

Introduction
Resistenza Myxovirus (Mx) proteine sono una parte importante della difesa immunitaria innata contro i patogeni virali. Queste proteine sono grandi GTPases Dynamin simili che sono indotti da tipo I e tipo III interferoni. I corrispondenti geni Mx sono presenti in quasi tutti i vertebrati in una o più copie e loro prodotti genici inibiscono un'ampia gamma di virus, compresi Orthomyxoviridae (ad es., Virus dell'influenza), Rhabdoviridae (ad es., Virus della stomatite vescicolare), Bunyaviridae (es. , virus crosse la) e Retroviridae (ad esempio, il virus dell'immunodeficienza umana-1) 1-4. Non è chiaro come queste proteine riconoscono una così ampia gamma di virus, senza alcuna sequenza primaria apparente condiviso motivi in questi virus. Analizzando l'interazione delle proteine Mx con i loro obiettivi virali, potenzialmente coinvolge complessi di ordine superiore con altri fattori della cellula ospite, aiuterà a comprendere i meccanismi molecolari tCappello si sono evoluti nella corsa agli armamenti tra virus e loro ospiti.
L'interazione tra le proteine di mammiferi Mx e target virali è stato studiato più ampiamente per MxA umana. MxA umana può inibire la replicazione di molti virus, tra cui il ortomixovirus influenza A e virus Thogoto. MxA lega complessi ribonucleoproteici virus Thogoto (vRNPs), impedendo così il loro ingresso nucleare, che si traduce nel blocco di infezione 5. Questa interazione tra MxA e Thogoto vRNPs virus è stata dimostrata con il co-sedimentazione e co-immunoprecipitazione esperimenti 6-9. Come proteine Mx ostacolano virus dell'influenza A è meno chiaro. Uno dei problemi principali è che non è semplice dimostrare una interazione tra una proteina Mx e un prodotto genico influenza. Un rapporto ha dimostrato un'interazione tra MxA umana e la proteina NP in influenza A virus cellule infettate 10. Questa interazione può essere visualizzato solo dal co-immunoprecipitation se le cellule erano stati trattati con le ditiobis reticolanti reagenti (succinimidil propionato) prima lisi, suggerendo che l'interazione è transitorio e / o debole. Studi più recenti hanno dimostrato che la sensibilità Mx differenziale di diversi ceppi di influenza A è determinata dall'origine della proteina NP 11,12. In linea con questo, i virus influenzali A possono sfuggire in parte dal controllo Mx mutando residui specifici nella proteina NP 13. Questo suggerisce che l'obiettivo principale del virus dell'influenza per host MX è la proteina NP, probabilmente NP assemblato in complessi vRNP. Tuttavia, nessuno di questi studi più recenti hanno dimostrato una interazione tra influenza NP o vRNPs e sia umana MxA o il mouse MX1.
Recentemente abbiamo mostrato, per la prima volta, una interazione tra l'influenza NP e la proteina Mx1 mouse con un protocollo ottimizzato co-immunoprecipitazione 14, che è qui descritto in dettaglio. In generale, i co-mmunoprecipitation è uno degli approcci biochimici più frequentemente utilizzati per indagare le interazioni proteina-proteina. Questa tecnica è spesso preferito su tecniche alternative, ad esempio, due ibridi di lievito, in quanto consente di studiare le interazioni proteina-proteina nel loro ambiente naturale. Co-immunoprecipitazione può essere eseguita su proteine espresse endogenamente se sono disponibili anticorpi contro le proteine di interesse. In alternativa, le proteine di interesse possono essere espressi nella cellula attraverso trasfezione o infezione e un tag di affinità possono essere utilizzati. Oltre ai vantaggi sopra menzionati, il protocollo di co-immunoprecipitazione descritto consente il rilevamento di interazioni deboli e / o transitori proteine. Il componente principale di questo protocollo ottimizzato è l'aggiunta di N-etilmaleimmide (NEM) nel tampone di lisi cellulare. NEM è un reagente alchilante che reagisce con i gruppi tiolici liberi come presente in cisteine, a pH 6,5-7,5, per formare una stabile tio-estere(Figura 1). Al più alto pH, NEM può anche reagire con gruppi amminici o subire l'idrolisi 15. NEM è in genere utilizzato per bloccare gruppi tiolo gratuito, al fine di prevenire la formazione di legami disolfuro o inibire l'attività enzimatica. Ad esempio, NEM è spesso usato per bloccare enzimi desumoylating, che sono cisteina proteasi. Nel protocollo co-immunoprecipitazione descritto, NEM è stato