW9ADXX /', tiponel comando in Figura S1A, e inserire la cartella dei dati.
- Installare Casava 1.8.2 nel sistema Linux. Supponiamo che il outputfolder è 'Unaligned', utilizzare il comando in Figura S1B per preparare il file di configurazione per la conversione. Utilizzare l'opzione --fastq-cluster-count 0 per garantire la si crea solo un file .fastq per ogni campione. Il file .fastq generato è in formato .gz. Decomprimerlo per l'analisi a valle (Figura S1 B).
- Dopo che la cartella 'Unaligned' è stato generato, passare alla cartella 'Unaligned' (Figura S1C).
- Utilizzare il comando nella Figura S1D per iniziare il processo di conversione. Il parametro '-j' fornisce il numero di CPU che verrà utilizzato.
- Dopo che il sistema finito il processo di conversione, passare alla cartella risultato sotto la cartella 'Unaligned' (Figura S1E).
- Utilizzare il comando nella Figura S1F </ Strong> per decomprimere il file .fastq.gz in .fastq file in ogni cartella del campione.
- Convertire .bcl (file chiamata di base) file per .fastq di file utilizzando il software Casava (Illumina, versione 1.8.2).
- Rileva nuove trascrizioni e valutare il livello di espressione utilizzando Tuxedo Suite 26:
- Mappa del-end accoppiato RNA-Seq legge al genoma di riferimento del mouse (UCSC versione MM9, ottenuto da http://cufflinks.cbcb.umd.edu/igenomes.html ) utilizzando il software Tophat (versione 1.3.3) 27, che utilizza la Bowtie leggere mapper (versione 0.12.7) 28. Tophat viene fornito con l'opzione "-no-romanzo-juncs" per migliorare la precisione stima del livello di espressione.
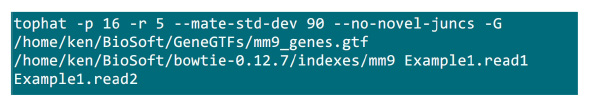
- Inserire i file .fastq in una cartella in cui sarà attuato il processo di mappatura. Supponiamo che ci siano 2 file .fastq (rinominare in Example1.read1, Example1.read2) per un campione di sequenziamento accoppiato-fine, utilizzare il comando in Figura S2 per fare la mappatura (regolare i parametri in base alle impostazioni di sistema).Il parametro "-p" fornisce il numero di CPU che verrà utilizzato. I parametri "r" e "-mate-STD-dev" può essere ottenuto dalla libreria di controllo di qualità o dedotte da un sottoinsieme di allineamento legge (Figura S2).
- Montare il mappato legge in trascrizioni di RNA utilizzando il software Gemelli (versione 1.3.0) 29. Gemelli Run utilizzando il file di annotazione di geni noti (stesso file .gtf usato da Tophat) e il file .bam prodotto da Tophat.
- Dopo Tophat terminato l'esecuzione, nella stessa cartella, utilizzare il comando in Figura S3A per eseguire gemelli di costruire trascrittoma e stima trascrizione livello di espressione. Il 'mm9_repeatMasker.gtf' e file di sequenze del genoma nella cartella 'GenomeSeqMM9' sono disponibili presso UCSC Genome Browser.
- I file genes.expr e transcripts.expr risultanti contengono il valore di espressione dei geni e trascritti (isoforme). Copia e incollail contenuto del file in un file Excel e manipolare con foglio di calcolo (Figura S3B).
- Utilizzare il comando nella Figura S3C per confrontare il file risultante 'transcripts.gtf' per il file di riferimento 'mm9_genes.gtf' al fine di identificare nuove trascrizioni.
- Il file .tmap risultante contiene il risultato del confronto. Copia e incolla il contenuto del file in un file Excel e manipolare con foglio di calcolo. Trascrizioni con il codice della classe 'u' possono essere considerati come 'romanzo' rispetto al riferimento .gtf file fornito (Figura S3D).
NOTA: Per la valle convenienza analisi, impostare i valori FPKM a 0,1 se i valori sono sotto 0.1.
NOTA: Punto 3.2.3 - 3.2.6 è facoltativo per coloro che desiderano migliorare la precisione di espressione stima nuove trascrizioni. Questo richiederà un tempo molto più lungo, perché la mappatura e la costruzione del trascrittoma devono essere rdelle Nazioni Unite più di una volta.
