

Summary
Se utilizaron análisis de ARN de secuenciación y bioinformática para identificar factores de transcripción expresados diferencialmente significativamente y en subpoblaciones Lin-CD34 + y Lin-CD34 de ratón EMLcells. Estos factores de transcripción que podrían desempeñar un papel importante en la determinación del cambio entre las células Lin-CD34- auto-renovación de Lin-CD34 + y parcialmente diferenciado.
Abstract
Las células madre hematopoyéticas (HSC) se utilizan clínicamente para el tratamiento de trasplante para reconstruir el sistema hematopoyético de un paciente en muchas enfermedades tales como la leucemia y el linfoma. Dilucidar los mecanismos que controlan la auto-renovación y diferenciación de las HSC es importante para la aplicación de las CMH para la investigación y aplicaciones clínicas. Sin embargo, no es posible obtener gran cantidad de HSCs debido a su incapacidad de proliferar in vitro. Para superar este obstáculo, se utilizó una línea celular derivada de la médula ósea de ratón, la EML (eritroide, mieloide y linfocítica) línea celular, como un sistema modelo para este estudio.
RNA-secuenciación (RNA-Seq) se ha utilizado cada vez más para sustituir microarray para estudios de expresión génica. Presentamos aquí un método detallado de la utilización de la tecnología de ARN-Seq para investigar los posibles factores clave en la regulación de las células EML auto-renovación y diferenciación. El protocolo previsto en el presente documento se divide en tres partes. El primer part explica cómo la cultura células EML y separada Lin-CD34 + y células Lin-CD34. La segunda parte del protocolo ofrece procedimientos detallados para la preparación de ARN total y de la construcción de la biblioteca para la posterior secuenciación de alto rendimiento. La última parte se describe el método para el análisis de datos de RNA-Seq y se explica cómo utilizar los datos para identificar los factores de transcripción expresados diferencialmente entre las células Lin-Lin-CD34- CD34 + y. Se identificaron los factores de transcripción más significativamente expresadas diferencialmente a ser los potenciales reguladores clave que controlan las células EML auto-renovación y diferenciación. En la sección de discusión de este artículo, se destacan los pasos clave para el desempeño exitoso de este experimento.
En resumen, este trabajo ofrece un método de uso de la tecnología de ARN-Seq para identificar posibles reguladores de la auto-renovación y diferenciación en células EML. Los factores clave identificados se someten a análisis funcional de aguas abajo in vitro e in vivo.
Introduction
Las células madre hematopoyéticas son células sanguíneas raras que residen principalmente en el nicho de la médula ósea de adultos. Son responsables de la producción de células requeridas para reponer la sangre y los sistemas inmunitarios 1. Como una especie de células madre, las CMH son capaces tanto de auto-renovación y diferenciación. Dilucidar los mecanismos que controlan la decisión destino de las HSC, ya sea hacia la auto-renovación o diferenciación, ofrecerá una valiosa orientación sobre la manipulación de las CMH para las investigaciones de enfermedades de la sangre y el uso clínico 2. Uno de los problemas que enfrentan los investigadores es que las CMH se pueden mantener y ampliar in vitro en una medida muy limitada; la gran mayoría de su progenie son parcialmente diferenciado en cultivo 2.
Con el fin de identificar los principales reguladores que controlan los procesos de auto-renovación y diferenciación en un genoma de gran escala, hemos utilizado un ratón primitiva línea de células progenitoras hematopoyéticas EML como un sistema modelo. Thse línea celular se deriva de médula ósea murina 3,4. Cuando se alimenta con diferentes factores de crecimiento, las células pueden diferenciarse en EML eritroide, mieloide, y las células linfoides in vitro 5. Es importante destacar que esta línea celular puede propagarse en gran cantidad en medio de cultivo que contiene el factor de células madre (SCF) y todavía conservan su pluripotencialidad. EML células se pueden separar en subpoblaciones de auto-renovación de Lin-SCA + CD34 + y células parcialmente diferenciadas Lin-SCA-CD34- basados en marcadores de superficie CD34 y SCA 6. Al igual que a corto plazo las CMH, SCA + células CD34 + son capaces de auto-renovación. Cuando se trata con células CD34 + SCF, Lin-SCA + puede regenerar rápidamente una población mixta de células CD34 + y Lin-SCA-CD34 + Lin-SCA y continuar a proliferar 6. Las dos poblaciones son similares en morfología y tienen niveles similares de ARNm de c-kit y la proteína 6. Células Lin-SCA-CD34 son capaces de propagarse en los medios de comunicación que contiene IL-3 en lugar de SCF 3. Unveiling los reguladores clave en la decisión del destino celular EML ofrecerá una mejor comprensión de los mecanismos celulares y moleculares en la transición del desarrollo temprano durante la hematopoyesis.
Con el fin de investigar las diferencias moleculares subyacentes entre las células Lin-SCA-CD34- parcialmente diferenciadas auto-renovación de Lin-SCA + CD34 + y, se utilizó ARN-Seq para identificar los genes expresados diferencialmente. En particular, nos centramos en los factores de transcripción, como factores de transcripción son cruciales en la determinación del destino celular. ARN-Sec es un enfoque recientemente desarrollado que utiliza las capacidades de secuenciación de próxima generación de tecnologías (NGS) para perfilar y cuantificar ARN transcrito a partir del genoma 7,8. En resumen, el ARN total es poli-A seleccionado y fragmentado como la plantilla inicial template.The ARN se convierte después en ADNc utilizando transcriptasa inversa. Con el fin de asignar transcripciones de ARN de longitud completa, utilizando ARN intacto, no degradado para la construcción de la biblioteca de ADNc es importante. Para el purpose de la secuenciación, se añaden secuencias adaptadoras específicas a ambos extremos del cDNA. Entonces, en la mayoría de los casos, las moléculas de ADNc se amplifican por PCR y se secuenciaron de una manera de alto rendimiento.
