

Summary
El protocolo de inmunofluorescencia indirecta descrito en este artículo permite la detección y la localización de las proteínas en la glándula mamaria del ratón. Un método completo se da para preparar muestras de glándula mamaria, para llevar a cabo la inmunohistoquímica, a la imagen las secciones de tejido mediante microscopía de fluorescencia, y para reconstruir imágenes.
Abstract
La inmunofluorescencia indirecta se utiliza para detectar y localizar proteínas de interés en un tejido. El protocolo que se presenta aquí describe un método completo y sencillo para la detección inmunológica de las proteínas, el ratón glándula mamaria lactante ser tomado como un ejemplo. Un protocolo para la preparación de las muestras de tejido, especialmente en relación con la disección de glándula mamaria de ratón, la fijación del tejido y de corte de tejido congelado, son detallada. Un protocolo estándar para realizar inmunofluorescencia indirecta, incluyendo una etapa de recuperación de antígeno opcional, también se presenta. La observación de las secciones de tejido marcados así como la adquisición de imagen y post-tratamientos también se indican. Este procedimiento da una visión completa, de la colección de tejido animal a la localización celular de una proteína. Aunque este método general se puede aplicar a otras muestras de tejido, debe ser adaptado a cada tejido / primaria pareja anticuerpo estudiado.
Introduction
La glándula mamaria es un órgano exocrino mamíferos atípica cuya función principal es producir leche para alimentar a los recién nacidos. El desarrollo del tejido mamario se produce principalmente después del nacimiento y se caracteriza por un proceso único en el que el epitelio invade el estroma circundante. Este tejido sufre muchos cambios (crecimiento, diferenciación y regresión), especialmente durante la vida adulta, de forma concomitante con las variaciones en el estado reproductivo (Figura 1). Además de la morfología general de los tejidos, las proporciones de diferentes tipos de células, así como su disposición dentro de la glándula mamaria cambian dramáticamente durante el desarrollo 1-5.
Durante la vida embrionaria, el epitelio mamario se deriva de las líneas de leche mamarias, que se define por un ligero engrosamiento y la estratificación del ectodermo, entre las extremidades delanteras y traseras a cada lado de la línea media alrededor de día embrionario 10,5 (E10.5) (Figura 1A ).En E11.5, la línea de leche se rompe en placodas individuales, que están simétricamente colocadas a lo largo de la línea de leche mamaria en lugares reproducibles, y el mesénquima que rodea comienza a condensarse. Los placodas comienzan a hundirse más profundamente en la dermis y el mesénquima mamaria organiza en capas concéntricas alrededor de la yema mamaria (E12.5-E14.5). A partir de E15.5, el epitelio mamario, comienza a proliferar y alargadas para formar el brote primario que empuja a través del mesénquima mamaria hacia la almohadilla de grasa. El brote primario desarrolla un lumen hueco con una abertura en la piel, caracterizada por la formación de la vaina pezón. En E18.5, el conducto alargando ha crecido hasta convertirse en la almohadilla de grasa y se ha ramificado en un pequeño sistema ductal arborized englobado en la almohadilla de grasa. El desarrollo es esencialmente detenido y la glándula mamaria sigue siendo rudimentaria morfogenéticamente reposo hasta la pubertad. En el embrión masculino, la activación de los receptores de andrógenos conduce a la degeneración de los brotes, que desaparecenpor E15.5. A partir de E18, desarrollo mamario cesa hasta la pubertad 6-9.
Al nacer, la glándula mamaria alberga un sistema ductal rudimentaria que se alarga y las ramas lentamente (crecimiento isométrico). En el inicio de la pubertad, estructuras esféricas situadas en las puntas de los conductos llamados los brotes extremas terminales (TEBs), están formados por una capa exterior de las células gorra y un núcleo interior de varias capas de células (células del cuerpo). Estas estructuras son altamente proliferativa y se infiltran en el tejido estromal que rodea en respuesta a señales hormonales. Proliferación dentro de los resultados TEBs en el alargamiento ductal, junto con la morfogénesis de ramificación. Este proceso conduce a la creación de una red arborized epitelial básica que emana de la boquilla (Figura 1B, la pubertad). En ~ 10-12 semanas después del nacimiento, cuando el epitelio ha invadido toda la almohadilla de grasa, su expansión se detiene y desaparece la TEBs. Desarrollo ductal entonces sufre cambios dinámicos, es decir, successive la proliferación y la regresión de las células epiteliales de acuerdo con los ciclos de estro 10 (Figura 1B, adulto).
