

Summary
Hier berichten wir über die doppelte Kennzeichnung von neuralen Kammzellen und Blutgefäßen mit KükenGFP Neuralröhre Intraspezies Transplantation kombiniert mit intravaskulären DiI-Injektion. Diese experimentelle Technik ermöglicht es uns, die Entwicklung des NCC-abgeleiteten (enterischen) Nervensystems und des Gefäßsystems während der Organogenese gleichzeitig zu visualisieren und zu untersuchen.
Abstract
Alle sich entwickelnden Organe müssen sowohl mit dem Nervensystem (zur sensorischen und motorischen Steuerung) als auch mit dem Gefäßsystem (für Gasaustausch, Flüssigkeits- und Nährstoffversorgung) verbunden sein. Folglich entwickeln sich sowohl das Nerven- als auch das Gefäßsystem nebeneinander und haben auffallende Ähnlichkeiten in ihrer verzweigten Architektur. Hier berichten wir embryonale Manipulationen, die es uns ermöglichen, die gleichzeitige Entwicklung von neuronalem Kamm-abgeleitetem Nervengewebe (in diesem Fall dem enterischen Nervensystem) und dem Gefäßsystem zu untersuchen. Dies wird erreicht, indem Hühnerchimäre durch Transplantation diskreter Segmente des Neuralrohrs und des zugehörigen Neuralkamms in Kombination mit einer vaskulären DiI-Injektion im selben Embryo erzeugt werden. Unsere Methode verwendet transgene KükenGFP-Embryonen für die Transplantation nach Denspezies, was die Transplantationstechnik leistungsfähiger macht als das klassische Wachtel-Küken-Interspezies-Transplantationsprotokoll, das seit den 1970er Jahren mit großer Wirkung verwendet wird. ChickGFP-chick Intraspecies Transplantation erleichtert die Bildgebung von transplantierten Zellen und deren Projektionen in intakten Geweben und eliminiert mögliche Verzerrungen in der Zellentwicklung im Zusammenhang mit Artenunterschieden. Diese Methode nutzt den einfachen Zugang des Vogelembryons (im Vergleich zu anderen Wirbeltierembryonen) voll aus, um die Ko-Entwicklung des enterischen Nervensystems und des Gefäßsystems zu untersuchen.
Introduction
Der Hühnerembryon ist ein unschätzbarer Modellorganismus in der Wirbeltierentwicklungsbiologie, nicht zuletzt, weil seine Entwicklung in ovo experimentelle Manipulationen ermöglicht, die sonst bei Wirbeltieren, die sich in uteroentwickeln, unmöglich sind. Diese Zugänglichkeit und einfache Manipulation hat dazu geführt, dass der Küken-Embryo eine Schlüsselrolle bei vielen bahnbrechenden Entdeckungen auf dem Gebiet der Entwicklungsbiologie spielt. Zu den mächtigsten Techniken gehörte die Verwendung von Wachtel-Küken-Chimer-Embryonen zur Untersuchung des Zellschicksals, eine Methode, die von Professor Nicole Le Douarin in den 1970er Jahren1-3entwickelt wurde. Insbesondere Wachtelküken-Chimären waren besonders nützlich, um während der frühen Entwicklung stark wandernde neurale Kammzellpopulationen (NCC) genetisch zu markieren und zu folgen. NCC sind eine multipotente Population von Zugzellen, die im dorsalen Ektoderm an den Rändern des Neuralrohrs entstehen und zu einer Vielzahl von Zelltypen im gesamten Wirbeltierembryon führen. Dazu gehören craniofacial Strukturen (Knorpel, Knochen, Muskeln), Neuronen und Glia (im sensorischen und autonomen Nervensystem), Melanozyten, und eine Subpopulation von Zellen des endokrinen Systems2,4,5. Einer der wichtigsten Faktoren, die das NCC-Schicksal beeinflussen, ist ihre ursprüngliche Position entlang der vorderen-hinteren Achse des Neuralrohrs. Zum Beispiel entstehen enterische NCC, die die Neuronen und Glia des enterischen Nervensystems (ENS) hervorrufen, aus zwei diskreten Subpopulationen: die erste in der vagalen (kaudalen Hinterhirn) Region und die zweite in der sakralen Region des Neuralrohrs6-13. Inter- oder Intra-Spezies-Transplantation der entsprechenden Regionen des Neuralrohrs waren die Techniken der Wahl, um diese Zellen dauerhaft zu kennzeichnen und anschließend die Verfolgung von ihrer Geburt an den Rändern des Neuralrohrs bis zu ihren endgültigen Bestimmungspunkten innerhalb des Verdauungstraktes zu ermöglichen6,7,10.
Eine weitere embryonale Manipulation, die bei Küken im Vergleich zu anderen Tiermodellen leichter durchzuführen ist, ist die lebenswichtige Kennzeichnung des Gefäßsystems. Tatsächlich liegt der Kükenembryon, während er sich entwickelt, auf einem extraembryonalen Gefäßnetz, das Sauerstoff und Nährstoffe aus dem Eigelb zirkuliert. Dieses zugängliche Gefäßnetz, das sich auf der Oberfläche des Dotters befindet, kann als Tor zur Kennzeichnung des sich entwickelnden Gefäßsystems des Embryos während der Organogenese12,14-17verwendet werden. Die intravaskuläre Injektion verschiedener Farbstoffe, wie z.B. des lipophilen Farbstoffs DiI, ermöglicht es, alle luminisierten Gefäße des entstehenden Gefäßnetzes abzuschrägen/zu färben.
Da sich entwickelnde Organe sowohl mit dem Nervensystem (für die sensorische und motorische Steuerung) als auch mit dem Gefäßsystem (für Gasaustausch, Flüssigkeits- und Nährstoffversorgung) verbunden sein müssen, entwickeln sich die beiden Netzwerke nebeneinander und haben in ihrer Verzweigungsarchitektur18-20frierende Ähnlichkeiten. Hier berichten wir von embryonalen Manipulationen, die es uns ermöglichen, die gleichzeitige Entwicklung des nCC-abgeleiteten ENS zusammen mit dem Gefäßsystem während der Organogenese zu untersuchen. Dies wird durch die Erzeugung von Hähnchenchimären durch Transplantation diskreter Segmente des Neuralrohrs, einschließlich des Neuralkamms, in Kombination mit einer vaskulären DiI-Injektion erreicht. Als Fortschritt von Wachtel-Huhn-Chimären verwendet unsere Methode transgene GFP-Küken-Embryonen für die Transplantation innerhalb der Spezies, wodurch die Transplantationstechnik in Bezug auf bildgebende Zellen und ihre Projektionen leistungsfähiger wird und mögliche Verzerrungen im Zusammenhang mit Artenunterschieden beseitigt werden.