inizialmente incluso nel tampone di lisi perché era stato riferito che la sumoilazione delle proteine influenza può influenzare l'interazione tra proteine virali 16. Inaspettatamente, l'aggiunta di NEM rivelata chiave per documentare l'interazione tra influenza NP e mouse Mx1 mediante co-immunoprecipitazione. Non è chiaro perché l'aggiunta di NEM è cruciale per rilevare l'interazione NP-MX1. Forse l'interazione è troppo transitoria e / o debole. NEM potrebbe stabilizzare l'interazione, ad esempio, conservando una specifica conformazione MX1 una proteina virale o anche un terzo compo sconosciutanente. Tale effetto stabilizzante di NEM è stato osservato in precedenza, ad esempio, per l'interazione tra la ribonucleotide reduttasi M1 e la sua gemcitabina inibitore (F2dC) 17. MX1 e NP entrambi contengono più residui di cisteina che potrebbero essere modificati da NEM. Ad esempio, un recente studio condotto da Rennie et al. Hanno dimostrato che un stalkless MxA variante contiene tre residui di solventi cisteina esposti che possono essere modificate da iodoacetamide. Mutando questi residui di serine non ha influenzato l'attività enzimatica di MxA, ma impedisce l'aggregazione disolfuro-mediata 18. Poiché questi cisteine sono conservati in MX1, questo suggerisce che le cisteine analoghe in MX1 possono essere modificati da NEM e come tale influenza la sua conformazione e solubilità. Inoltre, NEM potrebbe anche influenzare l'attività di GTPasi Mx1, che è essenziale per l'attività anti-influenzale di MX1, stabilizzando così l'interazione tra MX1 e NP. Tuttavia, un effetto diretto di NEM sul GTPase actività di Mx1 è improbabile, come NEM è necessario anche per rilevare l'interazione tra l'influenza NP e GTPase mutanti inattivi della proteina Mx1 14. Chiaramente, sono necessarie ulteriori ricerche per chiarire l'effetto del NEM sull'interazione NP-MX1.
In sintesi, il protocollo di co-immunoprecipitazione descritto consente di studiare l'interazione tra la proteina Mx1 antivirale e il suo bersaglio virale, la proteina influenza NP. Questo protocollo può essere utilizzato anche per studiare altre interazioni deboli o transitori che dipendono dalla stabilizzazione specifiche conformazioni proteiche. Interazione proteina-proteina che dipendono conformazioni specifiche sono state descritte in precedenza, ad esempio, per le proteine leganti il calcio come calmodulina 19. Infine, il ruolo benefico di NEM potrebbe essere utilizzato anche in altri metodi che rilevano interazioni proteina-proteina, come saggi co-sedimentazione.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Nota: La seguente protocollo trasfezione e co-immunoprecipitazione è stabilito per un formato piatto 9 centimetri Petri. Altri formati sono possibili anche dopo il ridimensionamento del protocollo.
1. Semina del rene embrionale umano (HEK) Cellule 293T
- Seme della cellule HEK293T un giorno prima trasfezione in 1,2 x 10 6 cellule per 9 centimetri piatto Petri in 12 ml di mezzo modificato Eagle di Dulbecco (DMEM) supplementato con siero fetale di vitello al 10%, 2 mM L-glutammina, 0,4 mM Na-piruvato, 0,1 mM aminoacidi non essenziali, 100 U / ml di penicillina e 0,1 mg / ml di streptomicina.
- Crescere le cellule 16 ore a 37 ° C e 5% CO 2.
- Ispezionare visivamente la morfologia e la vitalità delle cellule con un microscopio ottico invertito prima trasfezione. Le cellule devono essere sub-confluenti per una ottimale efficienza trasfezione.
2. calcio-fosfato Trasfezione delle cellule HEK293T
Note: Uso 0.5-1 mg di pCAXL-NP o vuoto plasmide pCAXL in combinazione con 1-3 mg di pCAXL-MX1 per 9 centimetri piatto. Utilizzare una pari quantità di DNA plasmidico totale in tutti i campioni; regolare con vuoto plasmide, se necessario.
- Preparare i seguenti buffer di trasfezione:
- Preparare Tris-EDTA (TE) con concentrazioni di 1,0 mm Tris-HCl pH 8,0 e 0,1 mM EDTA pH 8.0.
- Preparare BS / HEPES con concentrazioni di 25 mM HEPES (5,96 g / L, 4- (2-idrossietil) -1-acido piperazineethanesulfonic), 274 mM NaCl (16 g / L), KCl 10 mM (0,74 g / L), 1.5 mM NaHPO 4 · 12H 2 O (0,5 g / L) e destrosio mM 11,1 (2 g / L). Regolare il pH a 7,05.
- Preparare CaCl 2 / HEPES con concentrazioni di 1,25 M CaCl 2 · 2H 2 O (183,8 g / L) e 125 mM HEPES (29.79 g / L). Regolare il pH a 7,05 con NaOH.
- Riscaldare i buffer di transfezione a 37 ° C prima dell'uso.