Desde el inicio de la gestación, el tejido mamario se somete a un crecimiento importante y cambios morfológicos para prepararse para la lactancia. El epitelio mamario ampliamente proliferar y diferenciarse, lo que lleva a una red túbulo-alveolar altamente ramificado. Al mismo tiempo, las células epiteliales mamarias (MEC) se polarizan y capaz de sintetizar y secretar productos lácteos. MECs se organizan en numerosas estructuras alveolares (acinos) que están rodeados por las células mioepiteliales contráctiles e incorporados en un estroma compuesto por tejido conectivo y adiposo, vasos sanguíneos y terminaciones nerviosas (Figura 1B, embarazo). Además, el lado basal de MECs está en estrecho contacto con la membrana basal (matriz extracelular), y las interacciones entre estas dos entidades fuertemente regular tanto la morfogénesis y la secretora función de la mamary epitelio 11-13.
Todos estos procesos se basan en la acción de diversas señales ambientales, de los cuales los más importantes son hormones14, factores paracrinos y la matriz extracelular. Por ejemplo, la progesterona induce extensa 15 y alveologenesis que, en combinación con la prolactina (PRL) de ramificación lateral 16,17, promueve y mantiene la diferenciación de los alvéolos. Además de los esteroides y PRL18, citoquinas y vías de señalización asociadas con el desarrollo (Wnt y Notch vías de señalización) también están involucrados en el compromiso de linaje mamaria y el desarrollo 19 a 21. Al final del embarazo, las MECs luminales comienzan a producir una leche rica en proteínas conocida como calostro en el lumen de los alvéolos. Además, la progesterona actúa sobre la permeabilidad epitelial y desde las uniones estrechas están todavía abiertos, el calostro también se encuentra en la corriente de la sangre materna.
Después del parto, la Mammary epitelio ocupa casi todo el volumen de la glándula mamaria y está altamente organizada (Figura 2, el epitelio mamario). Unidades productoras de leche, es decir, los alvéolos (Figura 2, alveolus), están formados por una monocapa de células secretoras mamarias epiteliales polarizadas (mESCs), con su membrana plasmática apical delimita el lumen. Los alvéolos se disponen en lóbulos que se agrupan en lóbulos conectados a conductos que drenan la leche al medio exterior (Figura 2, lóbulo). Lactancia ocurre, es decir., MESCs comienzan a secretar cantidades abundantes de leche, provocadas principalmente por la caída de las hormonas placentarias (principalmente progesterona) (Figura 1B, la lactancia). Genes de la proteína de la leche se activan en un curso de tiempo temporal definido que van desde el embarazo hasta la lactancia 9,22,23, principalmente en respuesta a PRL pituitaria lanzado en el momento de la succión. Al mismo tiempo, los contactos entre mESCs y la matriz extracelular tanto estimulan la leche proteína syntHESIS a través de señales que están mediadas a través de las interacciones entre las integrinas celulares y laminina 24,25, y suprimen la apoptosis en mESCs 26,27. Estas vías de señalización conducen a la activación de la proteína de la leche promotores de genes 28 a través de la activación de la transcripción factores específicos 29. Contactos célula-célula también son importantes para algunos aspectos de la diferenciación entre ellos el establecimiento de la polaridad apical y la secreción vectorial de los productos lácteos. Las uniones estrechas rápidamente cerca después del comienzo de la lactancia y mESCs finamente orquestan la absorción de moléculas de la sangre, así como la síntesis, transporte y secreción de componentes de la leche, en respuesta a las necesidades nutricionales de los recién nacidos. En el momento de la succión, la contracción de las células mioepiteliales que rodean los alvéolos se produce en respuesta a la oxitocina y conduce a la eyección de la leche a través de los conductos y en el pezón. La leche es un fluido complejo que contiene proteínas (principalmentecaseínas), azúcares (principalmente lactosa), lípidos y minerales, así como moléculas bioactivas, tales como inmunoglobulinas A (IgA), factores de crecimiento y hormonas. Las caseínas son sintetizados, ensamblado en estructuras supramoleculares, es decir, micelas de caseína, que se transportan a lo largo de la ruta secretora, y luego liberados por exocitosis, es decir, la fusión de vesículas secretoras que contienen caseína (SV) con la membrana plasmática apical de MESC (Figura 2).