Protocol
1. Vorbereitung von Mikroskalpell für Neuralrohrablationen
- Gestalten Sie ein Mikroskalpell aus einer handelsüblichen Stahlnähnadel.
- Abflachen Sie zunächst die Nadel beidseitig mit einer Schleifscheibe, die auf einem angetriebenen Bankschleifer montiert ist.
- Beginnen Sie mit der Formgebung des Skalpells, zuerst auf einem groben Arkansas-Stein mit einer kontrollierten kreisförmigen Bewegung, in wechselnden Richtungen, auf beiden Seiten der Nadel.
- Tragen Sie auf den gleichen Schärfbewegungen auf einem extra feinen Grade Arkansas Stein, um ein ultrafeines Mikro-Skalpell zu formen, mit einer gut definierten Schneide (Abbildung 1A, B).
HINWEIS: Alternativen zum Mikroskalpell könnten elektrolitisch geschärfte Nadeln, handelsübliche Wolframnadeln oder gezogene Glasnadeln sein.
2. Wildtype und GFP-Eier in die gewünschte Phase eintauchen
- Dünge Hühnereier und transgene GFP-Hühnereier in einem gekühlten Inkubator bei 14 - 15 °C vor der Inkubation aufbewahren, da die Entwicklung bei dieser Temperatur gestoppt wird. Eier für ein paar Tage, bis zu einer Woche aufbewahren.
- Um mit der Entwicklung zu beginnen, legen Sie Wild-Typ und GFP-Eier horizontal auf ein Tablett und brüten gleichzeitig in einem befeuchteten (58 - 60%) Inkubator bei 37,5 oC, so dass sich Embryonen in einem entsprechenden Stadium für die Neuralrohrtransplantation befinden.
- Um Embryonen im 10 - 12- Entwicklungsstadium für die Durchführung von Vagal-Neuralrohr-Transplantationen zu erhalten, 1,5 Tage (33 - 38 Std.) und Stadium-Embryonen nach den Entwicklungstabellen von Hamburger und Hamilton21zu inkubieren.
3. Bereiten Sie Eier für Das Windowing und Grafting
- Bewegen Sie ein Ei nach dem anderen zu einem maßgeschneiderten Eierhalter zum Fenstern. Machen Sie ein kleines Loch in der Eierschale, indem Sie wiederholt mit gerader Schere auf die Oberseite des spitzen Endes des Eis tippen.
- 2 - 3 ml Albumin mit einer hypodermischen Nadel von 181,2 G und einer Spritze von 5 ml aus dem Ei entfernen. Das Entfernen des Albumins senkt das Eigelb und erleichtert das nachfolgende Fenster, ohne den Embryo zu beschädigen.
- Verwerfen Sie das Albumin. Versiegeln Sie das Loch mit einem kleinen Streifen aus klarem Klebeband, das mit einer feinen Schere auf Diegröße geschnitten ist.
- Tippen Sie mit einer gekrümmten Schere auf ein weiteres Loch in der Oberseite der Eierschale. Setzen Sie die Spitze der Schere in das Loch ein und arbeiten Sie in einer kreisförmigen Bewegung, um ein Fenster mit einem Durchmesser von 2 cm auf der Oberseite der Schale zu schneiden.
- Bewahren Sie die Schere in einer stationären Position auf und drehen Sie das Ei. Entsorgen Sie die entfernte Scheibe der Eierschale. Bei E1.5 ist der Embryo als dunklere gelbe Scheibe auf dem Dotter erkennbar.
- Entfernen Sie alle Schalenabfälle, die in das Ei gefallen sind, mit einer Pinzette. Entsorgen Sie alle unbefruchteten Eier (identifiziert durch einen kleinen weißen Fleck oben auf dem ansonsten hellgelben Eigelb).
4. Bereiten Sie den Wirtsembryon darauf vor, grafiertes Gewebe zu erhalten
- Stellen Sie das Stereomikroskop auf Augenhöhe ein und optimieren Sie die Ausrichtung der Schwanenhalslichtquelle, um den Embryo adäquat zu beleuchten, ohne Reflexionen zu verursachen.
- Um den Embryo selbstzu visualisieren, injizieren Sie eine kleine Menge indischer Tinte unter die Mitte der dunkleren gelben Scheibe, mit einem Mundrohr und einer gezogenen Glas-Mikropipette (Abbildung 1C, Oii).
- Bereiten Sie die Tinte 50:50 mit PBS mit Penicillin/Streptomycin in einer Endkonzentration von 100 g/ml vor. Setzen Sie die Mikropipette durch die Dottermembran außerhalb des Umfangs des Blastoderms und winkeln Sie dann vorsichtig ihre Spitze direkt unter dem Embryo.
- Geben Sie Tinte unter dem Embryo, indem Sie auf das Mundrohr blasen. Wenn Mundpipettieren nicht erlaubt ist, verwenden Sie stattdessen eine 1 ml Spritze. Achten Sie darauf, keine Luftblasen unter dem Embryo einzuführen, die zu einer Kontamination führen können, und entfernen Sie dann vorsichtig die Glas-Mikropipette. Dies ist ein heikler Schritt, der zum Tod des Embryos führen kann, wenn er nicht mit Präzision durchgeführt wird.
- Inszenieren Sie den Embryo unter Bezugnahme auf Hamburger und Hamilton21 und zeichnen Sie die Bühne in einem Laborbuch auf.
- Mit einem maßgeschneiderten Mikro-Skalpell (oder eine feine Wolframnadel) auf einem Nadelhalter montiert, machen eine sehr kleine Gash in der Vitelline-Membran, neben dem Bereich, wo die Mikro-Chirurgie durchgeführt wird.