- Eseguire Tophat con i parametri di default e quindi eseguire gemelli in file .gtf generato utilizzando il comando nella Figura S3E.
- Confrontare il file .gtf risultante al file .gtf genoma di riferimento utilizzando il comando nella Figura S3F.
- Analizzare il file .tmap portato come descritto nel passaggio 3.2.2.4. Copia e incolla il contenuto del file in un file Excel e manipolare con foglio di calcolo. Trascrizioni con il codice della classe 'u' possono essere considerati come 'romanzo' rispetto al riferimento .gtf file fornito.
- Dopo il punto 3.2.5, esiste un file .combined.gtf nella cartella che può essere usato come file di riferimento .gtf. Una seconda serie di Tophat e gemelli può essere eseguita come descritto nel passaggio 3.2.1 e 3.2.2 per ottenere una più accurata stima FPKM di nuovi trascritti.
- Mappa del-end accoppiato RNA-Seq legge al genoma di riferimento del mouse (UCSC versione MM9, ottenuto da http://cufflinks.cbcb.umd.edu/igenomes.html ) utilizzando il software Tophat (versione 1.3.3) 27, che utilizza la Bowtie leggere mapper (versione 0.12.7) 28. Tophat viene fornito con l'opzione "-no-romanzo-juncs" per migliorare la precisione stima del livello di espressione.
- Rileva differentially espresso geni utilizzando il pacchetto DESeq 30.
- L'ingresso di DESeq è una prima tabella di conteggi di lettura. Per ottenere tale tabella, utilizzare lo script htseq-count distribuito con il pacchetto HTSeq Python che può essere scaricato dal sito HTSeq ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Assicurarsi che samtools, pitone, e htseq-count programsare installati nel sistema. Ottenere i numeri di conteggio di lettura prime da produzione tophat utilizzando il comando nella Figura S4A.
- Preparare 'Raw_Count_Table.txt', file 'ExperimentDesign.txt' con Excel. Copiare e salvare il contenuto in formato .txt per il pacchetto DESeq R (figura 4J).
- Installare il programma R nel sistema. Nel terminale, tipo 'R' e premere ENTER.A messaggio sullo appearas mostrato nella Figura S4C.
- Leggi 'Raw_Count_Table.txt ',' ExperimentDesign.txt 'in R con il comando in Figura S4D.
- Caricare il pacchetto DESeq utilizzando il comando nella Figura S4E.
- Condizioni di fattorizzare in R (Figura S4F).
- Utilizzare il comando nella Figura S4G per eseguire il test binomiale negativo sul tavolo conteggio normalizzato.
- Utilizzare il comando nella Figura S4H di geni espressi in uscita significativi differenziali in un file .csv.
- L'ingresso di DESeq è una prima tabella di conteggi di lettura. Per ottenere tale tabella, utilizzare lo script htseq-count distribuito con il pacchetto HTSeq Python che può essere scaricato dal sito HTSeq ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Fattori di trascrizione Lookup '(TFS) valori FPKM attraverso campioni utilizzando Excel. Intersezione DE tavolo gene e tavolo TF. I geni appartengono a entrambi tabella sono differenzialmente espressi fattori di trascrizione.
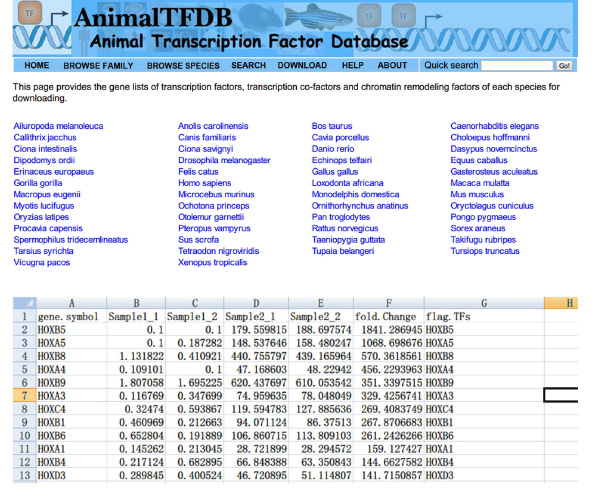
- Vai al sito http://www.bioguo.org/AnimalTFDB/download.php e scaricare i fattori di trascrizione. Poi ricercare i fattori di trascrizione DE in Excel (< strong> Figura S5).
- Generazione di file .bigwig per la visualizzazione del browser UCSC genoma.