- Preparare i campioni plasmide diluendo il DNA plasmide in 600 ml di TE.Preparare queste miscele in pozzetti di una piastra da 6 pozzetti.
- Aggiungere 150 ml di CaCl 2 / HEPES in modo saggio goccia ai campioni plasmide e mescolare pipettando 3 volte su e giù.
- Preparare la soluzione trasfezione goccia saggio aggiungendo la soluzione plasmide (TE + DNA + CaCl 2 / HEPES; 750 ml) a 750 ml di buffer di BS / HEPES forniti in un 6-pozzetti fresco. Distribuire la soluzione plasmide uniformemente sul BS buffer di completo benessere contenente / HEPES.
- Agitare la soluzione trasfezione su un agitatore per 90 secondi a 1.000 RPM.
- Incubare la miscela per 5 minuti a temperatura ambiente.
- Aggiungere la soluzione trasfezione (1,5 ml) gocciolamento alle cellule. Utilizzare una micropipetta P1000 a gocciolare la soluzione trasfezione sulle cellule. Disperdere il composto sulla completa 9 centimetri piastra di Petri e scuotere la piastra molto delicatamente.
- Incubare le cellule a 37 ° C e 5% CO 2 per 6 ore. Quindi rimuovere il supporto per aspirazione e sostituire immediatamente with 12 ml fresco medio, pre-riscaldata. Aggiungere delicatamente il mezzo fresco alle cellule per evitare il distacco delle cellule. Per questo, tenere la punta della pipetta contro il lato del pozzo e delicatamente spingere fuori il mezzo.
- Incubare le cellule per un ulteriore 16 ore a 37 ° C e 5% CO 2.
3. Co-immunoprecipitazione
Nota: Eseguire la co-immunoprecipitazione 24 ore dopo la trasfezione.
- Preparazione del tampone di lisi basso sale e tampone di lavaggio di sale.
- Preparare una soluzione madre di 2 M N-etilmaleimmide (NEM), pesando la quantità di NEM e sciogliere in etanolo assoluto. Preparare la soluzione di riserva NEM fresco prima dell'uso.
ATTENZIONE: NEM è molto tossico, preparare e utilizzare questa soluzione madre in una cappa aspirante. - Preparare basso tampone di lisi sale a concentrazioni di 50 mm Tris-HCl pH 8, 150 mM NaCl, 5 mM acido etilendiamminotetraacetico (EDTA), 1% NP40 e un cocktail inibitore della proteasi (sciogliere 1 tavolot in tampone di lisi 50 ml). Aggiungere NEM ad una concentrazione finale di 25 mM (cioè, diluire 1:80). Tenere sul ghiaccio dopo aver aggiunto gli inibitori della proteasi e NEM.
Nota: aggiungere sempre gli inibitori della proteasi e NEM momento, poco prima dell'uso. - Preparare un sale tampone di elevata lavaggio a concentrazioni di 50 mm Tris-HCl pH 8, NaCl 500 mm, 5 EDTA mm e 1% NP40. Si noti che il tampone di lavaggio di sale non contiene NEM.
- Preparare una soluzione madre di 2 M N-etilmaleimmide (NEM), pesando la quantità di NEM e sciogliere in etanolo assoluto. Preparare la soluzione di riserva NEM fresco prima dell'uso.
- Preparazione dei lisati cellulari.
- Rimuovere il supporto e lavare le cellule con 2 ml di ghiaccio freddo fosfato salina tamponata (PBS). Aggiungere molto delicatamente il tampone di lavaggio, come cellule HEK293T staccano facilmente.
- Rimuovere la PBS e aggiungere 600 ml di ghiaccio freddo tampone di bassa lisi sale per 9 centimetri piastra di Petri.
- Incubare le piastre per 20 minuti in ghiaccio. Assicurarsi che le piastre sono tenuti orizzontali, per assicurare una copertura completa della superficie della piastra con tampone di lisi. Agitare delicatamente le piastre ogni 5 min.
- Raccogliere il lisato cellulare in una microcentrifuga da 1,5 mlprovetta e centrifugare per 3 min a 4 ° C e 16.000 xg a pellet frazione insolubile.
- Trasferire la frazione solubile, cioè il lisato cellulare ad un nuovo 1,5 ml provetta e tenere in ghiaccio. Continuare immediatamente con il protocollo di co-immunoprecipitazione, per impedire la dissociazione delle proteine interagenti. Eseguire le seguenti operazioni per quanto possibile su ghiaccio oppure a 4 ° C per limitare l'attività proteolitica nei lisati.
- Generazione di immuno-complessi.
Nota: In questo passaggio, la proteina di interesse è vincolata dalla anticorpi appropriati. Per studiare l'interazione NP-MX1, utilizzare un anti-NP anticorpo monoclonale mouse.- Per ogni campione, miscelare 135 ml di lisato con 2 ml di anticorpo monoclonale anti-NP e 113 ml di tampone di lisi sale basso (volume totale di 250 microlitri). Conservare il lisato rimasto a -20 ° C per ulteriori analisi, come western blotting, per documentare i livelli di espressione del Intera ipotizzatopartner ction nelle cellule transfettate.