- Tragen Sie vorsichtig 2 - 3 Tropfen PBS über den Membranriss (mit einer Glas-Mikropipette und dem Mundrohr) auf, um Raum zwischen embryo und membran zu schaffen. Schneiden Sie ein größeres Fenster in der Membran, um den gesamten Bereich freizulegen, in dem die Mikrochirurgie stattfinden wird.
- Entfernen Sie den neuronalen Röhrenbereich von Interesse mit dem Mikro-Skalpell, beginnend mit rostralen und kaudalen Transversalschnitten über das gesamte dorsale Neuralrohr (auf der Ebene von 1 bis 7 im Video).
- Bilateral zwischen dem Neuralrohr und den Soden schneiden, um das Neuralrohr von umgebenden Geweben zu trennen, ohne die Soden zu beschädigen.
- Trennen Sie das Neuralrohr sehr sanft vom darunter liegenden Notochord, der intakt bleiben sollte. Beachten Sie, dass eine erfolgreiche Neuralrohrexzision alle umgebenden Gewebe vollkommen intakt lässt (Abbildung 2).
- Entfernen Sie das ausgeschnittene Neuralrohr, indem Sie es in eine Glas-Mikropipette zerkrümen und dann entsorgen.
- Zeichnen Sie den Grad der Neuralrohrablation in einem Laborbuch auf. Der Wirtsembryo ist nun bereit, das Spender-Neuralrohr zu erhalten.
5. Bereiten Sie das Spender-Graft-Gewebe vor
- Wählen Sie einen fensterförmigen, stufengerechten GFP-Embryo aus, indem Sie unter einem fluoreszierenden Stereomikroskop mit FITC-Filter betrachten. Die GFP-Fluoreszenz macht es sehr einfach, die Soden zu visualisieren und den Embryo zu inszenieren.
- Sobald ein stadium-matched Embryo identifiziert wurde, entfernen Sie den Embryo aus dem Ei durch 4 Schnitte, mit Pascheff-Wolff Federschere (Abbildung 1C, l) in einer Rechteckform um den Embryo und nehmen Sie ihn dann vorsichtig mit einem Embryolöffel auf.
- Legen Sie den Embryo in ein quadratisches Uhrenglas mit einer Sylgard-Polymerbasis. Schütteln Sie den Embryo vorsichtig mit Dumont #5 Pinzette, um jedes angebrachte Eigelb zu entfernen. Entfernen Sie die Vitelline-Membran und fixieren Sie den Embryo mit rostfreien Minutien-Pins auf die Polymerbasis (Abbildung 1C).
- Mit der Federschere machen Sie 4 Einschnitte in rechteckiger Form um das Neuralrohr und die umgebenden Soden, in der gleichen Region, die aus dem Wirtsembryon entfernt wurde.
- Übertragen Sie mit einer Kunststoff-Transferpipette das Neuralrohr und das Gewebe aus dem Spender-GFP-Embryo in ein Uhrenglas mit 0,2% Pankreatin in Pen/Strep PBS.
- Lassen Sie die enzymatische Verdauung 10 min bei RT fortsetzen, um die Gewebe zu trennen. Verwenden Sie nach der Inkubation im Enzym die an einem Griff montierten Rost-Minutien-Pins, um das Neuralrohr manuell von allen angrenzenden Geweben zu trennen.
- Mit einer Glas-Mikropipette das dissoziierte Neuralrohr auf ein anderes Uhrenglas übertragen, das DMEM + 10% Serum(z.B.Ziege, Pferd oder fetales Kalb) auf Eis enthält, um die überschüssige Pankreatin zu spülen und die enzymatische Verdauung zu stoppen. Nach 5 min ist das sezierte Neuralrohr bereit, orthotopisch in den Kükenwirt eingepfropft zu werden (Abbildung 2 und S1).
6. Graft das Gewebe
- Übertragen Sie mit einer Glas-Mikropipette das sezierte Neuralrohr vorsichtig vom Uhrenglas auf den Wirtsembryon. Positionieren Sie das Neuralrohr in der korrekten vorderen-hinteren Ausrichtung und schieben Sie das Explant neben dem ausgeschnittenen Bereich des Kükenwirts vorsichtig mit dem Mikroskalpell. Lassen Sie ein kleines Fragment des Ektoderms an der dorsalen Oberfläche befestigt oder durch Schneiden eines kleinen Nicks ein, um die Ausrichtung des Neuralrohrs zu identifizieren.
- Verwenden Sie bei Bedarf das Mikroskalpell, um die Explantation auf die exakte Größe der ausgeschnittenen Region zu trimmen.
- Führen Sie das Neuralrohr vorsichtig in den abierten Bereich und positionieren Sie es so, dass die Dorsalseite richtig ausgerichtet ist. Verwenden Sie eine Glas-Mikropipette, die an einem Mundrohr befestigt ist, um PBS und/oder Flüssigkeit, die das Transplantat umgibt, zu entfernen. Dies hilft dem Spender und Wirtgewebe zu haften und das Transplantat zu etablieren.
- Versiegeln Sie das gesamte Fenster mit 24 mm breitem Klarband, um Austrocknung und Kontamination zu verhindern.
- Beschriften Sie den chimeric embryo durch Markierung mit einem Bleistift auf der Eierschale und notieren Sie seine Nummer im Laborbuch. Das Ei zur weiteren Entwicklung in den Inkubator zurückbringen.
7. Injizieren Sie DiI in die Blutgefäße des Wirtsembryons
- Zum gewünschten versuchsreichen Zeitpunkt (hier 3 - 10 Tage später) den chimeric embryo aus dem Inkubator holen und das klare Klebeband mit einer geraden Schere entfernen, um Zugang zum Embryo innerhalb des Eis zu erhalten.
- Bei Bedarf vergrößern Sie das Fenster in der Schale mit der Schere. Achten Sie darauf, die chorioallantoische Membran nicht zu beschädigen, wenn sie an der Schale befestigt ist, was zu Blutungen führen und die Kennzeichnung des Blutgefäßes gefährden würde.
- Wählen Sie eine zugängliche Vene auf dem Dotter, um sicherzustellen, dass der Blutfluss auf den Embryo gerichtet ist. Wählen Sie einen Verzweigungspunkt einer der Vitelline-Venen (Abbildung 3B, C).