Después de la secuenciación, la resultante lee se puede alinear a un genoma de referencia y una base de datos transcriptoma. El número de lecturas que mapa para el gen de referencia se cuenta y esta información se puede utilizar para estimar el nivel de expresión génica. El lee también puede ser ensamblado de novo sin un genoma de referencia, lo que permite el estudio de la transcriptomes en organismos no modelo 9. Tecnología de RNA-seq también se ha utilizado para detectar isoformas de empalme 10-12, nuevas transcripciones 13 y 14 fusiones de genes. Además de la detección de genes codificadores de proteínas, RNA-Seq también se puede utilizar para detectar y analizar novela nivel de transcripción de los ARN no codificantes, tales como RNA largo 15,16, microRNA 17, 18 etc. siRNA no codificante. Debido a tque la precisión de este método, se ha utilizado para la detección de variaciones de un solo nucleótido 19,20.
Antes de la llegada de la tecnología de ARN-Seq, microarray fue el principal método utilizado para el análisis de perfil de expresión génica. Sondas pre-diseñadas se sintetizan y posteriormente unidos a una superficie sólida para formar un microarray de diapositivas 21. ARNm se extrae y se convirtió en ADNc. Durante el proceso de transcripción inversa, los nucleótidos marcados con fluorescencia se incorporan en el ADNc y el ADNc se pueden hibridó en las diapositivas de microarrays. La intensidad de la señal obtenida de un punto específico depende de la cantidad de ADNc de unión a la sonda específica en ese lugar 21. En comparación con la tecnología de RNA-Seq, microarray tiene varias limitaciones. En primer lugar, los microarrays se basa en el conocimiento preexistente de anotación de genes, mientras que la tecnología de ARN-Seq es capaz de detectar nuevas transcripciones en alto nivel de fondo relativa, lo que limita su uso cuando genivel de expresión ne es baja. Además, la tecnología de ARN-Seq tiene mucho mayor rango dinámico de detección (8000 veces) 7, mientras que, debido a fondo y la saturación de las señales, la exactitud de microarrays es limitada para ambos genes altamente expresados y humilde 7,22. Por último, las sondas de microarrays difieren en su eficiencia de hibridación, que hacen que los resultados menos fiables al comparar los niveles de expresión relativos de diferentes transcripciones dentro de una muestra de 23. Aunque RNA-Seq tiene muchas ventajas sobre microarrays, su análisis de los datos es complejo. Esta es una de las razones por las que muchos investigadores todavía utilizan microarrays en lugar de RNA-Seq. Se requieren varias herramientas bioinformáticas para el procesamiento de datos de ARN-Seq y análisis 24.
Entre varios de secuenciación de próxima generación (NGS) plataformas, 454, Illumina, SOLID y Ion Torrent son los más ampliamente usados. 454 fue la primera plataforma comercial NGS. En contraste con las otras plataformas de secuenciaciónlongitud como Illumina y sólido, la plataforma 454 genera ya leer (promedio base de 700 lecturas) 25. Longer lecturas son mejores para la caracterización inicial de transcriptiome debido a su mayor eficiencia montar 25. La principal desventaja de la plataforma 454 es su alto coste por megabase de secuencia. La iluminación y plataformas SÓLIDOS generar lee con el aumento del número y longitudes cortas. El coste por megabase de la secuencia es mucho menor que la plataforma 454. Debido a la gran cantidad de cortos lee para la iluminación y plataformas sólidas, el análisis de datos es mucho más computacionalmente intensivas. El precio del instrumento y los reactivos para la secuenciación para la plataforma Ion Torrent es más barato y el tiempo de la secuencia es más corta 25. Sin embargo, la tasa de error y el costo por megabase de secuencia son más altos en comparación con la iluminación y plataformas SÓLIDOS. Las diferentes plataformas tienen sus propias ventajas y desventajas, y requieren diferentes métodos para el análisis de datos. El plaTForm debe ser elegido sobre la base de la finalidad de secuenciación y la disponibilidad de fondos.
En este trabajo, tomamos plataforma Illumina ARN-Seq como ejemplo. Utilizamos células EML como un sistema modelo para investigar los reguladores clave en EML células auto-renovación y diferenciación, y proporciona un métodos detallados de construcción de la biblioteca de ARN-Seq y análisis de datos para el cálculo de nivel de expresión y la novela detección transcripción. Hemos demostrado en nuestra publicación anterior que el estudio RNA-seq en el sistema de modelo de EML 2, cuando se combina con la prueba funcional (por ejemplo desmontables shRNA) proporcionar un poderoso enfoque en la comprensión del mecanismo molecular de las primeras etapas de la diferenciación hematopoyética, y puede servir como una modelo para el análisis de células auto-renovación y diferenciación en general.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Activada por fluorescencia 1. EML cultivo celular y separación de las células Lin-CD34 + y Lin-CD34- Uso de clasificación celular magnética Sistema y método de clasificación de la célula
- Preparación de riñón de hámster bebé (BHK) medio de cultivo celular para la recogida de factor de células madre:
- Cultura células BHK en medio DMEM que contenía FBS al 10% en 25 cm 2 frasco (Tabla 1) a 37 ° C, 5% de CO2 en un incubador de cultivo celular.
- Cuando las células crecen a un 80 - 90% de confluencia, lavar las células una vez con 10 ml de PBS. Añadir 5 ml de solución de tripsina-EDTA al 0,25% a la monocapa y se incuban las células durante 1-5 min a temperatura ambiente (RT) hasta que se separan las células.
- Pipetear la solución de arriba a abajo suavemente para romper los grumos de células. Añadir 5 ml de DMEM completo al matraz para detener la actividad de la tripsina. Recoger las células por centrifugación a 200 xg durante 5 min a RT.
- Retirar el medio y volver a suspender el sedimento celular en 10 ml de BHK fresco medio de cultivo celular. Transferencia de 2 ml de la suspensión celular de la etapa 1.1.4 a un nuevo matraz de 75 cm 2 y añadir 48 ml de medio de cultivo de células BHK fresco al matraz.
- Cultura de las células BHK durante dos días y recogen el medio de cultivo. Passage el medio a través de un filtro de 0,45 micras. Almacenar el medio en -20 ° C hasta su uso posterior.