- Scaricare il pacchetto software 'bedtools' dal sito https://github.com/arq5x/bedtools2 e installare il software nel sistema 31. Scarica gli strumenti UCSC 'bedGraphToBigWig' dal sito http://hgdownload.cse.ucsc.edu/admin/exe/ e installare il software nel sistema.
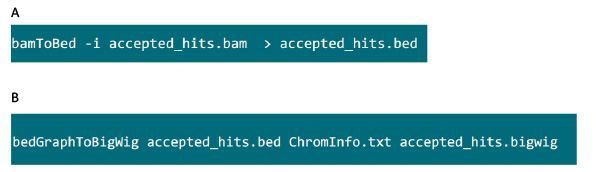
- Nella cartella che contiene il file .bam, utilizzare il comando in Figura S6A per convertire file .bam generato da tophat in un file .bed.
- Dopo che il file .bed viene prodotto, utilizzare il comando in Figura S6B per generare file di .bigwig. Il file 'ChromInfo.txt' può essere ottenuto dal seguente URL:Arget = "_blank"> http://hgdownload.cse.ucsc.edu/goldenPath/mm9/database/chromInfo.txt.gz.
- Osservare una traccia personalizzata su UCSC Genome Browser. Consultare il sito Web http://genome.ucsc.edu/goldenPath/help/customTrack.html su come visualizzare un brano personalizzato utilizzando il browser genoma UCSC.

Figura S1: Conversione di file .bcl a .fastq di file utilizzando il software Casava.

Figura S2: Mapping legge di riferimento genoma utilizzando Tophat.
Figura S3: Individuazione di nuovi trascrizioni e la stima livello di espressione.

Figura S4: Richiamo differenziale dei geni espressi utilizzando DESeq pacchetto.

Figura S5: Identificazione dei fattori di trascrizione differenzialmente espressi.

Figura S6: Conversione risultato della mappatura per la visualizzazione dei dati.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Al fine di analizzare i geni differenzialmente espressi in Lin-CD34 + e le cellule Lin-CD34- EML, abbiamo utilizzato la tecnologia RNA-Seq. La figura 1 mostra l'iter delle procedure. Dopo l'isolamento di cellule negative lignaggio di separazione delle cellule magnetiche, ci siamo separati Lin-SCA + CD34 + e le cellule Lin-SCA-CD34- mediante FACS Aria. Cellule EML Lin-arricchiti sono state colorate con anticorpi anti-CD34, anti-Sca1 e anticorpi cocktail lignaggio. Solo le cellule lin- stati gated per l'analisi di espressione Sca1 e CD34. Due popolazioni (SCA + CD34 + e cellule SCA-CD34- EML) potrebbe essere riscontrata dal FACS analisi (Figura 2) 6.
Dopo la separazione delle cellule, abbiamo estratto l'RNA totale da cellule CD34 + e CD34-, rispettivamente, e analizzato la qualità dell'RNA. La precisione dei dati RNA-Seq dipende in grande misura la qualità della libreria di RNA-Seq e la qualità di RNA totale è fondamentale per la preparazione di una libreria di alta qualità. Campione di RNA di alta qualità dovrebbe avere un valore di 260/280 OD sono compresi tra 1.8 e 2.0. Oltre a utilizzare lo spettrofotometro, qualità RNA è stato ulteriormente valutato con maggiore accuratezza Bioanalyzer. La Figura 3 mostra il risultato di un campione di RNA di alta qualità con il RIN pari a 9.4. Solo di alta qualità del campione RNA totale con valore RIN superiore a 9 è stato utilizzato per l'estrazione di mRNA e le successive procedure di costruzione di libreria.
L'RNA ribosomiale è il tipo più abbondante di RNA in cellule. Attualmente due strategie principali, esaurimento di rRNA o positivamente selezione di poliadenilato mRNA (poli-A mRNA), sono utilizzati per l'arricchimento del target RNA prima della costruzione biblioteca. Specie di RNA poliadenilato non vengono persi durante la selezione di poli-A mRNA. Al contrario, rRNA metodi di esaurimento, come RiboMinus potrebbero preservare le specie di RNA non poliadenilato. Lo scopo del nostro studio è quello di cercare differenzialmente espressi geni che codificano in due tipi di cellule, quindi abbiamo usato il poli-A metodo di selezione mRNA per l'arricchimento di RNA di destinazione prima constru libreriaction. Quando la costruzione biblioteca era finita, la dimensione di frammenti di DNA della libreria è stata verificata prima sequenziamento usando Bioanalyzer. La Figura 4 mostra una biblioteca di buona qualità con picchi dimensioni frammento di circa 300 bp.