HINWEIS: Bei E6.5 - E7.5 muss die chorioallantoische Membran möglicherweise sanft mit einer Pinzette zur Seite geschoben werden, um auf die Dottervenen zuzugreifen. Nach E8.5 besteht die einzige Möglichkeit darin, eine der chorioallantoischen Membranvenen zu injizieren, da die chorioallantoische Membran zu diesem Zeitpunkt den Embryo vollständig abdeckt. - Entfernen Sie die Vitelline-Membran über dem gewählten Injektionspunkt mit zwei Dumont-#5 Pinzette, indem Sie in entgegengesetzte Richtungen reißen.
- Brechen Sie eine gezogene Glasnadel mit einem Dumont-#5 und passen Sie ihren Durchmesser vor dem Laden mit CellTracker CM-DiI an die ungefähre Größe der Vene an. Machen Sie die DiI-Lagerlösung in DMSO bei 40 g/l und lagern Sie sie bei -20 °C. Bereiten Sie die Arbeitslösung in 0,3 M Saccharose/PBS in einer Konzentration von 4 g/l vor.
- Aspirieren Sie zwischen 5 - 10 l DiI in 0,3 M Saccharose/PBS in die Nadel mit Absaugung mit einem Mundrohr. Ältere Embryonen können bis zu 25 l oder mehr benötigen. Ab E8.5 haben Embryonen größere, muskulösere Venen, die möglicherweise mit einem Dumont-#5 in Position gehalten werden müssen, bevor sie mit der DiI-belasteten Glasnadel stechen.
- Setzen Sie die Nadel schnell in die Vene ein und blasen Sie sie stetig mit dem Mundrohr, damit der DiI den Blutfluss langsam verbinden kann, ohne ein Gerinnsel zu bilden. Alternativ können Sie einen Druckinjektor für die DiI-Lieferung verwenden.
8. Ernte Embryonen für Schnitt oder Wholemount-Untersuchung
- Um so viel DiI wie möglich im Embryo zu behalten, ernten Sie den Embryo unmittelbar nach der Injektion, indem Sie ihn auf einen perforierten Löffel schaufeln und die Blutgefäße und Bindegewebe mit einer geraden Schere schneiden, um den Embryo vom Eigelb zu befreien.
- Entfernen Sie alle losen Membranen und sezieren Sie die Organe von Interesse(d.h.die Lunge und den Verdauungstrakt in diesem Tutorial), mit großer Sorgfalt nicht das Gewebe zu komprimieren, die Diffusion des DiI schafft. Beheben Sie das Gewebe sofort durch Eintauchen in 4% PFA für 1 - 2 Stunden bei RT.
- Spülen Sie das Gewebe für 5 min in PBS, dann 15 min in PBS mit 5 g/ml DAPI. Montieren Sie die Proben auf einem überbrückten Mikroskopschlitten für die gesamte Montageuntersuchung oder betten Sie sie für Kryo-Sektionen ein.
Representative Results
Abbildung 1 zeigt typische Instrumente, die für die mikrochirurgische Isolierung und Transplantation des Neuralrohrs erforderlich sind. Abbildung 2 zeigt das Transplantationsverfahren. Nach der Transplantation werden Embryonen auf Transplantationserfolg untersucht. Dabei wird der Embryo unter einem Stereofluoreszenzmikroskop, typischerweise am Morgen nach der Mikrochirurgie, auf das Vorhandensein von transplantatabgeleiteten (GFP+) NCC untersucht. Wenn die Transplantation erfolgreich war, kann GFP+ NCC in der Nähe des Neuralrohrs und in frühen Migrationswegen beobachtet werden, die zum Vorgut führen. Wenn das Verfahren nicht erfolgreich war, wird GFP+ NCC nicht außerhalb des Neuralrohrs beobachtet, oder wenn sie im Wirt vorhanden sind, können sie in kleineren Zahlen vorhanden sein. Diese erfolglosen Embryonen werden verworfen. In der Regel werden 5-8 Neuralrohrtransplantationen an einem Tag durchgeführt, und von diesen 80% sind erfolgreich. Gründe für eine erfolglose Neuralrohrtransplantation sind der Tod des Embryos aufgrund von Gewebeschäden, die während der Mikrochirurgie entstanden sind, oder das Versagen des Neuralrohrs, sich in den Wirtsembryon zu integrieren. Letzteres kann auf eine schlechte Platzierung des Neuralrohrs innerhalb des Wirts oder durch ein neuronales Rohr von schlechter Qualität aufgrund einer schlechten Dissektionstechnik oder durch übermäßige Exposition gegenüber Dissoziationsenzym zurückzuführen sein. Der erste Screening-Schritt sowie ähnliche spätere Untersuchungen für GFP+-Zellen sind nützlich, da dies bedeutet, dass Zeit und Ressourcen nicht durch Experimente an Embryonen verschwendet werden, die keine GFP-markierte NCC im Darm haben.
Abbildung 3 zeigt das Verfahren zur DiI-Injektion der Blutgefäße. Die Effizienz/Erfolg der DiI-Injektionstechnik hängt davon ab: erstens, die Injektionsnadel auf den optimalen Durchmesser für die zielgerichtete Vene zu schneiden, zweitens eine präzise Geste beim Einsetzen der Nadel in die Vene (also nicht durch die andere Seite zu durchbohren), und drittens, um zu vermeiden, dass die Nadel während der Injektion durch eine konstante Rate verstopft wird. Wenn einer dieser drei Parameter falsch ausgeführt wird, wird der Embryo ausbluten oder mehrere Stunden brauchen, um sich zu erholen, bevor ein zweiter Versuch unternommen wird, da die Blutung es fast unmöglich macht, sofort wieder injizieren zu können. Erfolgreiche Embryonen sollten sofort unter einem Stereofluoreszenzmikroskop ausgewählt und schnell seziert werden. Bei erfolgreichen Embryonen sind DiI-gekennzeichnete Blutgefäße im gesamten Embryo vorhanden (Abbildung 3C,D) einschließlich Kapillarbetten (Abbildung 3D).