Nota: In alternativa, misurare la concentrazione di proteine del lisato (ad esempio con reagente di Bradford) e usare una quantità fissa di proteine totali, per esempio, 400 mg, per ciascun lisato. - Incubare la miscela anticorpi lisato per 3 ore su una ruota che gira a 4 ° C. Questo passaggio può essere esteso ad una notte di incubazione.
- Per ogni campione, miscelare 135 ml di lisato con 2 ml di anticorpo monoclonale anti-NP e 113 ml di tampone di lisi sale basso (volume totale di 250 microlitri). Conservare il lisato rimasto a -20 ° C per ulteriori analisi, come western blotting, per documentare i livelli di espressione del Intera ipotizzatopartner ction nelle cellule transfettate.
- Preparazione della proteina G perline.
Nota: Le perle di proteine G sono spediti e conservati in etanolo al 20% per la conservazione. Il tallone-slurry normalmente consiste di 50% perle e perline questi devono essere lavati prima di essere utilizzati per immunoprecipitare i complessi immuni.- Usare 50 ml di perline, cioè 100 ml di bead-liquami, per ogni campione. Lavare la quantità di perline necessari per tutti i campioni del test co-immunoprecipitazione in un tubo. Tagliare la punta di una punta di pipetta 1 ml per facilitare pipettato la bead-sospensione.
- Centrifugare la proteina G bead-liquami a 8.000 xge 4 ° C per 30 sec. Rimuovere la soluzione di etanolo ed aggiungere un volume uguale di tampone di lisi basso sale. Centrifugare la proteina G tallone-slurry a 8.000 xg e 4 ° C per 30 sec e rimuovere il surnatante delicatamente. Ripetere questa operazione di lavaggio per 3 volte.
Nota: Il buffer bassa lisi sale usato per lavare le perline non deve contenere inibitori della proteasi o NEM. - Stimare il volume di proteine G perline e aggiungere un uguale volume di tampone bassa lisi di sale per fare un nuovo 50% bead-sospensione in tampone di lisi basso sale.
- Per ciascun campione, trasferire 100 ul di tallone-slurry in una nuova provetta da 1,5 ml e memorizzare in ghiaccio fino al momento dell'uso. Attenzione per risospendere il tallone-slurry prima di dividere, come queste perle sedimentano rapidamente al fondo della provetta.
- Immunoprecipitazione dei complessi immuni da proteine G perline e la loro eluizione.
- Prima di utilizzare le proteine G perline per immunoprecipitazione, centrifuga tutte le provette 30 sec a 8000 xg e 4 ° C everificare mediante ispezione visiva che c'è una quantità uguale di perline presente in tutti i campioni. Se necessario regolare la quantità di perline in alcuni dei campioni e centrifugare di nuovo. Scartare il surnatante. Fare attenzione a non disturbare le proteine G perline pellet.
- Centrifugare brevemente i complessi immuni (es., Lisati con anticorpi, 250 microlitri) per 30 secondi a 8000 xg e 4 ° C per raccogliere il campione completa sul fondo della provetta. Trasferire i complessi immuni alla proteina G perline (50 microlitri).
- Incubare 60 min su una ruota che gira a 4 ° C. Non incubare questi complessi immunitari più di 75 minuti con le perline per ridurre legame aspecifico di proteine alle proteine G perline.
- Centrifugare le proteine G (con perle legate complessi immuni) per 30 sec a 8000 xg e 4 ° C e rimuovere il surnatante. Fare attenzione a non disturbare le proteine G perline pellet. Opzionale: memorizzare questo supernatanti a 4 ° C o -20 ° C per una successiva analisi,ad esempio, per stimare la quantità di proteina non legata.
- Lavare le proteine G perline per circa 5 minuti con 900 ml di tampone di lisi di sale. Assicurarsi che le perle sono completamente risospese nel tampone di lavaggio per il lavaggio ottimale. Centrifugare le proteine G perline per 30 sec a 8000 xg e 4 ° C e scartare il surnatante. Ripetere questa operazione di lavaggio 4 volte. Fare attenzione a non disturbare le perline proteine G pellet per evitare la perdita di materiale immunoprecipitato.
- Dopo l'ultima fase di lavaggio, aggiungere 50 ml di tampone campione 2x Laemmli ai talloni, riscaldare la sospensione per 10 min a 95 ° C per eluire i (co) proteine immunoprecipitate.