- Cultivo de células EML:
- Cultura células EML (en suspensión) en medio básico EML que contienen medio de cultivo de células BHK (Tabla 1) a 37 ° C, 5% de CO2 en un incubador de cultivo celular.
- Mantener las células EML a baja densidad celular (0,5-5 x 10 5 células / ml) con la densidad de pico de menos de 6 x 10 5 células / ml. Dividir las células cada 2-3 días en la proporción de 1: 5. Células Pasaje EML suavemente y deseche la cultura después de pases para 10 generaciones.
- El agotamiento de las células positivas de linaje:
- Recoger las células por centrifugación a EML 200 xg for 5 min y lavar las células una vez con PBS. Recoger las células por centrifugación a 200 xg durante 5 min.
- Resuspender las células con PBS y contar las células con un hemocitómetro. Determinar la concentración de anticuerpos en la etapa de separación celular posterior de acuerdo con el número de las células (por favor, consulte las instrucciones ofrecidas por el proveedor del sistema de aislamiento de células).
- Aislar la negativa linaje (Lin-) células utilizando cóctel de anticuerpos linaje (cóctel de anticuerpos monoclonales conjugados con biotina CD5, CD45R (B220), CD11b, Anti-Gr-1 (Ly-6G / C), 7-4 y Ter-119 ) y un sistema magnético de separación de células activadas de acuerdo con las instrucciones del fabricante.
- La separación de las células Lin-CD34 + y Lin-CD34:
- Centrifugar las células Lin- de la etapa 1.3.3 a 200 xg durante 5 min. Resuspender el sedimento celular con PBS y contar las células con un hemocitómetro.
- Lavar las células dos veces con tampón FACS y sedimentar las células a 200 xgdurante 5 min.
- Etiqueta de cinco 1,5 ml tubos de microcentrífuga con el número 1, 2, 3, 4, 5, respectivamente. Resuspender las células con 100 l de tampón FACS por 10 6 células (10 6 células por tubo).
- Añadir 1 g de anticuerpo anti-ratón CD34 FITC al tubo 1 y el tubo 2 y mezclar suavemente los tubos.
- Incubar todos los tubos a 4 ° C durante 1 hora en la oscuridad.
- Añadir 0,25 g de anticuerpo anti-Sca1 conjugado con PE y 20 l de anticuerpos de cóctel Lineage APC conjugado al tubo 1, 0,25 g de PE-conjugado anticuerpo anti-Sca1 al tubo 3, y 20 l de anticuerpos de cóctel Lineage APC-conjugado con tubo 4.
- Mezclar todos los tubos suavemente e incubar las células a 4 ° C durante 30 min adicionales en la oscuridad.
- Añadir 300 l de tampón de FACS a las células y centrifugar las células a 200 xg durante 5 min.
- Lavar las células con 500 l de tampón de FACS para tres veces.
- Resuspender el sedimento celular en 500 l de FACS buffer.
- Usar las células en tubos de 2, 3, 4, y 5 para el establecimiento de la compensación. Aislar las células Lin-SCA + CD34 + y Lin-SCA-CD34 en tubo 1 utilizando FACS Aria.
2. Preparación de ARN y la Biblioteca de Construcción en secuenciación de alto rendimiento
- Aislamiento, análisis de la calidad y cuantificación de ARN:
- Extraer el ARN total de las células Lin-CD34 respectivamente utilizando TRIzol Lin-CD34 + y siguiendo el protocolo de fabrica ".
- Retire el ADN usando desoxirribonucleasa contaminada I (DNasa I) siguiendo el protocolo del fabricante. Opcionalmente, el ARN almacenar a -80 ° C en este paso para su uso posterior.
- Evaluar la calidad de RNA total usando Bioanalyzer acuerdo con las instrucciones ofrecidas por el proveedor. Utilice la muestra de ARN con Número Integridad ARN (RIN) del 9 lager.
- Biblioteca de construcción y secuenciación de alto rendimiento:
NOTA: Este protocolo describe ARN-Seq utilizando la plataforma Illumina. Paraotras plataformas de secuenciación, se requieren diferentes métodos de preparación de la biblioteca.- Utilice 0,1-4 g de ARN total por muestra de alta calidad para la preparación de la biblioteca. Normalmente 2 g de RNA total se pueden extraer de 10 5 células EML.
- Utilice un sistema de preparación de muestras de ARN-secuenciación para la purificación de ARN y la fragmentación, primera y segunda síntesis de ADNc, la reparación final, 3 'adenylation, la ligadura de adaptador y la amplificación por PCR, siguiendo los procedimientos estándares detallados de las instrucciones del proveedor.
- Positivamente seleccionar poliA mRNA utilizando oligo-dT perlas magnéticas y fragmentar el ARNm.
- Realizar la transcripción inversa usando cebadores aleatorios para obtener el ADNc y, posteriormente, sintetizar la segunda cadena de cDNA para generar ADNc bicatenario.
- Retire los 'domina y llenar el 5' 3 voladizos por la ADN polimerasa. Ciclasa 3 'extremos para evitar fragmentos de ADNc de ligando entre sí.
- Añadir adaptadores de indexación múltiplex a ambos extremos de la dscDNA. Lleve a cabo la PCR para el enriquecimiento de fragmentos de ADN.
- Mida el A260 / A280 para obtener información acerca de la concentración de la biblioteca usando un espectrofotómetro.
- Evaluar la calidad de la biblioteca y medir el rango de tamaño de los fragmentos de ADN utilizando un Bioanalyzer.
Análisis 3. Datos
Para tener una referencia de software utilizado en esta parte, por favor ver (Tabla 2).
- El procesamiento de archivos de datos para el análisis de aguas abajo:
- Convertir .bcl (archivo llamado base) presentar a .fastq archivo usando el software de yuca (Illumina, versión 1.8.2).