El tráfico intracelular se basa en el intercambio de material entre compartimentos membranosos e implica soluble N-etilmaleimida-Sensible Fusion (NSF) Fijación de proteínas (SNAP) Receptor (SNARE) 30,31. La familia de proteínas SNARE se subdivide en trampas vesiculares (v-SNAREs), presentes en la membrana de la vesícula, y trampas objetivo (t-SNAREs), localizadas en las membranas de destino. Por comprimir a través de sus dominios de doble arrollamiento, V y T-SNAREs ensamblan para formar una gran estabilidad complejo haz de cuatro hélices, conocidos como XXcomplejo SNARE e. Este complejo promueve la fusión de dos bicapas lipídicas opuestas llevando gradualmente en estrecha proximidad 30,32. Después, complejos SNARE se disocian por la adenosintrifosfatasa NSF y sus proteínas adaptador de proteínas SNAP y SNARE se reciclan de nuevo a su compartimento de origen 33. Curiosamente, cada proteína SNARE reside predominantemente en compartimentos celulares distintos y SNARE emparejamiento puede contribuir a la especificidad de los eventos de fusión intracelulares 34. Estudios previos sugieren que al menos proteína 23 (SNAP23) y asociada a vesículas de membrana de proteínas 8 (VAMP8) y syntaxins (STX) sinaptosomal asociada -7 y -12 juegan un papel en la caseína exocitosis 35,36. Estas proteínas también se han encontrado en asociación con la fracción lipídica de la leche, es decir, la grasa de la leche glóbulos (MFG) 37. El modelo imperante actual postula que las gotas de lípidos citoplasmáticos (CLD) se forman por la acumulación de l neutralipids (principalmente triglicéridos y ésteres de esterol) y el colesterol derivado de la dieta materna entre las dos valvas de la membrana 38 a 41 retículo endoplasmático (ER). Grandes CLD se forman, al menos en parte, por la fusión de CLD más pequeños mientras son transportados a la parte apical de mESCs donde son liberados como MFG (1-10 micras de diámetro) por gemación, siendo envuelto por la membrana plasmática apical MESC 40-42. Lactancia cesa después cachorros son destetados y los mESCs mueren progresivamente por apoptosis, lo que lleva a la regresión del tejido mamario de nuevo a un estado puberal (Figura 1B, la involución).
La inmunofluorescencia (IF) es un método de laboratorio de análisis común que se utiliza en casi todos los aspectos de la biología, tanto en investigación como en el diagnóstico clínico. SI técnicas se pueden realizar en las secciones de tejidos (inmunohistoquímica, IHC) o celulares (inmunocitoquímica, ICC) muestras. Este enfoque poderoso basa en el uso de fluorescent-anticuerpos marcados que se unen específicamente (directa o indirectamente) para el antígeno de interés, permitiendo así la visualización de su distribución tejido a través de microscopía de fluorescencia. Las señales de fluorescencia dependen principalmente de la calidad y la concentración de los anticuerpos y el manejo adecuado de la muestra. Un protocolo simple de inmunofluorescencia indirecta (IFI) se presenta para detectar los productos lácteos (caseínas y MFG) y proteínas implicadas en la secreción de producto lácteo (butyrophilin (btn1), proteínas SNARE) en secciones congeladas de tejido mamario de ratón (Figura 3). Aunque este protocolo proporciona una visión completa de IHC, que van desde la recogida de tejidos a la imagen post-tratamiento, crítica y pasos opcionales, así como algunas recomendaciones técnicas también son presentados y discutidos.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Ratones CD1 fueron criados en el INRA (UE0907 IERP, Jouy-en-Josas, Francia). Todos los aspectos éticos del cuidado de los animales cumplen las directrices pertinentes y los requisitos de licencia establecidos por el Ministerio de Agricultura francés. Los procedimientos utilizados fueron aprobados por el comité de ética local (acuerdo 12/097 de la Comethea Jouy-en-Josas / AgroParisTech).