- Preparare 10 ml di tampone Laemmli 6x con 1 g di sodio dodecil solfato, 3,5 ml di glicerolo, 3,5 ml di 1 M Tris-HCl pH 6,8 e 420 ml di β-mercaptoetanolo. Regolare ad un volume totale di 10 ml aggiungendo acqua distillata. Diluire 3 volte in acqua distillata per ottenere tampone 2x Laemmli.
ATTENZIONE: &# 946; -mercaptoethanol è tossico, preparare e usare tampone Laemmli in una cappa aspirante.
- Preparare 10 ml di tampone Laemmli 6x con 1 g di sodio dodecil solfato, 3,5 ml di glicerolo, 3,5 ml di 1 M Tris-HCl pH 6,8 e 420 ml di β-mercaptoetanolo. Regolare ad un volume totale di 10 ml aggiungendo acqua distillata. Diluire 3 volte in acqua distillata per ottenere tampone 2x Laemmli.
- Dopo il riscaldamento, centrifugare le proteine G perline per 30 sec a 8000 xg e conservare i campioni a 4 ° C (a breve termine) o -20 ° C (a lungo termine).
4. Analizzare il (co) immunoprecipitati Proteine
- Visualizzare le proteine presenti nel lisato cellulare e eluato della co-immunoprecipitazione mediante SDS-PAGE e Western blotting 20 21,22. Caricare Tipicamente metà eluato Laemmli su gel. Fare attenzione a non disturbare le perline G proteine pellet quando il prelievo di campioni per il gel di carico. MX1 ed espressione NP sono stati rivelati con un anti-MX1 e anticorpi-NP contro, rispettivamente, 14. Le bande sono state rilevate con chemiluminescenza a base di HRP ed uno sviluppatore pellicola X-ray.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
N-etilmaleimmide è un composto organico che può essere utilizzato per modificare irreversibilmente gruppi tiolici liberi, ad esempio per inibire proteasi cisteina (Figura 1).
La proteina antivirale Mx1 inibisce la replicazione del virus influenzale A interagendo con la nucleoproteina virale. Il protocollo di co-immunoprecipitazione ottimizzato qui descritto permette di studiare questa interazione NP-MX1. HEK293T cellule sono state trasfettate con vettori di espressione per la proteina antivirale Mx1 in assenza o presenza della proteina influenza NP. Successivamente, la proteina NP stato tirato giù da lisati cellulari totali con un anticorpo monoclonale NP-specifica. La Figura 2 mostra che la proteina Mx1 è solo co-immunoprecipitato in presenza di co-espressi NP. Possibile aspecifica co-immunoprecipitazione di Mx1 in assenza di NP è causato da aspecifica pull-down della proteina Mx1 dal anticorpo anti-NP o legame non specifico della Mx1 alproteine G perline. Pertanto, comprendono sempre un controllo negativo per valutare questo aspecifica co-immunoprecipitazione. La figura 3 mostra che l'interazione NP-MX1 può essere rilevata solo in presenza di NEM. In questo esperimento, legame non specifico di Mx1 alla proteina G perline è stato valutato con una reazione di controllo co-immunoprecipitazione in assenza di anticorpi anti-NP.
Questo protocollo può anche essere usato per studiare l'interazione tra MX1 dell'influenza NP proteina isolata da cellule infette o da virioni. Per questa applicazione, il protocollo di cui sopra è stato leggermente modificato combinando lisati di cellule che esprimono con Mx1 lisati contenenti la proteina virale NP prima di iniziare il protocollo di co-immunoprecipitazione. Figura 4 mostra la co-immunoprecipitazione di Mx1 con NP isolato da cellule transfettate, cellule infettate o virioni.
In conclusione, questi risultati mostrano che questa co-immunoprecipitazione protocol può essere usato per studiare l'interazione tra una proteina antivirale e la sua destinazione virale.