- Arranca la 'Terminal' en el sistema Linux. Ir a la carpeta de datos que contiene el archivo de datos de una máquina de secuenciación de Illumina HiSeq2000. Supongamos que la carpeta resultado es 'NASboy1 / JiaqianLabData / HiSeq_RUN / 2013_07_11 / 130627_SN860_0309_A_2013-166_H0PW9ADXX /', tipoen el comando en la figura S1A, y entrar en la carpeta de datos.
- Instale Casava 1.8.2 en el sistema Linux. Supongamos que el outputFolder es 'Unaligned', utilice el comando en la Figura S1B para preparar el archivo de configuración para la conversión. Utilice la opción --fastq-cluster-0 Cuenta para asegurar que se crea sólo un archivo .fastq para cada muestra. El archivo .fastq generado es en formato .gz. Descomprimir que para el análisis de aguas abajo (Figura S1B).
- Después de que se ha generado en la carpeta 'Unaligned', vaya a la carpeta 'Unaligned' (Figura S1C).
- Utilice el comando en la figura S1D para iniciar el proceso de conversión. El parámetro '-j' suministra el número de CPU que se utilizará.
- Después de que el sistema termine el proceso de conversión, vaya a la carpeta de resultado en la carpeta 'Unaligned' (Figura S1E).
- Utilice el comando en la figura S1F </ Strong> para descomprimir el archivo .fastq.gz en .fastq archivo en cada carpeta muestra.
- Convertir .bcl (archivo llamado base) presentar a .fastq archivo usando el software de yuca (Illumina, versión 1.8.2).
- Detectar nuevas transcripciones y evaluar el nivel de expresión utilizando Tuxedo Suite de 26:
- Asignar el extremo emparejado ARN-Seq lee a la referencia del genoma del ratón (UCSC versión mm9, obtenido a partir de http://cufflinks.cbcb.umd.edu/igenomes.html ) utilizando software de Tophat (versión 1.3.3) 27, que utiliza el Bowtie leer mapeador (versión 0.12.7) 28. Tophat se suministra con la opción "-no-nuevos-juncs" para mejorar la precisión de la estimación del nivel de expresión.
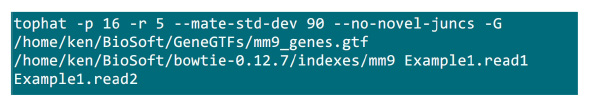
- Ponga los archivos en una carpeta .fastq donde se llevará a cabo el proceso de mapeo. Supongamos que hay dos archivos .fastq (cambiar el nombre a Example1.read1, Example1.read2) para una muestra de la secuencia de extremo emparejado, utilice el comando en la figura S2 para hacer el mapeo (ajustar los parámetros de acuerdo a la configuración del sistema).El parámetro "-p" suministra el número de la CPU que se utilizará. Los parámetros "r" y "-mate-STD-dev" se pueden obtener a partir de la biblioteca de control de calidad o inferirse de un subconjunto de alineado lee (Figura S2).
- Montar el mapeado lee en las transcripciones de ARN utilizando el software de Gemelos (versión 1.3.0) 29. Gemelos de ejecución utilizando el archivo de anotación de genes conocidos (mismo archivo .gtf utilizado por Tophat) y el archivo de .bam producido por Tophat.
- Después de Tophat termina de ejecutarse, en la misma carpeta, utilice el comando en la Figura S3A para ejecutar las mancuernas para construir el nivel de expresión del transcriptoma y estimación transcripción. El 'mm9_repeatMasker.gtf' y los archivos de la secuencia del genoma de la carpeta 'GenomeSeqMM9' se pueden obtener a partir de la UCSC Genome Browser.
- Los archivos genes.expr y transcripts.expr resultantes contienen el valor de la expresión de los genes y las transcripciones (isoformas). Copiar y pegarel contenido del archivo a un archivo Excel y manipular con la aplicación de hoja de cálculo (Figura S3B).
- Utilice el comando en la figura S3C para comparar el archivo resultante 'transcripts.gtf' en el fichero de referencia 'mm9_genes.gtf' con el fin de identificar nuevas transcripciones.
- El archivo .tmap resultante contiene el resultado de la comparación. Copie y pegue el contenido del archivo a un archivo de Excel y manipular con la aplicación de hoja de cálculo. Las transcripciones con código de clase 'u' pueden ser considerados como 'novela' en comparación con la referencia .gtf archivo proporcionado (Figura S3D).
NOTA: Para el análisis de aguas abajo de conveniencia, establezca los valores FPKM a 0,1 si los valores están bajo 0,1.
NOTA: El paso 3.2.3 - 3.2.6 es opcional para aquellos que desean mejorar la precisión de la estimación de expresión nuevas transcripciones. Esto tomará un tiempo mucho más largo, ya que la cartografía y la construcción transcriptoma deben ser rONU más de una vez.
- Ejecute Tophat utilizando los parámetros por defecto y luego ejecutar las mancuernas a .gtf archivo generado con el comando en la figura S3E.
- Compare el archivo resultante .gtf al archivo .gtf genoma de referencia con el comando en la figura S3F.
- Analizar el archivo .tmap resultado como se describe en el paso 3.2.2.4. Copie y pegue el contenido del archivo a un archivo de Excel y manipular con la aplicación de hoja de cálculo. Las transcripciones con código de clase 'u' pueden ser considerados como 'novela' en comparación con la referencia .gtf archivo proporcionado.
- Después de la etapa 3.2.5, hay un archivo .combined.gtf en la carpeta que se puede utilizar como el archivo de .gtf referencia. Una segunda aplicación de Tophat y gemelos se puede realizar como se describe en el paso 3.2.1 y 3.2.2 para obtener una estimación más precisa de FPKM nuevas transcripciones.
- Asignar el extremo emparejado ARN-Seq lee a la referencia del genoma del ratón (UCSC versión mm9, obtenido a partir de http://cufflinks.cbcb.umd.edu/igenomes.html ) utilizando software de Tophat (versión 1.3.3) 27, que utiliza el Bowtie leer mapeador (versión 0.12.7) 28. Tophat se suministra con la opción "-no-nuevos-juncs" para mejorar la precisión de la estimación del nivel de expresión.