Bei der Ernte von Embryonen und der Untersuchung von Gewebeabschnitten oder ganzen Typische Ergebnisse zeigen GFP+ NCC innerhalb des primitiven ENS und die feine Struktur der DiI-markierten Darm-Blutgefäßnetze (Abbildung 4) Ganzmontagepräparate können mittels konfokaler Mikroskopie untersucht werden, wobei Bildstapel dreidimensionale (3D) Rekonstruktionen erzeugen, die die Zusammenhänge zwischen den feinen Projektionen der GFP+ ENS-Zellen und dem DiI-gebeizten Gefäßsystem zeigen ( Abbildung4 A-C; G-I; Videos 1 und 2).

Abbildung 1. Empfohlene Mikrochirurgie-Instrumente. (A) Mikroskalpell formiert aus einer Nähnadel. (B) feiner Arkansas-Stein zur Gestaltung eines Mikroskalpells. (C) a) gerade Schere, b) gebogene Schere, c) 5 ml Spritze mit 181,2 G hypodermischer Nadel, d) Kunststoffpipette, e) maßgefertigter Eierhalter, f) schwarze Tinte, g) quadratische Uhr Glas, h) quadratische Uhr Glas mit schwarzer Sylgard Basis, i) Mikro-Skalpell auf Nadelhalter, j) minutien Stifte, k) Minutien oder Wolframnadel auf Nadelhalter, l) Pascheff-Wolff Federschere, m) Dumont #5 Pinzette, n) perforiert Löffel, oi) kurze Feuer-gezogene Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 2. Intraspecies Neuralrohrtransplantation. Kükenembryo/GFP Neuralröhrenbilder wurden von Delalande et al.modifiziert. 12. Die Vaskularisation ist für die Darmbesiedlung durch enterische Neuralkammzellen nicht notwendig. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 3. Intravenöse DiI-Injektion. (A) Empfohlene Instrumente: a) CellTracker CM-DiI Tropfen auf Parafilm, b) gezogene Glasinjektionsnadel, c) Mundrohr. (B) Schematische Darstellung der intravenösen DiI-Injektion in E4 chimäre Kükenembryon. (C) in der intravenösen Injektion von ovo DiI mit feiner Glasnadel, die DiI enthält, die in die Vene (Pfeil) eingeführt wird. (D) E4 chimeric embryo post DiI injection (red) with GFP+ neural tube (arrow). (E) DiI gefärbtes Feines Blutgefäßnetz in einem lebenden Embryo, 24 Stunden nach der Injektion. Br: Gehirn; H: Herz; LB: Gliedmaßenknospe; A: Allantois. Die Bilder in (C) und (D) wurden von Delalande et al. 12 Die Vaskularisation ist für die Darmbesiedlung durch enterische neurale Kammzellen nicht notwendig. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 4: Repräsentative Ergebnisse im Magen und Caecum eines E5.5-Küken-Embryos. (A-C) 3dimensionale (3D) Rekonstruktion eines konfokalen Bildstapels im Bereich des Magens, der (D) die GFP+ enterischen Neuralkammzellen (ENCC) (E) das DiI-gefärbte Gefäßsystem und (F) ein zusammengeführtes Bild beider Netzwerke D-F histologische Abschnitte auf der Ebene des Magens zeigt (G) das GFP+ ENCC (H) das DiI-gebeiztes Gefäßsystem und (I) ein zusammengeführtes Bild beider Netzwerke D-F histologische Abschnitte auf der Ebene des Magens zeigt (G) das GFP+ ENCC (H) das DiI-gebeiztes Gefäßsystem und (I) ein zusammengeführtes Bild beider Netzwerke D-F histologische Abschnitte auf der Ebene des Magens, das (G) das GFP+ ENCC (H) das DiI-gebeiztes Gefäßsystem und (I) ein zusammengeführtes Bild beider Netzwerke zeigt. Kerne sind mit DAPI (Cyan) gefärbt. (G-H) 3D-Rekonstruktion eines konfokalen Bildstapels im Caecum-Bereich, der (A) die GFP+ ENCC-Migrationsfront in Grün, (B) das DiI-gebeiztes Gefäßsystem in Rot und (C) ein zusammengeführtes Bild beider Netzwerke zeigt. Bilder (A-F) wurden von Delalande et al.modifiziert. 12 Die Vaskularisation ist für die Darmbesiedlung durch enterische neurale Kammzellen nicht notwendig. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

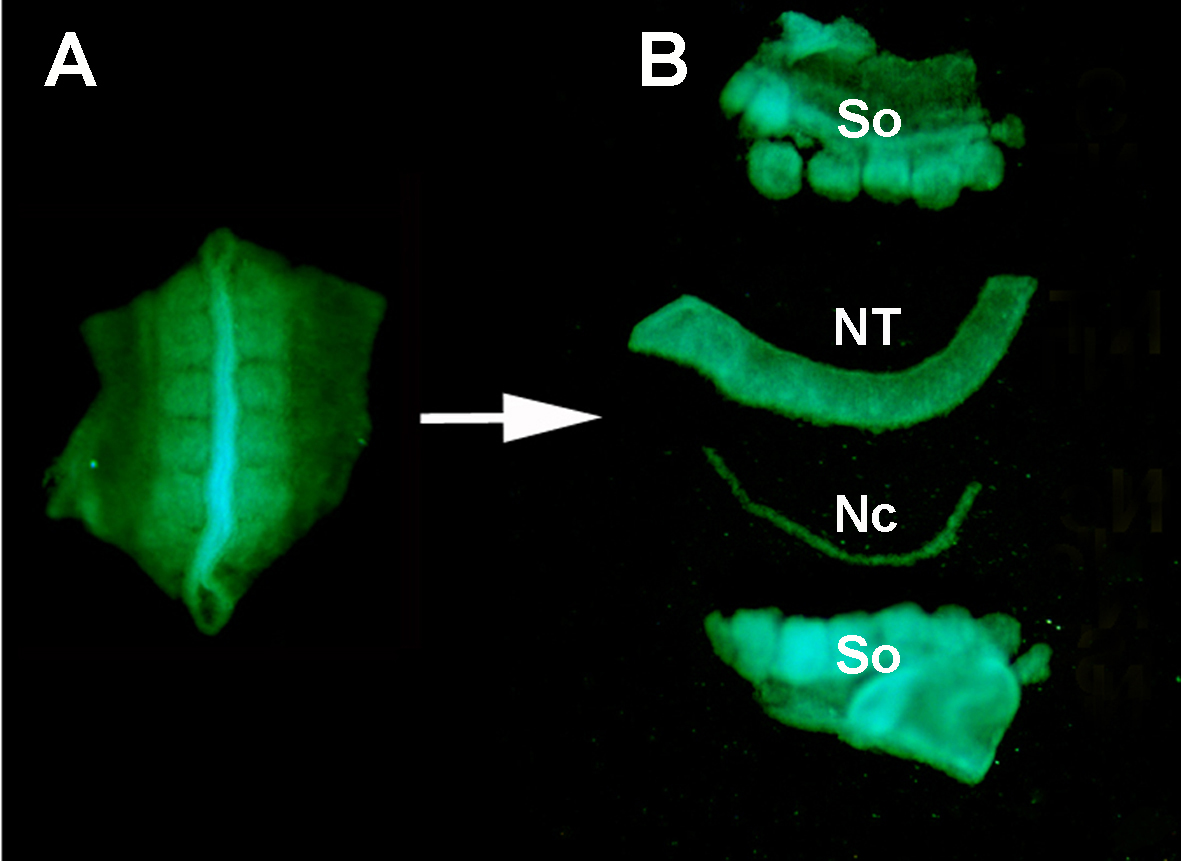
Abbildung S1. Isolierung eines Spenders GFP+ Neuralrohr aus dem umgebenden Gewebe durch enzymatische Verdauung und Mikrosektion. (A) GFP+ Neuralrohr und benachbarte Sos, die aus dem Spenderembryon seziert wurden. (B) Isoliertes Neuralrohr nach Pankreatinverdauung und Mikrosektion mit rostfreien Minutienstiften. Also: das ist so; NT: Neuralrohr; Nc: Notochord.