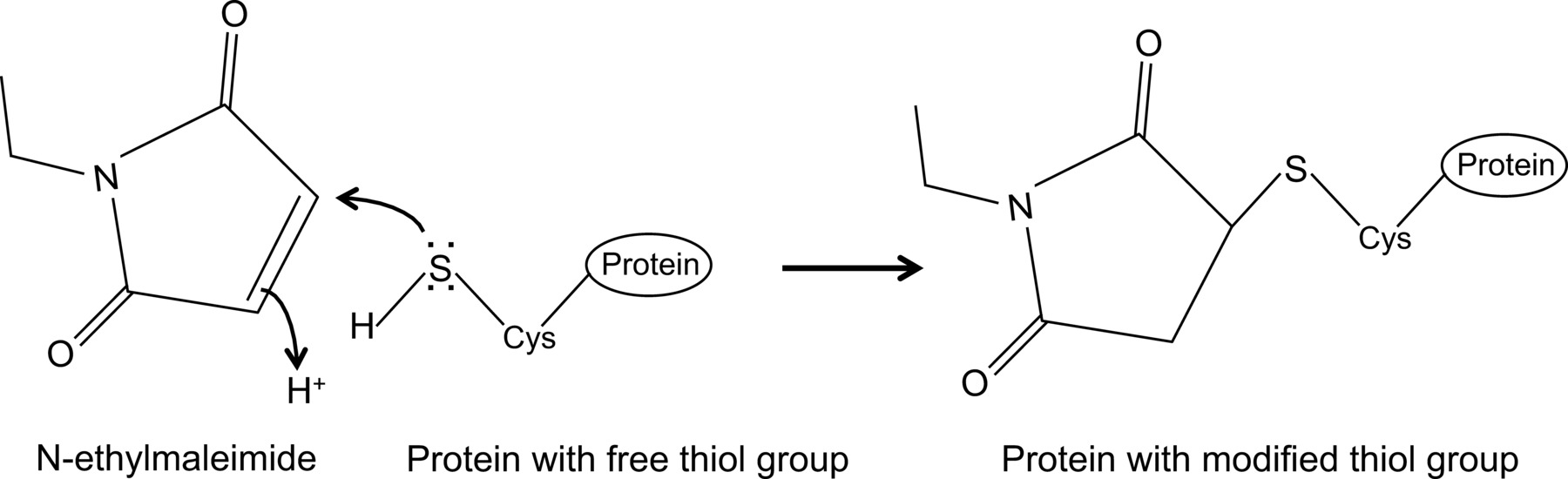
Figura 1:. Modifica irreversibile dei gruppi tiolici liberi da N-etilmaleimmide Cliccate qui per vedere una versione più grande di questa figura.

Figura 2:. Mx1 interagisce con NP analisi Western blot di un esperimento di co-immunoprecipitazione con due campioni: una in cui sia MX1 e NP sono presenti e una configurazione di controllo in cui la proteina NP è assente. NP è stata immunoprecipitata con anti-NP e NP e Mx1 sono stati visualizzati mediante western blotting. Questa cifra è stata modificata from 14.

Figura 3: N-etilmaleimmide è importante rilevare la NP - interazione Mx1 analisi Western blot di un esperimento di co-immunoprecipitazione eseguita in presenza o assenza di N-etilmaleimmide.. NP è stata immunoprecipitata con anti-NP e Mx1 e NP sono stati visualizzati mediante western blotting.

Figura 4:. Mx1 interagisce con NP isolato dalle cellule infette o da virioni lisati contenente Mx1 sono stati combinati con lisati contenenti NP da fonti diverse: un lisato di controllo (senza vRNPs), transfettate vRNPs, vRNPs dalle cellule infette o vRNPs isolati da virioni dell'influenza A . Dopo la miscelazione lisati, co-immunoprecipitation con anti-NP è stata eseguita e Mx1 e NP sono stati visualizzati mediante western blotting.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Studiare l'interazione tra le proteine antivirali e ai loro obiettivi virale è molto importante per comprendere i dettagli del meccanismo antivirale di queste proteine. Questo può dare nuove intuizioni come i virus ei loro ospiti co-evoluti e sarà la base per lo sviluppo di nuove strategie antivirali. Il protocollo di co-immunoprecipitazione ottimizzata qui descritto consente di studiare l'interazione tra la proteina del mouse MX1 suo bersaglio virale, la proteina influenza NP. L'aspetto più importante di questo protocollo è l'aggiunta di NEM nel tampone di lisi, come l'interazione NP-MX1 è rilevabile in assenza di NEM (Figura 3). Fino ad oggi, non è noto perché la presenza di NEM è essenziale per rilevare questa interazione. Tuttavia, questo protocollo potrebbe essere utile per studiare altre interazioni deboli e / o transitorie che dipendono dalla stabilizzazione specifiche conformazioni proteiche, specialmente se sono coinvolti cisteine.