Nella fase successiva, la libreria è stata sottoposta a high-throughput sequencing. In linea di principio, la lunghezza più leggere sarà utile per la mappatura di lettura. Si può ridurre la probabilità che la lettura è mappato in più posizioni a causa della somiglianza tra i geni duplicati o membri della famiglia genica. Poiché le sequenze di sequenziamento pair-end sono da entrambe le estremità dei frammenti, la lunghezza di lettura prescelta deve essere inferiore alla metà della lunghezza media frammenti. Se l'obiettivo principale dell'esperimento è quello di misurare il livello di espressione invece di costruire struttura di trascrizione, single-end leggere (75 o 100 punti base) in grado di ridurre i costi senza perdere troppe informazioni. Accoppiato-end sequenziamento è più utile per la costruzione di strutture trascrizione e più breviLeggere lunghezza può essere utilizzata per ridurre i costi. Certo, quando di finanziamenti sufficienti, la lunghezza più leggere è preferito.
Per l'analisi di espressione differenziale, ci sono molti algoritmi alternativi diversi dai DESeq. C'è anche uno incluso nel pacchetto gemelli di nome cuffdiff 32. DESeq è uno degli algoritmi di analisi genetiche DE più ampiamente usato conteggio basato. Metodo DESeq si basa su un modello di statistiche ben caratterizzato - distribuzione binomiale negativa. Nella nostra esperienza, DESeq è più stabile confronta con cuffdiff. Le prime versioni di cuffdiff spesso danno significativamente diverso numero di geni DE. Per questo abbiamo usato DESeq per l'analisi DE qui.
Poiché i fattori di trascrizione sono cruciali per la determinazione del destino cellulare, ci siamo concentrati sulla trascrizione significativamente differenzialmente espressi fattori di 33. Il TF ha cambiato> 1,5 volte tra Lin-CD34 + e Lin-CD34- sono stati trovati e sono mostrati sulla mappa termica (Figure 5) 2. In particolare, il livello di espressione relativa di Tcf7 a CD34-Lin + cellule è più di 100 volte superiore a quella in cellule Lin-CD34-. Così Tcf7 è stato scelto per l'ulteriore ChIP-Sequencing (immunoprecipitazione della cromatina e sequenziamento) analisi e test funzionali per confermare la funzione Tcf7 s 'nella regolazione di EML cellule auto-rinnovamento e differenziazione 2.

Figura 1:. Flusso di lavoro delle procedure di Lin-CD34 + e CD34- Lin-cellule sono state separate dal sistema di separazione cellulare magnetica e la fluorescenza delle cellule attivate metodo di ordinamento. L'RNA totale è stato estratto seguita da purificazione di mRNA e costruzione della libreria. Dopo l'analisi della qualità biblioteca, i campioni sono stati sottoposti a sequenziamento ad alta velocità. I dati sono stati analizzati e differenzialmente espressi fattori di trascrizione sono stati identificati.

Figura 2: Separazione delle cellule 6 celle Lin- EML sono stati arricchiti da cell sorting magnetico Lin-CD34 + e CD34- Lin-EML.. Cellule lin- sono state colorate con anti-CD34, anti-Sca1 e anticorpi miscela lignaggio. Cellule lin- stati gated per l'espressione di CD34 e Sca1. Lin-CD34 + SCA + e popolazioni di cellule Lin-CD34-SCA- EML sono stati ordinati.

Figura 3:. Un rappresentante di alta qualità del campione di RNA totale La qualità di RNA totale è stato valutato da Bioanalyzer. L'RNA Integrity Number è 9.4 (FU, Fluorescenza unità).

Figura 4:. Gamma di dimensioni Frammenti di libreria associati-End DNA distribuzione delle dimensioni della biblioteca è stato analizzato utilizzando Bioanalyzer. La maggior parte dei frammenti sono all'interno della gamma di dimensioni di 250-500 bp.

Figura 5:. Differenzialmente espressi fattori di trascrizione (> 1,5 volte) tra le cellule Lin-CD34 + e le cellule Lin-CD34- 2 Per ogni tipo di cellula, sono stati eseguiti due esperimenti indipendenti. Up-regolati i geni sono indicati come il colore rosso e geni down-regolati sono indicati come colore verde.