Video 1. 3-dimensionale 360°-Drehung des Bildes in Abbildung 4C, die das Gefäßsystem und das ENCC im Magen bei E5.5 (HH27-28) zeigt. Bitte klicken Sie hier, um dieses Video anzuzeigen.

Video 2. 3-dimensionale 360°-Drehung des Bildes in Abbildung 4I, die das Gefäßsystem und die ENCC-Migrationsfront im Bereich des Caecums bei E5.5 (HH27-28) zeigt. Bitte klicken Sie hier, um dieses Video anzuzeigen.
Discussion
Die Hier beschriebene Methode der neuralröhreninternen Neuralrohrtransplantation in Kombination mit der hier beschriebenen Blutgefäßkennzeichnung nutzt den einfachen Zugang des Aviären Embryos innerhalb des Eis (im Vergleich zu anderen Wirbeltierembryonen) voll aus, um die gemeinsame Entwicklung eines Elements des autonomen Nervensystems (ENS) und des Gefäßsystems zu untersuchen.
Für die Kennzeichnung von NCC-Derivaten hat die von uns beschriebene Küken-GFP-Küken-Intraspezies-Transplantationsmethode eine Reihe von Vorteilen gegenüber der klassischen Wachtel-Küken-Chimäre-Methode, die vor über 40 Jahren1-3eingeführt wurde. Erstens ist die GFP-Fluoreszenz unter FITC-Licht extrem hell, da GFP+-Zellen in lebenden chimer Embryonen leicht erkennbar sind. Dies ermöglicht es, den Erfolg des Transplantats in ovozu überprüfen, während die Quail-Küken-Transplantation erfordert, dass der Embryo mit QCPN getötet, verarbeitet und immungefärbt wird, bevor der Erfolg des Transplantats ermittelt werden kann2. Zweitens ist die GFP-Expression im transgenen KükenGFP zytoplasmisch, daher werden nicht nur Zellkörper gelabelet, sondern auch die Projektionen der transplantierten Zellen visualisiert22. Dies ermöglicht die Beobachtung komplizierter neuronaler Netzwerke bei hoher Auflösung (beachten Sie, dass feine Projektionen am besten visualisiert werden, wenn die Probe mit Anti-GFP-Antikörpern immunisiert wird). Da die QCPN-Kennzeichnung auf den Wachtelzellkern beschränkt ist, werden solche Netzwerke nicht mit Wachtel-Küken-Chimären aufgedeckt. Drittens eliminiert die Transplantation nach Innenarten mögliche Artenunterschiede zwischen den Zellen innerhalb des chimären Embryos. Da Wachtelembryonen eine kürzere Inkubationszeit haben als Küken (19 Tage versus 21 Tage) wurde vorgeschlagen, dass Wachtelzellen eine höhere Proliferationsrate als Kükenzellen haben, was potenziell die Entwicklung der chimaschigen Gewebe beeinflussen könnte23. Interessanterweise hat sich auch in Pflanzen gezeigt, dass die Veredelung zwischen Arten umfangreiche Veränderungen in den DNA-Methylierungsmustern im Wirt 24hervorrufen könnte. Viertens erleichtert das KükenGFP Back-Transplantationsexperimente, um Themen wie NCC-Schicksal und Zellbindung25anzusprechen. Fünftens ist das transgene KükenGFP auch für viele andere Techniken nützlich, einschließlich FACS-Sortierung von GFP+-Zellsubpopulationen, organotypische Kultur von Organen, die GFP+-Zellen enthalten, genetische Manipulation von GFP+-veredeltem Gewebe durch Elektroporation von Expressionsplasmiden26und anderen bildgebenden Technologien wie der optischen Projektionstomographie27.
Der Neuronistentransplantationsansatz kann durch mikrochirurgisches Ersetzen kürzerer Mengen an Neuralrohr enthaton verändert werden. Durch die Verwendung kleinerer Segmente von Neuralrohren ist die Mikrochirurgie potenziell weniger schädlich für den Embryo und das Überleben kann verbessert werden. Der Nachteil der Transplantation weniger Neuralrohr ist jedoch, dass die Anzahl der GFP+ NCC im Wirt reduziert werden. Die Benutzer könnten versuchen, ein Gleichgewicht zwischen der Menge der neuronalen Röhrchen transplantiert zu erreichen, um ein optimales Überleben der Embryonen zu geben, und die Anzahl der GFP+ NCC im Wirtsdarm ausreichend, um informative Ergebnisse zu liefern.