Unimportante limitazione di saggi di co-immunoprecipitazione in generale è la disponibilità di anticorpi specifici e di alta qualità che riconoscono epitopi conformazionali in uno dei partner di interazione ad alta affinità. L'interazione NP-MX1 non è stato possibile dimostrare con il nostro in casa prodotti anti-Mx1 policlonale. Questo antisiero immunoprecipita anche la proteina influenza NP, anche in assenza di MX1. Inoltre, l'anticorpo monoclonale anti-NP che è stato utilizzato, riconosce la proteina NP del / Puerto Rico / 8/34 ceppo di influenza A, ma purtroppo non è adatto a discesa NP di ceppi del virus dell'influenza aviaria. Un altro limite di questo protocollo co-immunoprecipitazione è il legame non specifico di Mx1 alle proteine G perline. Quest'ultimo legame può essere superato aumentando la concentrazione di sale nel tampone di lavaggio e riducendo il tempo di contatto tra il lisato e la proteina G perline. Inoltre, riducendo la quantità di perline utilizzato per ogni immunoprecipitazionereazione da 50 ml a 25 ml, può ridurre ulteriormente legame non specifico della MX1 a queste perle. In generale, la volute pull-down di proteine contaminanti, causata dal legame non specifico alla proteina G perline, può essere ridotto anche altre strategie. Ad esempio, queste proteine possono essere rimossi in una fase di pre-chiaro, in cui queste proteine vengono rimosse incubando il lisato con proteina G perline in assenza di anticorpi. Le proteine contaminanti vengono poi rimossi con le perline e il lisato pre-autorizzato viene utilizzato per co-immunoprecipitazione. Questa strategia è solo vantaggioso se le proteine contaminanti sono differenti dalla proteina (s) in fase di studio. In alternativa, i siti di legame non specifici sulla proteina G perline possono essere bloccate con BSA. Tuttavia, questa strategia è consigliata solo se le perline vengono prima rivestiti con l'anticorpo, come BSA può anche ridurre la precipitazione dei complessi immuni (cioè anticorpi ridotta vincolanti dalle perline rivestite). Presoinsieme, è molto importante includere controlli adeguati per escludere legame non specifico delle proteine di interesse per le proteine G perline o agli anticorpi utilizzati.
Il protocollo di co-immunoprecipitazione descritto può essere modificato per studiare l'interazione di Mx1 con la proteina NP presenti in un ambiente diverso, per esempio, in cellule infettate o in vRNPs purificati. Mx1 inibisce l'espressione di proteine virali, e NP, durante l'infezione da virus influenzale 23. Pertanto, è tecnicamente estremamente difficile studiare l'interazione NP-MX1 in cellule trasfettate Mx1 che successivamente sono infettati. Tuttavia, questo protocollo co-immunoprecipitazione può essere eseguita anche dopo la combinazione lisati di cellule esprimenti Mx1 e cellule infettate, permettendo il rilevamento successo dell'interazione NP-MX1 (Figura 4). Poiché l'obiettivo fondamentale di NEM non è noto, NEM stato aggiunto durante la lisi delle due popolazioni cellulari. Se lo si desidera, il pHil tampone di lisi può essere modificata. Gli esperimenti qui descritti sono effettuati a pH 8 (come in Turan et al. 10), ma la NP-MX1 co-immunoprecipitazione stata inoltre eseguita con successo con un tampone di lisi pH 7,2. In realtà un pH di 7,2 è preferibile, perché a questo pH NEM reagisce esclusivamente con gruppi tiolici liberi e questo pH aumenta anche la resa di estrazione della proteina MX1. Inoltre, le proteine G perline potrebbero essere sostituiti da proteina A perline, a seconda delle specie ospiti da cui gli anticorpi utilizzati sono derivati. Tuttavia, i risultati più pulite si ottengono con le proteine G perline. Infine, questo protocollo può anche essere usato per studiare l'interazione tra MX1 e altre proteine influenza, ad esempio, 14 PB2. In questo caso un epitopo V5 etichettato PB2 stato utilizzato, che potrebbe in linea di principio essere combinato con disponibile in commercio anti-V5 gel di affinità di agarosio.
In esperimenti futuri, questo protocollo potrebbe essere prezioso per identificare le regioni in MX1 eNP che sono importanti per l'interazione NP-MX1. Una tale regione in MX1 potrebbe essere ciclo L4, come questo ciclo è stato dimostrato essere critico per l'interazione tra MxA e virus Thogoto NP 8,9. Se gli anticorpi NP-specifici sono diventate disponibili idonee, questo protocollo permetterà anche di determinare se l'aumento della sensibilità dei ceppi A dell'influenza aviaria per Mx1 correla con una più forte interazione NP-MX1 o no, che potrebbe aiutare a comprendere il meccanismo di sensibilità Mx. Inoltre, l'interazione tra MX1 e componenti di virus appartenenti a famiglie diverse dalla Orthomyxoviridae, può anche essere risolto questo protocollo co-immunoprecipitazione adattato. Infine, ulteriori studi per svelare l'effetto di NEM sulla NP-MX1 sarà molto utile per comprendere meglio questa interazione specifica, ma anche al fine di conoscere nelle applicazioni più ampie della aggiunta di questo composto durante lisi cellulare e co-immunoprecipitazione esperimenti.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Gli autori dichiarano di non avere interessi finanziari in competizione.
Acknowledgments
Questo lavoro è stato sostenuto da FWO-Vlaanderen, il progetto IOF IOF10 / stARTT / 027 e Università di Gand Special Research Grant BOF12 / GOA / 014.