- Detectar differentially expresó genes usando DESeq paquete de 30.
- La entrada de DESeq es una tabla de conteos de lectura cruda. Para obtener una tabla de este tipo, utilice el script htseq recuento distribuido con el paquete HTSeq Python que se puede descargar desde el sitio web HTSeq ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Asegúrese de que samtools, Python y programsare htseq recuento instalados en el sistema. Obtener los números de conteo de lectura primas de salida sombrero de copa con el comando en la figura S4A.
- Prepara 'Raw_Count_Table.txt', los archivos de los ExperimentDesign.txt 'utilizando Excel. Copiar y guardar el contenido en el formato .txt para el paquete DESeq R (Figura S4B).
- Instalar programa de I en el sistema. En la terminal, la voluntad appearas mensaje pantalla de tipo 'R' y pulse ENTER.A mostraron en la figura S4C.
- Leer 'Raw_Count_Table.txt ',' ExperimentDesign.txt 'en R utilizando el comando en la figura S4D.
- Cargar paquete DESeq usando el comando en la figura S4E.
- Condiciones factorizar en R (Figura S4F).
- Utilice el comando en la figura S4G para ejecutar la prueba binominal negativa sobre la mesa recuento normalizado.
- Utilice el comando en la figura S4H a genes expresados salida diferencial significativo en un archivo .csv.
- La entrada de DESeq es una tabla de conteos de lectura cruda. Para obtener una tabla de este tipo, utilice el script htseq recuento distribuido con el paquete HTSeq Python que se puede descargar desde el sitio web HTSeq ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Factores de transcripción de búsqueda '(TFS) a través de los valores FPKM muestras usando Excel. Intersección de table gen y mesa de TFS. Los genes pertenecen a ambas tabla se expresan diferencialmente factores de transcripción.
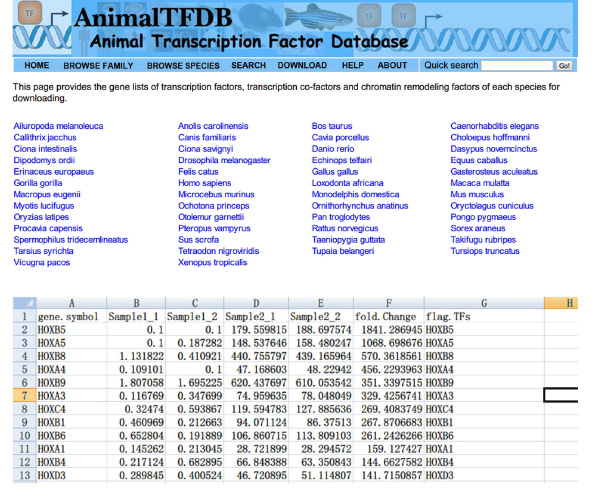
- Ir a la página web http://www.bioguo.org/AnimalTFDB/download.php y descargar los factores de transcripción. A continuación, las operaciones de búsqueda de los factores de transcripción DE en el Excel (< strong> Figura S5).
- Generación de archivo .bigwig para visualización UCSC genoma navegador.
- Descargar paquete de software 'bedtools' de la página web https://github.com/arq5x/bedtools2 e instalar el software en el sistema 31. Descargue las herramientas UCSC 'bedGraphToBigWig' de la página web http://hgdownload.cse.ucsc.edu/admin/exe/ e instalar el software en el sistema.
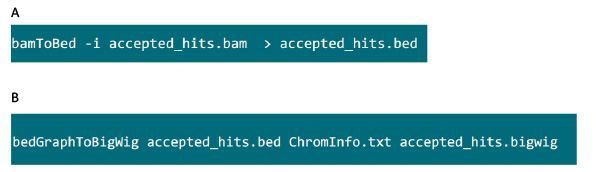
- En la carpeta que contiene el archivo .bam, utilice el comando en la figura S6A para convertir archivos .bam generada por el sombrero de copa en el archivo .bed.
- Después se produce el archivo .bed, utilice el comando en la figura S6B para generar archivo .bigwig. El archivo 'ChromInfo.txt' se puede obtener del siguiente url:arget = "_blank"> http://hgdownload.cse.ucsc.edu/goldenPath/mm9/database/chromInfo.txt.gz.
- Observar una pista personalizada en la UCSC Genoma del navegador. Consulte el sitio web http://genome.ucsc.edu/goldenPath/help/customTrack.html sobre cómo mostrar una pista personalizada utilizando la UCSC genoma navegador.

Figura S1: Convertir archivo .bcl a .fastq archivo usando el software de yuca.

Figura S2: Mapeo lee para hacer referencia genoma usando Tophat.
Figura S3: La detección de nuevas transcripciones y estimación del nivel de expresión.

Figura S4: Llamando gen expresado diferencial usando DESeq paquete.

Figura S5: Identificación de factores de transcripción expresados diferencialmente.

Figura S6: Conversión resultado de la asignación para la visualización de datos.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Con el fin de analizar los genes expresados diferencialmente en las células Lin-Lin-EML CD34- CD34 + y, se utilizó la tecnología de ARN-Seq. La figura 1 muestra el flujo de trabajo de los procedimientos. Después del aislamiento de células negativas de linaje por clasificación celular magnética, nos separamos células Lin-SCA-CD34 utilizando FACS Aria Lin-SCA + CD34 + y. Células enriquecidas EML-Lin se tiñeron con anti-CD34, anti-Sca1 y los anticuerpos de cóctel de linaje. Sólo las células Lin- fueron cerrada para el análisis de Sca1 y CD34 expresión. Dos poblaciones (células CEA-CD34- EML SCA + CD34 + y) se pudieron observar por análisis FACS (Figura 2) 6.