Für die Gefäßmalerei hat DiI den Vorteil, dass seine Fluoreszenz sehr hell und robust ist. Außerdem hat es die Fähigkeit, während der Fixierung zu diffundieren und die Färbung der feinsten geöffneten Kapillaren zu sichern. Da es sich um einen lebenswichtigen Farbstoff handelt, können Embryonen das Injektionsverfahren überleben und sich mit einem gebeizten Gefäßsystem weiterentwickeln (bis zu 24 Stunden in unseren Händen, obwohl die Färbung im Laufe der Zeit punktueller wird, siehe Abbildung 3E). Die Kombination von KükenGFP-Veredelung mit DiI Gefäßmalerei ist daher kompatibel mit Live-Bildgebung. Neben all diesen Vorteilen ist zu beachten, dass die Gefäßinjektion nur luminisierte Gefäße kennzeichnet und daher keine ungeöffneten Kapillaren, Endothelspitzenzellen oder isolierte Endothelzellen identifiziert. Weitere Fortschritte bei der Aviären Transgenese könnten jedoch neue Wege zur Umgehung solcher Probleme bieten, wie Experimente mit Tg(tie1:H2B-eYFP) Wachtelembryonen zur Untersuchung der vaskulären Morphogenese28zeigen. Eine weitere Einschränkung dieser Technik besteht darin, dass für eine effektive Gefäßkennzeichnung in Embryonen bei E7.5 und darüber hinaus größere Mengen an Farbstoff injiziert werden müssen, was Experimente teuer machen kann. Eine Modifikation der Technik könnte jedoch eine kostengünstige Blutgefäßkennzeichnung mit Hervorhebungstinte14beinhalten, obwohl dieser Ansatz nicht in unseren Händen erprobt wurde.
Kritische Schritte der Verfahren sind der Prozess der Visualisierung des Embryos durch Injektion von Tinte unter die Blastodisc. Wenn die Membran, die das Eigelb bedeckt, in diesem Stadium von der mit Tinte gefüllten Nadel gerissen wird, ist das Überleben des Embryos stark gefährdet. Auch ist es wichtig, bei der Vorbereitung eines Spender-Neuralrohrs, dass das Gewebe nicht für eine übermäßig lange Zeit in Pankreatin gelassen wird (betrachten sie etwa 10 min als Maximum). Längere Exposition gegenüber Pankreatin schädigt das Gewebe und das Neuralrohr ist dann schwer zu handhaben und es wird nicht gut in den Wirt integrieren. Die Erlangung von Erfahrungen mit der DiI-Injektionstechnik an wilden Embryonen ist vor der Injektion von chimer Embryonen unerlässlich, da für jeden Embryo im Allgemeinen nur ein Injektionsversuch möglich ist. DiI-Volumen und Nadeldurchmesser sind kritische Parameter für jeden Embryo und sollten anhand von Wildtyp-, Stufen-Matched-Kontrollen bewertet werden.
Zusammenfassend lässt sich sagen, dass unsere Dual-Labeling-Methode der Neuralrohrtransplantation und DiI-Gefäßmalerei in lebenden Kükenembryonen verwendet werden kann, um die Wechselbeziehungen zwischen NCC und Blutgefäßnetzen während der Organogenese zu untersuchen. Angesichts der Noch weitgehend unbekannten Mechanismen, die für die Festlegung der korrekten Zielinnervation und Vaskularisation während der Organentwicklung verantwortlich sind, birgt diese Methode Potenzial für zukünftige Entdeckungen in diesem Bereich.
Disclosures
Die Autoren erklären, dass sie keine konkurrierenden finanziellen Interessen haben.
Acknowledgments
Befruchtete GFP-Hühnereier wurden von Prof. Helen Sang, dem Roslin Institute und der University of Edinburgh, UK, geliefert. Die Roslin Transgenic Chicken Facility wird vom Wellcome Trust und vom Biotechnology and Biological Sciences Research Council (BBSRC) finanziert. Die Arbeit wurde teilweise von Great Ormond Street Hospital Children es Charity, London, UK, finanziert und NT unterstützt. Die Autoren danken Ben Jevans vom UCL Institute of Child Health für die Hilfe bei der Vorbereitung von Embryonen für die Pfropfung.
Materials
| Name | Company | Catalog Number | Comments |
| Fertilised chick eggs | Henry Stewart and Co, Louth, UK | ||
| Fertilised GFP chick eggs | The Transgenic Chicken Facility, The Roslin Institute, The University of Edinburgh | ||
| Egg incubator (Profi-H Hatcher) | Lyon Technologies, CA, USA | 910-033 | |
| 14C Incubator | Precision Cooled Incubator, Leec Ltd., Nottingham, UK | Model LT2 | |
| Stereo-microscope | LEICA | Model MZ 12.5 | |
| Digital Camera | LEICA | DC500 | |
| Image acquisition software | LEICA | IM50 | |
| Goose neck halogen cold light source | Advanced Imaging Concepts, Inc | KL 1500 LCD | |
| 181⁄2 G hypodermic needle | SIGMA - ALDRICH | HSWNH181 | |
| Pancreatin | SIGMA - ALDRICH | P3292 | |
| DMEM | SIGMA - ALDRICH | D5030 | |
| Goat serum | SIGMA - ALDRICH | G6767 | |
| 5 ml syringe | SIGMA - ALDRICH | Z248010 | |
| Mouth tube | SIGMA - ALDRICH | A5177 | |
| Sigma Pasteur pipettes non-plugged, L 5 3/4 in. | SIGMA - ALDRICH | S6018 | |
| Transfer pipettes, polyethylene | SIGMA - ALDRICH | Z350796 | |
| Borosillicate glass capillaries, thin wall without filament | Harvard apparatus | PY8 30-0035 | |
| Iris Scissors - ToughCut | Fine Science Tools | 14058-09 | |
| Curved Iris Scissors - ToughCut | Fine Science Tools | 14059-09 | |
| Needle holders (Nickel-plated pin holder) | Fine Science Tools | 26018-17 | |
| Pascheff-Wolff Spring Scissors | Fine Science Tools | 15371-92 | |
| Dumont #5 forceps | Fine Science Tools | 11251-30 | |
| Minutien pins | Fine Science Tools | 26002-15 | |
| Dumont AA forceps, Inox Epoxy- coated | Fine Science Tools | 11210-10 | |
| Perforated spoon | Fine Science Tools | 10370-18 | |
| Tungsten needles (0.125mm diameter) | Fine Science Tools | 10130-05 | |
| Sellotape (clear, 24 mm width) | Any Supplier | ||
| Pen/Strep (Penicillin, Streptomycin) Solution | VWR international | 101447-068 | |
| Sylgard 184 silicone elastomer kit | Dow Corning | S09 512 516 | |
| Pelikan black ink | Pelikan | 211-169 | |
| CellTracker CM-DiI | Molecular Probes | C-7001 | |
| DAPI (4',6-Diamidino-2-Phenylindole, Dihydrochloride) | Molecular Probes | D1306 | |
| Settings for glass needle puller | Sutter Instruments | Flaming/Brown micropipette puller model P-86 | |
| Heat 950; Pull 150; Velocity 100; Time 200; Pressure 500 |
References
- Le Douarin, N. A biological cell labeling technique and its use in expermental embryology. Developmental Biology. 30, 217-222 (1973).