Tabella 1: Buffer e colture cellulari medium.
| Software | Uso | Riferimento | |||
| Bowtie 1.2.7 | Utilizzato da Tophat per la mappatura | [28] | |||
| 1.3.3 tophat | Mappatura legge torna a riferimento genoma | [27] | |||
| Gemelli 1.3.0 | Trascrizioni costruzione e la stima livello di espressione | [29] | |||
| DESeq 1.16.0 | Analisi di espressione differenziale | [30] | Bedtools 2.18 | Convertire file di .bam in file di .bed | [31] |
| bedGraphToBigWig | Convertire file di .bed a .bigwig di file | http://genome.ucsc.edu/ |
Tabella 2: Elenco di software per l'analisi dei dati.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Trascrittoma mammiferi è molto complessa 34-38. La tecnologia RNA-Seq svolge un ruolo sempre più importante negli studi di analisi del trascrittoma, romanzo di rilevamento trascrizioni e singolo nucleotide variante scoperta ecc Ha molti vantaggi rispetto ad altri metodi di analisi di espressione genica. Come accennato nell'introduzione, supera i manufatti ibridazione di microarray e può essere utilizzato per identificare nuovi trascritti de novo. Una limitazione di RNA-sequenza è breve lunghezza lettura relativa confronto con Sanger sequenziamento. Tuttavia, con il rapido miglioramento della tecnologia di sequenziamento, leggere la lunghezza è in costante aumento. In questo lavoro, forniamo i dettagli sui metodi di utilizzo di questa tecnologia per identificare i potenziali regolatori chiave nel topo cellule EML self-renewal e la differenziazione.
Il primo passo fondamentale per questo protocollo è coltura cellulare EML. Anche se EML è una linea di cellule precursori ematopoietici e può esserepropagato in grande quantità con SCF. La condizione di coltura di cellule EML richiede più attenzione rispetto alle solite linee cellulari immortalizzate. Le cellule devono essere nutriti e diversi passaggi in maniera regolare con il funzionamento dolce; in caso contrario le cellule potrebbero cambiare le loro proprietà di auto-rinnovamento e differenziazione e subire la morte delle cellule. Come primo passo dopo aver raccolto un numero sufficiente di cellule, abbiamo isolato cellule negative lignaggio con un sistema di smistamento delle cellule attivate magnetico. Poi ci siamo separati CD34 + e cellule CD34- mediante fluorescenza-attivato l'ordinamento delle cellule. Le cellule EML sono normalmente diversi passaggi meno di 10 generazioni prima di utilizzare per l'estrazione dell'RNA e il numero di cellule CD34 + e le cellule CD34- dovrebbe essere simile dopo la separazione. Se le due popolazioni variano notevolmente in numero di cellule, si consiglia di eliminare la cultura e ri-scongelare un altro tubo di cellulare stock per la cultura.
Dopo la separazione delle cellule CD34 + e cellule CD34-, l'estrazione dell'RNA totale è stata eseguita, un altro passo importante per questa stUdy. RNA di alta qualità è la base per la costruzione di una libreria di alta qualità, che promette l'accuratezza dei dati di sequenziamento. In questa fase critica, qualsiasi contatto con RNasi deve essere evitato. Tutti i reagenti devono essere RNasi gratuito. È importante indossare guanti in ogni momento durante la manipolazione dell'RNA. Campione di RNA di alta qualità ha un valore di 260/280 OD sono compresi tra 1.8 e 2.0. Quando si raccolgono la fase acquosa contenente l'RNA, fare attenzione a non trasportare qualsiasi fase organica con il campione di RNA. Solventi organici residui, come fenolo e cloroformio nel RNA si tradurrebbe in un valore di OD260 / 280 inferiore a 1,65. Se il valore OD260 / 280 è inferiore a 1,65, precipitare nuovo RNA con etanolo. Dopo aver lavato con etanolo al 75%, non fare asciugare eccessivamente pellet di RNA. Asciugatura pellet di RNA completamente influenzerà la solubilità di RNA e portare a bassa resa di RNA.