Después de la separación de células, se extrajo el ARN total de las células CD34 + CD34 y, respectivamente, y analizaron la calidad del ARN. La exactitud de los datos de ARN-Seq depende en gran medida de la calidad de la biblioteca de ARN-Seq y la calidad de RNA total es de vital importancia para la preparación de una biblioteca de alta calidad. Muestra de ARN de alta calidad debe tener un valor de DO 260/280 entre 1.8 y 2,0. Además de utilizar el espectrofotómetro, la calidad del ARN se evaluó adicionalmente con mayor precisión por Bioanalyzer. Figura 3 muestra un resultado de una muestra de ARN de alta calidad con el RIN igual a 9,4. Sólo de alta calidad de la muestra total de ARN con valor RIN superior a 9 se utilizó para la extracción de ARNm y procedimientos de construcción de bibliotecas posteriores.
Ribosomal RNA es el tipo más abundante de ARN en la célula. Actualmente dos estrategias principales, el agotamiento de rRNA o positivamente selección de ARNm poliadenilado (poli-A de ARNm), se utilizan para el enriquecimiento de ARN diana antes de la construcción de la biblioteca. Especies no ARN poliadenilado se pierden durante la selección de poli-A mRNA. Por el contrario, los métodos de agotamiento rRNA como RiboMinus podrían preservar especies de ARN no polyadenylated. El objetivo de nuestro estudio es la búsqueda de los genes expresados diferencialmente de codificación en dos tipos de células, por lo que se utilizó el método de poli-A la selección de ARNm para el enriquecimiento de ARN diana antes de constru bibliotecacción. Cuando la construcción de la biblioteca fue terminado, el tamaño de fragmentos de ADN en la biblioteca se comprobó antes de la secuenciación utilizando Bioanalyzer. La Figura 4 muestra una biblioteca de buena calidad con los picos de tamaño de los fragmentos a aproximadamente 300 pb.
En la etapa siguiente, la biblioteca se sometió a secuenciación de alto rendimiento. En principio, la duración más larga de leer será útil para la cartografía de lectura. Se puede reducir la probabilidad de que la lectura se asigna a varias ubicaciones debido a la similitud entre los genes duplicados o miembros de la familia de genes. Como las secuencias de secuenciación par de gama son de ambos extremos de los fragmentos, la longitud de lectura elegido debe ser menos de la mitad de la longitud media de fragmentos. Si el objetivo principal del experimento es medir el nivel de expresión en lugar de la construcción de la estructura de la transcripción, de extremo solo leer (75 o 100 pb) puede reducir el coste sin perder demasiada información. Secuenciación de extremo emparejado es más útil para la estructura de la transcripción de la construcción y más cortoleer longitud puede ser utilizado para reducir los costos. Ciertamente, cuando se dispone de suficiente financiación, se prefiere la longitud más larga de leer.
Para el análisis de expresión diferencial, hay muchos algoritmos alternativos que no sean DESeq. También hay uno incluido en el paquete gemelos llamado cuffdiff 32. DESeq es uno de los más ampliamente utilizado de recuento basado en los algoritmos de análisis de genes DE. DESeq método se basa en un modelo bien caracterizado estadísticas - distribución binomial negativa. En nuestra experiencia, DESeq es más estable en comparación con cuffdiff. Las primeras versiones de cuffdiff menudo dan significativamente diferente número de genes DE. Por lo tanto se utilizó para el análisis DESeq DE aquí.
Debido a que los factores de transcripción son cruciales para la determinación del destino celular, nos centramos en la transcripción significativamente expresadas diferencialmente factores de 33. El TFS cambió> 1,5 veces entre Lin-CD34 + y Lin-CD34- fueron encontrados y se muestran en el mapa de calor (Figure 5) 2. En particular, el nivel de expresión relativa de TCF7 en células lin-CD34 + es más de 100 veces mayor que en las células Lin-CD34-. Así TCF7 fue elegido para su posterior chip-secuenciación (cromatina inmunoprecipitación y secuenciación) el análisis y la prueba de funcionamiento para confirmar la función TCF7 's en la regulación de EML células auto-renovación y diferenciación 2.

Figura 1:. Flujo de trabajo de los procedimientos de lin-CD34 + y CD34- Lin-células fueron separadas por el sistema de separación celular magnética y método de clasificación de células activadas por fluorescencia. El ARN total se extrajo seguido de purificación de ARNm y construcción de la biblioteca. Después de análisis de la calidad de la biblioteca, las muestras se sometieron a secuenciación de alto rendimiento. Se analizaron los datos y diferencialmente expresados factores de transcripción fueron identificados.

Figura 2: Separación de células 6 células Lin- EML se enriquecieron mediante clasificación celular magnética Lin-CD34 + y Lin-CD34- EML.. Células Lin- se tiñeron con anti-CD34, anti-Sca1 y los anticuerpos de la mezcla linaje. Células Lin- fueron cerrada para la expresión de CD34 y Sca1. Lin-CD34 + SCA + y poblaciones de células Lin-CD34-CEA-EML fueron ordenados.

Figura 3:. Un representante de la alta calidad de la muestra total de ARN La calidad del ARN total fue evaluada por Bioanalyzer. El Número de la integridad del ARN es de 9,4 (FU, Fluorescencia unidades).

Figura 4:. Rango de tamaño de los fragmentos de la biblioteca-End vinculado el Se analizó la distribución de tamaños de ADN de la biblioteca utilizando Bioanalyzer. La mayoría de los fragmentos se encuentran dentro del rango de tamaño de 250-500 pb.

Para cada tipo de células, se realizaron expresados diferencialmente los factores de transcripción (> 1,5 veces) entre las células Lin-CD34 + y células CD34- Lin-2 de dos experimentos independientes: la Figura 5.. Hasta reguladas genes se indican como el rojo y los genes regulados hacia abajo se indican como de color verde.