1. Glándula Mamaria Preparación de la muestra
- Ratón disección de la glándula mamaria
- La eutanasia a los ratones en el día 10 de la lactancia por dislocación cervical y el pin al animal con su abdomen hacia arriba.
- Mojar la zona ventral con etanol y secar con una toalla de papel.
- Con unas pinzas, tire hacia arriba de la piel del abdomen entre las dos patas traseras y hacer una incisión (sólo a través de la piel) de aproximadamente 1 cm con tijeras afiladas. A partir de esta primera incisión, a continuación, utilizar tijeras para cortar la piel hasta el cuello en el ratón. Tire de la piel de la peritoneo y pin abajo de un lado de la piel a la vez, que se extiende enseñó.
- Recoger el abdominal y las glándulas mamarias inguinales empujándolos lejos de la piel con un hisopo y finalmente tirar o de corte lejos del peritoneo.
Nota: En este paso tinción Carmine se puede realizar con el fin de visualizar el epitelio mamario dentro de toda la glándula 43. Este enfoque puede ser útil para analizar la morfología global de la glándula mamaria en diversas condiciones fisiológicas (etapas de desarrollo, enfermedades, in vivo tratamientos). - Retire el ganglio linfático se encuentra en el cruce de la abdominal y las glándulas inguinales 44.
- La fijación del tejido mamario
- Cortar el tejido mamario en 3 mm 3 fragmentos con un bisturí e inmediatamente enjuagar estos fragmentos en una solución salina tamponada con fosfato (PBS), pH 7,4, con el fin de eliminar la mayor cantidad de leche posible.
- Secar rápidamente los fragmentos en un papeltoalla y ponerlos en una solución de PBS frío que contenía 4% de paraformaldehído (PFA, HCHO, 32% de solución de formaldehído, PRECAUCIÓN) de 10 a 15 minutos en hielo.
Nota: Este es el tiempo suficiente para permitir el posterior análisis en cortes de tejido mamario por IIF36 y / o hibridación in situ 45. Sin embargo, como fijadores aldehído penetran bastante lentamente en trozos de tejido (~ 1-3 mm por hora), esta vez podrá ampliarse para asegurar una fijación óptima de la muestra de tejido. Alternativamente, fijar tejidos in vivo por perfusión un animal anestesiado con una solución de fijación (no se detalla en el presente estudio).
- Infusión de sacarosa
- Rápidamente enjuagar los fragmentos mamarias en PBS frío y sumergirlos en una solución de PBS frío que contiene 40% de sacarosa (D-sacarosa, C12H22O11, Sr. 342.3 g / mol) de 16 a 48 horas a 4 ° C bajo agitación suave.
- Incrustación de tejidos
Nota: En este paso, los fragmentos mamarias se puede volver a cortar a fin de que fragmentos más pequeños (2-3 mm 3) O para ajustar su forma.- Adecuadamente etiquetar los moldes de plástico y llenar un tercio del volumen del molde con compuesto OCT, se mantiene a RT. Coloque un fragmento (2-3 mm 3) de tejido mamario por molde y cubrir con compuesto de octubre
- Coloque los moldes en la superficie del nitrógeno líquido (en una hoja de aluminio o utilizando un tamiz metálico) y permitir que el producto se congele.
Nota: Debe convertirse en sólido y blanco antes de sumergir el molde en nitrógeno líquido.
- Almacenar las muestras congeladas a -80 ° C hasta que se realizan cortes de tejido.
2. Seccionamiento tejido congelado
Nota: un criostato, que es esencialmente un microtomo dentro de un congelador, se requiere para hacer cortes de tejido congelados. Una temperatura más baja se requiere a menudo para los tejidos grasos o ricos en lípidos, como la glándula mamaria virginal.