Il prossimo passo fondamentale per questo protocollo è la preparazione biblioteca. Dopo l'estrazione di RNA totale, una fase di utilizzo DNasi per la rimozione di DNA contaminato is altamente raccomandato, in quanto la contaminazione del DNA potrebbe comportare la stima errata della quantità di RNA totale utilizzato. Si consiglia di eseguire la procedura a valle subito dopo l'isolamento di RNA, dal momento che dopo lo stoccaggio a lungo termine e congelamento-scongelamento procedura, l'RNA si degrada in una certa misura. Se i passaggi successivi, dopo l'isolamento di RNA non possono essere eseguite immediatamente, conservare l'RNA a -80 ° C. Prima di RNA totale viene utilizzata per la purificazione di mRNA e la sintesi del cDNA, la qualità dovrebbe essere sempre controllata. Solo alta qualità RNA può essere utilizzato per la preparazione della libreria. Utilizzo di bassa qualità o RNA degradato potrebbe portare a un eccesso di rappresentazione di 3 'finisce. Prima di sequenziamento, qualità biblioteca è stata valutata al fine di garantire la massima efficienza di sequenziamento.
Nella parte di analisi dei dati, dopo aver effettuato una corsa di Gemelli senza trascrittoma di riferimento, abbiamo unito le nuove trascrizioni con trascrizioni noti per formare un riferimento .gtf file ed eseguirlo Tophat e gemelli per la seconda volta.Si raccomanda Questa procedura in due run, dal momento che questo fornisce la stima più accurata FPKM che correre una sola volta. Dopo l'analisi dei dati, sono stati identificati i geni differenzialmente espressi. Esperimenti a valle possono essere eseguite per convalidare la funzione dei geni in vitro e in vivo. Nella nostra precedente pubblicazione 2, abbiamo scelto i fattori di trascrizione significativamente differenzialmente espressi e identificato il sito di legame del genoma di questi fattori eseguendo immunoprecipitazione della cromatina e la sequenza (ChIP-Seq). Inoltre, abbiamo applicato shRNA test atterramento per testare l'effetto funzionale di Tcf7. Abbiamo scoperto che nelle cellule Tcf7 smontabile, geni regolati up-sono stati i geni altamente arricchito in cellule CD34-, mentre sono risultati essere significativamente arricchito in cellule CD34 + geni down-regolati. Pertanto, il profilo di espressione genica di cellule smontabili Tcf7 spostato verso una parte differenziata CD34- state.Overall, utilizzando cellule EML come sistema modelloaccoppiato con la tecnologia RNA-Sequencing e saggi funzionali, abbiamo identificato e confermato Tcf7 come un importante regolatore di cellula EML self-renewal e la differenziazione.
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| Antibiotic-Antimycotic | Invitrogen | 15240-062 | BHK cell culture |
| Anti-Mouse CD34 FITC | eBioscience | 11-0341-81 | FACS sorting |
| Anti-Mouse Ly-6A/E (Sca-1) PE | eBioscience | 12-5981-81 | FACS sorting |
| APC Mouse Lineage Antibody Cocktail | BD Biosciences | 558074 | FACS sorting |
| BD FACSAria Cell Sorter | BD Biosciences | Special offer sysmtem | FACS sorting |
| Corning™ Cell Culture Treated Flasks 75 cm2 | Corning incorporated | 430641 | Cell culture |
| Corning™ Cell Culture Treated Flasks 25 cm2 | Corning incorporated | 430639 | Cell culture |
| Deoxyribonuclease I, Amplification Grade | Invitrogen | 18068-015 | Library preparation |
| DMEM | Invitrogen | 11965-092 | BHK cell culture |
| DPBS | Gibco | 14190 | Cell culture |
| HI FBS | Invitrogen | 16140071 | BHK cell culture |
| Horse Serum | Invitrogen | 16050-122 | EML cell culture |
| IMDM | HyClone | SH30228.02 | EML cell culture |
| L-Glutamine | Invitrogen | 25030-081 | Cell culture |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | 130-090-858 | Isolation of lineage negative cells |
| NanoVue Plus spectrophotometer | GE Healthcare | 28-9569-62 | Quality control |
| Thermo Scientific™ Napco™ 8000 Water-Jacketed CO2 Incubators | Thermo Scientific | 15-497-002 | Cell culture |
| Penicillin-Streptomycin | Invitrogen | 15140-122 | EML cell culture |
| TRIzol® Reagent | Invitrogen | 15596-018 | RNA exraction |
| TruSeq™ RNA Sample Prep Kit v2 -Set B (48 rxn) | Illumina | RS-122-2002 | Library preparation |
| 2100 Electrophoresis Bioanalyzer Instrument | Agilent | G2939AA | Quality control |
| 0.25% Trypsin-EDTA | Gibco | 25200 | Cell culture |
| 0.45 µm Syringe Filters | Nalgene | 190-2545 | Cell culture |
References
- Chambers, S. M., Goodell, M. A. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 3, 201-211 (2007).