Materials
| Name | Company | Catalog Number | Comments |
| DMEM high glucose | Gibco | 52100-047 | |
| N-Ethylmaleimide | Sigma | E-3876 | Toxic |
| Igepal CA-630 | Sigma | I-30212 | also known as NP40 |
| Protease inhibitor cocktail | Roche | 11 873 580 001 | |
| anti-NP monoclonal antibody | NIH Biodefense and Emerging Infections Research Resources Repository | NR-4282 | ascites blend of clones A1 and A3 |
| anti-RNP polyclonal serum | NIH Biodefense and Emerging Infections Research Resources Repository | NR-3133 | directed against A/Scotland/840/74 (H3N2) |
| Protein G Sepharose 4FF | GE Healthcare | 17-0618-01 | |
| Hyperfilm ECL 18 x 24 cm | GE Healthcare | 28-9068-36 | |
| ECL western blotting substrate | Pierce | 32106 |
References
- Verhelst, J., Hulpiau, P., Saelens, X. Mx proteins: antiviral gatekeepers that restrain the uninvited. Microbiol Mol Biol Rev. 77 (4), 551-566 (2013).
- Goujon, C., et al. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature. 502 (7472), 559-562 (2013).
- Kane, M., et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature. 502 (7472), 563-566 (2013).
- Liu, Z., et al. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe. 14 (4), 398-410 (2013).
- Kochs, G., Haller, O. Interferon-induced human MxA GTPase blocks nuclear import of Thogoto virus nucleocapsids. Proc Natl Acad Sci U S A. 96 (5), 2082-2086 (1999).
- Flohr, F., Schneider-Schaulies, S., Haller, O., Kochs, G. The central interactive region of human MxA GTPase is involved in GTPase activation and interaction with viral target structures. FEBS Lett. 463 (1-2), 24-28 (1999).
- Kochs, G., Haller, O. GTP-bound human MxA protein interacts with the nucleocapsids of Thogoto virus (Orthomyxoviridae). J Biol Chem. 274 (7), 4370-4376 (1999).
- Mitchell, P. S., et al. Evolution-guided identification of antiviral specificity determinants in the broadly acting interferon-induced innate immunity factor MxA. Cell Host Microbe. 12 (4), 598-604 (2012).
- Patzina, C., Haller, O., Kochs, G. Structural requirements for the antiviral activity of the human MxA protein against Thogoto and influenza A virus. J Biol Chem. 289 (9), 6020-6027 (2014).
- Turan, K., et al. Nuclear MxA proteins form a complex with influenza virus NP and inhibit the transcription of the engineered influenza virus genome. Nucleic Acids Res. 32 (2), 643-652 (2004).
- Dittmann, J., et al. Influenza A virus strains differ in sensitivity to the antiviral action of Mx-GTPase. J Virol. 82 (7), 3624-3631 (2008).
- Zimmermann, P., Manz, B., Haller, O., Schwemmle, M., Kochs, G. The viral nucleoprotein determines Mx sensitivity of influenza A viruses. J Virol. 85 (16), 8133-8140 (2011).
- Manz, B., et al. Pandemic influenza A viruses escape from restriction by human MxA through adaptive mutations in the nucleoprotein. PLoS Pathog. 9 (3), e1003279 (2013).
- Verhelst, J., Parthoens, E., Schepens, B., Fiers, W., Saelens, X. Interferon-inducible protein Mx1 inhibits influenza virus by interfering with functional viral ribonucleoprotein complex assembly. J Virol. 86 (24), 13445-13455 (2012).
- Brewer, C. F., Riehm, J. P. Evidence for possible nonspecific reactions between N-ethylmaleimide and proteins. Anal Biochem. 18 (2), 248-255 (1967).
- Wu, C. Y., Jeng, K. S., Lai, M. M. The SUMOylation of matrix protein M1 modulates the assembly and morphogenesis of influenza A virus. J Virol. 85 (13), 6618-6628 (2011).
- Chen, Z., Zhou, J., Zhang, Y., Bepler, G. Modulation of the ribonucleotide reductase M1-gemcitabine interaction in vivo by N-ethylmaleimide. Biochem Biophys Res Commun. 413 (2), 383-388 (2011).
- Rennie, M. L., McKelvie, S. A., Bulloch, E. M., Kingston, R. L. Transient dimerization of human MxA promotes GTP hydrolysis, resulting in a mechanical power stroke. Structure. 22 (10), 1433-1445 (2014).
- Gifford, J. L., Walsh, M. P., Vogel, H. J. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem J. 405 (2), 199-221 (2007).
- Separating Protein with SDS-PAGE. , JoVE. Available from: http://www.jove.com/science-education/5058/separating-protein-with-sds-page (2014).
- Gallagher, S., Chakavarti, D. Immunoblot analysis. J Vis Exp. (16), (2008).
- Eslami, A., Lujan, J. Western blotting: sample preparation to detection. J Vis Exp. (44), (2010).
- Pavlovic, J., Haller, O., Staeheli, P. Human and mouse Mx proteins inhibit different steps of the influenza virus multiplication cycle. J Virol. 66 (4), 2564-2569 (1992).