- Ajuste la temperatura del criostato a -26 ° C y espere hasta que tenga stabilizado. Mantener el bloque de tejido congelado a -26 ° C durante todo el proceso de corte. Absolutamente evitar la descongelación del tejido en cualquier momento durante el procedimiento.
- Se enfría la cuchilla de afeitar, el soporte de corte, el dispositivo anti-balanceo y el cepillo a -26 ° C por su inclusión en el criostato durante al menos 10 min. También colocar una caja de portaobjetos en el interior del criostato con el fin de ser capaz de almacenar portaobjetos de vidrio como se hacen las secciones.
- Adecuadamente etiquetar los portaobjetos de vidrio que se utilizan para recoger las secciones de tejido y mantenerlas a temperatura ambiente; de otro modo las secciones de tejido no se adhieren a ellos. Retire la muestra del molde dentro del criostato.
Nota: El uso de láminas de vidrio con carga positiva favorecerá enormemente la adherencia de las secciones de frescos de tejido congelado debido a la mayor atracción electrostática. - Cubrir la superficie de un disco de tejido de metal con compuesto OCT (mantenida a temperatura ambiente) y empuje la muestra congelada en la misma. Coloque el montaje húmedo dentro del criostato y deje que se cool durante al menos 15 min.
- Colocar la preparación en fresco en el soporte del disco del criostato. Ajuste del grosor de corte de 5-6 micras y, si es posible, use una nueva hoja afilada o al menos cambiar la zona en la hoja utilizada para cortar cada muestra ya algunos tejidos rápidamente se opaca ella.
- Ajuste la posición del dispositivo anti-vuelco la hoja de afeitar, mediante cortes del medio de montaje hasta que las rodajas se forman de manera uniforme y correcta. Idealmente, el dispositivo antivuelco intervendrá sobre la hoja de afeitar en alrededor de 1 mm.
- Una vez que los ajustes son correctos, realice cortes de tejido girando la rueda en un movimiento uniforme continua. A menos que la temperatura es ideal, una sección de tejido será, por naturaleza, tratar de acurrucarse.
- Use un cepillo para agarrar y maniobrar la sección por el escenario con el fin de colocarlo como desee en el portaobjetos de vidrio. Utilice el cepillo para limpiar los restos posiblemente presentes en el bloque de tejido congelado y / o la hoja de afeitar.
- Halarla sección de tejido hacia el usuario y evitar presionarla sobre la etapa de criostato. Evite presionar la sección de tejido en el escenario criostato ya que puede conducir a la adherencia de la lámina de tejido en el escenario y por lo tanto la incapacidad para recuperar con el portaobjetos de vidrio.
- Recuperar secciones de tejido de uno en uno a recogerlos en la superficie de un portaobjetos de vidrio, sosteniéndolo por encima de la sección y en ángulo hacia abajo para tocar la sección de tejido.
Nota: Las secciones de tejido se adhieren rápidamente al vidrio caliente debido a la atracción estática. Si varias secciones de tejido se colocan en la misma diapositiva, tenga cuidado de no solape ellos y el espacio lo suficiente como para ser capaz de encerrar de forma individual en un círculo hidrofóbica (ver sección 3.1.1.).
3. Inmunofluorescencia indirecta
- Localización de las secciones
- Use una barrera hidrofóbica lápiz para dibujar un círculo alrededor del tejido hidrófobo diapositivas montadas. Deje que el círculo seco durante aproximadamente 1 min a TA. Trace una línea alrededor de la tsecciones problema con un marcador permanente negro fino, así, pero en el lado de la corredera de vidrio opuesto al uno donde las secciones de tejido son.
Nota: Este círculo es repelente al agua y acetona y alcohol insoluble. Por lo tanto, proporciona una barrera a soluciones acuosas utilizadas durante el procedimiento de IHC y reduce el volumen de reactivos requeridos. - Rehidrate secciones de tejido cubriéndolos con una gota de ~ 250 l de PBS durante unos pocos minutos a TA. Fijar secciones de tejido cubriéndolos con ~ 250 ml de una solución de PFA 3% recién preparado en PBS durante 10 a 15 min.