- Teillet, M. A., Ziller, C., Le Douarin, N. M. Quail-chick chimeras. Methods in Molecular Biology. 461, 337-350 (2008).
- Teillet, M. A., Ziller, C., Le Douarin, N. M. Quail-chick chimeras. Methods in Molecular Biology. 97, 305-318 (1999).
- Garcia-Castro, M., Bronner-Fraser, M. Induction and differentiation of the neural crest. Current Opinion In. Cell Biology. 11, 695-698 (1999).
- Bhatt, S., Diaz, R., Trainor, P. A. Signals and switches in Mammalian neural crest cell differentiation. Cold Spring Harbor Perspectives In Biology. 5, (2013).
- Burns, A. J., Douarin, N. M. The sacral neural crest contributes neurons and glia to the post-umbilical gut: spatiotemporal analysis of the development of the enteric nervous system. Development. 125, 4335-4347 (1998).
- Burns, A. J., Le Douarin, N. M. Enteric nervous system development: analysis of the selective developmental potentialities of vagal and sacral neural crest cells using quail-chick chimeras. The Anatomical Record. 262, 16-28 (2001).
- Burns, A. J., Delalande, J. M., Le Douarin, N. M. In ovo transplantation of enteric nervous system precursors from vagal to sacral neural crest results in extensive hindgut colonisation. Development. 129, 2785-2796 (2002).
- Burns, A. J., Champeval, D., Le Douarin, N. M. Sacral neural crest cells colonise aganglionic hindgut in vivo but fail to compensate for lack of enteric ganglia. Developmental Biology. 219, 30-43 (1006).
- Wang, X., Chan, A. K., Sham, M. H., Burns, A. J., Chan, W. Y. Analysis of the sacral neural crest cell contribution to the hindgut enteric nervous system in the mouse embryo. Gastroenterology. 141, 992-1002 (2011).
- Goldstein, A. M., Hofstra, R. M., Burns, A. J. Building a brain in the gut: development of the enteric nervous system. Clinical Genetics. 83, 307-316 (1111).
- Delalande, J. M., et al. Vascularisation is not necessary for gut colonisation by enteric neural crest cells. Developmental Biology. 385, 220-229 (2014).
- Anderson, R. B., Stewart, A. L., Young, H. M. Phenotypes of neural-crest-derived cells in vagal and sacral pathways. Cell And Tissue Research. 323, 11-25 (2006).
- Takase, Y., Tadokoro, R., Takahashi, Y. Low cost labeling with highlighter ink efficiently visualizes developing blood vessels in avian and mouse embryos. Development, Growth & Differentiation. 55, 792-801 (2013).
- Bates, D., Taylor, G. I., Newgreen, D. F. The pattern of neurovascular development in the forelimb of the quail embryo. Developmental Biology. 249, 300-320 (2002).
- Mayes, P., Dicker, D., Liu, Y., El-Deiry, W. Noninvasive vascular imaging in fluorescent tumors using multispectral unmixing. BioTechniques. 45, 459-460 (2008).
- Li, Y., et al. Direct labeling and visualization of blood vessels with lipophilic carbocyanine dye DiI. Nature Protocols. 3, 1703-1708 (2008).
- Eichmann, A., Thomas, J. L. Molecular parallels between neural and vascular development. Cold Spring Harbor Perspectives In Medicine. 3, a006551 (2013).
- Weinstein, B. M. Vessels and nerves: marching to the same tune. Cell. 120, 299-302 (2005).
- Carmeliet, P., Tessier-Lavigne, M. Common mechanisms of nerve and blood vessel wiring. Nature. 436, 193-200 (2005).
- Hamburger, V., Hamilton, H. L. A series of normal stages in the development of the chick embryo. Journal of Morphology. 88, 49-92 (1951).
- Barraud, P., et al. Neural crest origin of olfactory ensheathing glia. Proceedings of the National Academy of Sciences of the United States of America. 107, 21040-21045 (2010).
- Senut, M. C., Alvarado-Mallart, R. M. Cytodifferentiation of quail tectal primordium transplanted homotopically into the chick embryo. Brain Research. 429, 187-205 (1987).
- Wu, R., et al. Inter-species grafting caused extensive and heritable alterations of DNA methylation in Solanaceae plants. PLoS One. 8, e61995 (2013).
- Freem, L. J., Delalande, J. M., Campbell, A. M., Thapar, N., Burns, A. J. Lack of organ specific commitment of vagal neural crest cell derivatives as shown by back-transplantation of GFP chicken tissues. The International Journal Of Developmental Biology. 56, 245-254 (2012).
- Delalande, J. M., et al. The receptor tyrosine kinase RET regulates hindgut colonization by sacral neural crest cells. Developmental Biology. 313, 279-292 (2008).
- Freem, L. J., et al. The intrinsic innervation of the lung is derived from neural crest cells as shown by optical projection tomography in Wnt1-Cre;YFP reporter mice. Journal of Anatomy. 217, 651-664 (2010).
- Sato, Y., et al. Dynamic analysis of vascular morphogenesis using transgenic quail embryos. PloS One. 5, e12674 (2010).

{kind=link}