- Wu, J. Q., et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS genetics. 8, (2012).
- Ye, Z. J., et al. Complex interactions in EML cell stimulation by stem cell factor and IL-3. Proceedings of the National Academy of Sciences of the United States of America. 108, 4882-4887 (2011).
- Tsai, S., Bartelmez, S., Sitnicka, E., Collins, S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 8, 2831-2841 (1994).
- Weiler, S. R., et al. D3: a gene induced during myeloid cell differentiation of Linlo c-Kit+ Sca-1(+) progenitor cells. Blood. 93, 527-536 (1999).
- Ye, Z. J., Kluger, Y., Lian, Z., Weissman, S. M. Two types of precursor cells in a multipotential hematopoietic cell line. Proc Natl Acad Sci U S A. 102, 18461-18466 (2005).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10, 57-63 (2009).
- Chu, Y., Corey, D. R. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic acid therapeutics. 22, 271-274 (2012).
- Hornett, E. A., Wheat, C. W. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC genomics. 13, 361 (2012).
- Eswaran, J., et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific reports. 3, 1689 (2013).
- Wang, E. T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470-476 (2008).
- Wu, J. Q., et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proceedings of the National Academy of Sciences of the United States of America. 107, 5254-5259 (2010).
- Loraine, A. E., McCormick, S., Estrada, A., Patel, K., Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant physiology. 162, 1092-1109 (2013).
- Edgren, H., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome biology. 12, 6 (2011).
- Ilott, N. E., Ponting, C. P. Predicting long non-coding RNAs using RNA sequencing. Methods. 63, 50-59 (2013).
- Sun, L., et al. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC bioinformatics. 13, 331 (2012).
- Luo, S. MicroRNA expression analysis using the Illumina microRNA-Seq Platform. Methods in molecular biology. 822, 183-188 (2012).
- Bolduc, F., Hoareau, C., St-Pierre, P., Perreault, J. P. In-depth sequencing of the siRNAs associated with peach latent mosaic viroid infection. BMC molecular biology. 11, 16 (2010).
- Chepelev, I., Wei, G., Tang, Q., Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic acids research. 37, 106 (2009).
- Djari, A., et al. Gene-based single nucleotide polymorphism discovery in bovine muscle using next-generation transcriptomic sequencing. BMC genomics. 14, 307 (2013).
- Murphy, D. Gene expression studies using microarrays: principles, problems, and prospects. Advances in physiology education. 26, 256-270 (2002).
- Chen, K., et al. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS one. 8, 72567 (2013).
- Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 18, 1509-1517 (2008).
- Ramskold, D., Kavak, E., Sandberg, R. How to analyze gene expression using RNA-sequencing data. Methods in molecular biology. 802, 259-274 (2012).
- Glenn, T. C. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 11, 759-769 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 7, 562-578 (2012).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25, 1105-1111 (2009).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 10, 25 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature. 28, 511-515 (2010).
- Anders, S., Huber, W. Differential expression analysis for sequence count data. Genome biology. 11, 106 (2010).
- Quinlan, A. R., Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841-842 (2010).
- Cheranova, D., et al. RNA-seq analysis of transcriptomes in thrombin-treated and control human pulmonary microvascular endothelial cells. J Vis Exp. , (2013).
- Zhang, H. M., et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research. 40, 144-149 (2012).
- Wu, J. Q., et al. Systematic analysis of transcribed loci in ENCODE regions using RACE sequencing reveals extensive transcription in the human genome. Genome Biol. 9, 3 (2008).
- Wu, J. Q., et al. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques. 36, 690-696 (2004).
- Wu, J. Q., Shteynberg, D., Arumugam, M., Gibbs, R. A., Brent, M. R. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 14, 665-671 (2004).
- Dewey, C., et al. Accurate identification of novel human genes through simultaneous gene prediction in human, mouse, and rat. Genome Res. 14, 661-664 (2004).
- Wu, J. Characterize Mammalian Transcriptome Complexity. , LAP Lambert Academic Publishing. (2011).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}