Tabla 1: tampones y medios de cultivo de células.
| Software | Uso | Referencia | |||
| Bowtie 1.2.7 | Utilizado por Tophat para la cartografía | [28] | |||
| 1.3.3 Tophat | Mapeo lee de nuevo a genoma de referencia | [27] | |||
| Gemelos 1.3.0 | Transcripciones construcción y estimación del nivel de expresión | [29] | |||
| DESeq 1.16.0 | Análisis de expresión diferencial | [30] | Bedtools 2.18 | Convertir archivo .bam en archivo .bed | [31] |
| bedGraphToBigWig | Convertir archivo .bed a .bigwig archivo | http://genome.ucsc.edu/ |
Tabla 2: Lista de software para el análisis de datos.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Transcriptoma de mamíferos es muy compleja 34-38. Tecnología de ARN-Seq juega un papel cada vez más importante en los estudios de análisis de transcriptoma, novela detección transcripciones y solo nucleótido variación descubrimiento etc. Tiene muchas ventajas sobre otros métodos para el análisis de expresión de genes. Como se mencionó en la introducción, que supera los artefactos de hibridación de microarrays y se puede utilizar para identificar nuevas transcripciones de novo. Una limitación de ARN-secuenciación es relativa corta longitud de lectura en comparación con la secuenciación de Sanger. Sin embargo, con la rápida mejora de la tecnología de secuenciación, leer longitud está aumentando constantemente. En este artículo, ofrecemos métodos detallados del uso de esta tecnología para identificar potenciales reguladores clave en el ratón células EML auto-renovación y diferenciación.
El primer paso clave para este protocolo es el cultivo de células EML. Aunque EML es una línea celular precursor hematopoyético y se puedepropagado en gran cantidad con SCF. La condición de cultivo de células EML requiere más atención que las líneas de células inmortalizadas habituales. Las células deben ser alimentados y se pasaron a una base regular con un funcionamiento suave; de otro modo las células podrían cambiar en sus propiedades de auto-renovación y diferenciación y someterse a la muerte celular. Como el primer paso después de recoger suficientes células, se aislaron células negativas de linaje utilizando un sistema de clasificación de células activadas magnético. Entonces nos separamos células CD34- CD34 + y el uso de fluorescencia de clasificación de células activadas. Las células EML son normalmente passaged menos de 10 generaciones antes de usar para la extracción de ARN y el número de células CD34 + y células CD34- debe ser similar después de la separación. Si las dos poblaciones varían en gran medida del número de células, es aconsejable para descartar la cultura y volver a descongelar otro tubo de celular stock para la cultura.
Después de la separación de células CD34 + y células CD34-, se realizó la extracción de ARN total, un paso importante para este ptUdy. ARN de alta calidad es la base para la construcción de una biblioteca de alta calidad, que promete la exactitud de los datos de secuenciación. En este paso crítico, cualquier contacto con RNasa debe ser evitado. Todos los reactivos deben ser libre de RNasa. Es importante usar guantes en todo momento durante la manipulación de ARN. Muestra de ARN de alta calidad tiene un valor de DO 260/280 entre 1.8 y 2.0. Al recoger la fase acuosa que contiene ARN, tenga cuidado de no realizar ninguna fase orgánica con la muestra de ARN. Cualquier disolventes orgánicos residuales, tales como fenol o cloroformo en el ARN se traducirían en un OD260 / 280 valor inferior a 1,65. Si el valor OD260 / 280 es inferior a 1,65, precipitar el ARN de nuevo con etanol. Después de lavar con etanol al 75%, no hacer pellet de ARN Overdry. El secado de ARN precipitado completamente afectará a la solubilidad de ARN y dar lugar a un bajo rendimiento de ARN.
El siguiente paso clave para este protocolo es la preparación de la biblioteca. Después de la extracción de RNA total, usando un paso de DNasa para la eliminación del ADN contaminado is muy recomendable, ya que la contaminación de ADN podría dar lugar a la estimación errónea de la cantidad de ARN total utilizado. Se recomienda realizar el procedimiento aguas abajo inmediatamente después de aislamiento de ARN, ya que el almacenamiento después de largo plazo y procedimiento de congelación-descongelación, el ARN se degrada en cierta medida. Si los pasos siguientes después de aislamiento de ARN no pueden llevarse a cabo inmediatamente, almacenar el ARN en -80 ° C. Antes de RNA total se utiliza para la purificación de ARNm y la síntesis de ADNc, la calidad siempre debe ser revisado. Sólo alta calidad del ARN se puede utilizar para la preparación de la biblioteca. El uso de baja calidad o ARN degradado puede llevar a extremos sobre-representación de 3 '. Antes de la secuenciación, la calidad de la biblioteca se evaluó para asegurar la máxima eficiencia de la secuenciación.
En la parte de análisis de datos, después de realizar una carrera de Gemelos sin transcriptoma de referencia, hemos combinado las nuevas transcripciones con las transcripciones conocidas para formar una referencia .gtf archivo y correr Tophat y gemelos por segunda vez.Se recomienda este procedimiento de dos carreras, ya que esto proporciona la estimación más precisa FPKM que correr sólo una vez. Tras el análisis de datos, se identificaron los genes expresados diferencialmente. Experimentos intermedios se pueden realizar para validar la función de los genes in vitro e in vivo. En nuestra publicación anterior 2, se optó por los factores de transcripción significativamente expresadas diferencialmente e identificamos el sitio de unión del genoma de estos factores mediante la realización de inmunoprecipitación de la cromatina y secuenciación (chip-Sec). Además, se aplicó ensayo de caída shRNA para probar el efecto funcional de TCF7. Se encontró que en las células TCF7 desmontables, los genes regulados hasta-eran los genes altamente enriquecidas en células CD34, mientras que se encontraron los genes regulados hacia abajo para ser enriquecido significativamente en las células CD34 +. Por lo tanto, el perfil de expresión génica de células desmontables TCF7 desplaza hacia un parcialmente diferenciado CD34- state.Overall, utilizando células EML como un sistema modelojunto con la tecnología de ARN-Secuenciación y ensayos funcionales, hemos identificado y confirmado TCF7 como un importante regulador de la célula EML auto-renovación y diferenciación.
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| Antibiotic-Antimycotic | Invitrogen | 15240-062 | BHK cell culture |
| Anti-Mouse CD34 FITC | eBioscience | 11-0341-81 | FACS sorting |
| Anti-Mouse Ly-6A/E (Sca-1) PE | eBioscience | 12-5981-81 | FACS sorting |
| APC Mouse Lineage Antibody Cocktail | BD Biosciences | 558074 | FACS sorting |
| BD FACSAria Cell Sorter | BD Biosciences | Special offer sysmtem | FACS sorting |
| Corning™ Cell Culture Treated Flasks 75 cm2 | Corning incorporated | 430641 | Cell culture |
| Corning™ Cell Culture Treated Flasks 25 cm2 | Corning incorporated | 430639 | Cell culture |
| Deoxyribonuclease I, Amplification Grade | Invitrogen | 18068-015 | Library preparation |
| DMEM | Invitrogen | 11965-092 | BHK cell culture |
| DPBS | Gibco | 14190 | Cell culture |
| HI FBS | Invitrogen | 16140071 | BHK cell culture |
| Horse Serum | Invitrogen | 16050-122 | EML cell culture |
| IMDM | HyClone | SH30228.02 | EML cell culture |
| L-Glutamine | Invitrogen | 25030-081 | Cell culture |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | 130-090-858 | Isolation of lineage negative cells |
| NanoVue Plus spectrophotometer | GE Healthcare | 28-9569-62 | Quality control |
| Thermo Scientific™ Napco™ 8000 Water-Jacketed CO2 Incubators | Thermo Scientific | 15-497-002 | Cell culture |
| Penicillin-Streptomycin | Invitrogen | 15140-122 | EML cell culture |
| TRIzol® Reagent | Invitrogen | 15596-018 | RNA exraction |
| TruSeq™ RNA Sample Prep Kit v2 -Set B (48 rxn) | Illumina | RS-122-2002 | Library preparation |
| 2100 Electrophoresis Bioanalyzer Instrument | Agilent | G2939AA | Quality control |
| 0.25% Trypsin-EDTA | Gibco | 25200 | Cell culture |
| 0.45 µm Syringe Filters | Nalgene | 190-2545 | Cell culture |
References
- Chambers, S. M., Goodell, M. A. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 3, 201-211 (2007).
- Wu, J. Q., et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS genetics. 8, (2012).
- Ye, Z. J., et al. Complex interactions in EML cell stimulation by stem cell factor and IL-3. Proceedings of the National Academy of Sciences of the United States of America. 108, 4882-4887 (2011).
- Tsai, S., Bartelmez, S., Sitnicka, E., Collins, S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 8, 2831-2841 (1994).
- Weiler, S. R., et al. D3: a gene induced during myeloid cell differentiation of Linlo c-Kit+ Sca-1(+) progenitor cells. Blood. 93, 527-536 (1999).
- Ye, Z. J., Kluger, Y., Lian, Z., Weissman, S. M. Two types of precursor cells in a multipotential hematopoietic cell line. Proc Natl Acad Sci U S A. 102, 18461-18466 (2005).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10, 57-63 (2009).
- Chu, Y., Corey, D. R. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic acid therapeutics. 22, 271-274 (2012).
- Hornett, E. A., Wheat, C. W. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC genomics. 13, 361 (2012).
- Eswaran, J., et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific reports. 3, 1689 (2013).
- Wang, E. T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470-476 (2008).
- Wu, J. Q., et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proceedings of the National Academy of Sciences of the United States of America. 107, 5254-5259 (2010).
- Loraine, A. E., McCormick, S., Estrada, A., Patel, K., Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant physiology. 162, 1092-1109 (2013).
- Edgren, H., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome biology. 12, 6 (2011).
- Ilott, N. E., Ponting, C. P. Predicting long non-coding RNAs using RNA sequencing. Methods. 63, 50-59 (2013).
- Sun, L., et al. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC bioinformatics. 13, 331 (2012).
- Luo, S. MicroRNA expression analysis using the Illumina microRNA-Seq Platform. Methods in molecular biology. 822, 183-188 (2012).
- Bolduc, F., Hoareau, C., St-Pierre, P., Perreault, J. P. In-depth sequencing of the siRNAs associated with peach latent mosaic viroid infection. BMC molecular biology. 11, 16 (2010).
- Chepelev, I., Wei, G., Tang, Q., Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic acids research. 37, 106 (2009).
- Djari, A., et al. Gene-based single nucleotide polymorphism discovery in bovine muscle using next-generation transcriptomic sequencing. BMC genomics. 14, 307 (2013).
- Murphy, D. Gene expression studies using microarrays: principles, problems, and prospects. Advances in physiology education. 26, 256-270 (2002).
- Chen, K., et al. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS one. 8, 72567 (2013).
- Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 18, 1509-1517 (2008).
- Ramskold, D., Kavak, E., Sandberg, R. How to analyze gene expression using RNA-sequencing data. Methods in molecular biology. 802, 259-274 (2012).
- Glenn, T. C. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 11, 759-769 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 7, 562-578 (2012).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25, 1105-1111 (2009).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 10, 25 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature. 28, 511-515 (2010).
- Anders, S., Huber, W. Differential expression analysis for sequence count data. Genome biology. 11, 106 (2010).
- Quinlan, A. R., Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841-842 (2010).
- Cheranova, D., et al. RNA-seq analysis of transcriptomes in thrombin-treated and control human pulmonary microvascular endothelial cells. J Vis Exp. , (2013).
- Zhang, H. M., et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research. 40, 144-149 (2012).
- Wu, J. Q., et al. Systematic analysis of transcribed loci in ENCODE regions using RACE sequencing reveals extensive transcription in the human genome. Genome Biol. 9, 3 (2008).
- Wu, J. Q., et al. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques. 36, 690-696 (2004).
- Wu, J. Q., Shteynberg, D., Arumugam, M., Gibbs, R. A., Brent, M. R. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 14, 665-671 (2004).
- Dewey, C., et al. Accurate identification of novel human genes through simultaneous gene prediction in human, mouse, and rat. Genome Res. 14, 661-664 (2004).
- Wu, J. Characterize Mammalian Transcriptome Complexity. , LAP Lambert Academic Publishing. (2011).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}