Nota: De forma opcional, en este caso, utilice una solución de enfriamiento aldehído cloruro de amonio 50 mM ((NH 4 Cl, Sr. 53,5 g / mol) en PBS o glicina 0,1 M (C 2 H 5 NO 2, el Sr. 75,07 g / mol) en PBS ) para detener la reacción de fijación. Lavado simple y abundante PBS es generalmente suficiente para eliminar aldehído sin reaccionar.
- Use una barrera hidrofóbica lápiz para dibujar un círculo alrededor del tejido hidrófobo diapositivas montadas. Deje que el círculo seco durante aproximadamente 1 min a TA. Trace una línea alrededor de la tsecciones problema con un marcador permanente negro fino, así, pero en el lado de la corredera de vidrio opuesto al uno donde las secciones de tejido son.
- La recuperación de antígenos (opcional)
- Coloque la solución AR (mM Tris 100 (C 4 H 11 NO 3, Mr 121,14) 5% de urea (NH 2 CONH 2, el Sr. 60,06) pH 9,6) en un vaso de precipitados. El volumen de solución AR debe ser suficiente para cubrir completamente los portaobjetos de vidrio colocadas en un soporte de vidrio.
- Precalentar la solución AR a 95 ° C mediante el control de la temperatura con un termómetro y luego colocar los portaobjetos de vidrio en un bastidor adecuado, sumergir el bastidor en el tampón caliente, cubierta para limitar la evaporación e incubar durante 10 min a 95 ° C.
- Retire el vaso de precipitados del baño de agua y dejar los portaobjetos de vidrio durante otros 10 minutos en el búfer.
- Enjuague secciones de tejido con PBS (~ 250 l / sección) y saturar con una soluciónde 3% de albúmina de suero bovino (BSA, ~ 250 l / sección) en PBS durante al menos 30 min a TA.
- Poner un 30-50 l de anticuerpo primario diluido en PBS que contenía 2% de BSA en cada sección de tejido.
Nota: Este volumen es suficiente para formar una gota que cubre completamente la sección de tejido. - Coloque el mismo volumen de diluyente (2% de BSA en PBS) por sí sola en una sección de tejido para realizar un control negativo sin anticuerpo primario.
- Sistemáticamente incluir este control negativo en cada experimento IHC y llevar a cabo para cada anticuerpo secundario utilizado para estimar el fondo del experimento (etiquetado no específica debido a la secundaria de anticuerpos y / o el tejido auto-fluorescencia). Otros tipos de controles positivos o negativos también se pueden realizar para asegurar la especificidad de la etiqueta (ver discusión).
- Coloque los portaobjetos de vidrio en una caja humidificado O / N a 4 ° C.
Nota: Los anticuerpos primarios utilizados fueron monoclonal de ratón anti-citoqueratina8 (CK8, dilución 1:50), monoclonal de ratón anti-citoqueratina 14 (CK14, dilución 1:50), policlonal de conejo caseína anti-ratón (# 7781, 1:50 dilución, generosamente proporcionada por MC Neville, Universidad de Colorado Health Centro de Ciencias, CO, EE.UU.), policlonal de conejo anti-btn1 (1: 300 dilución, generosamente proporcionado por IH Mather, Departamento de Ciencias Animales y aviar, de la Universidad de Maryland, College Park, MD, EE.UU.), policlonal de conejo anti-Stx6 ( dilución 1:50, generosamente proporcionado por S. Tooze, Cancer Research UK, Instituto de Investigación de Londres, Londres, Reino Unido) y conejo policlonal anti-VAMP4 (dilución 1:50). - Lávese bien las secciones de tejido con PBS al menos cuatro veces durante 10 min a RT.
- Diluir el anticuerpo apropiado secundario (conjugado con rodamina de cabra anti-IgG de conejo (H + L), 1: 300 dilución) en PBS que contenía 2% de BSA, colocar el 30-50 l de esta solución en todas las secciones de tejido, y se incuba durante 1,5 hr a TA.
- Desde fluorocromos son moléculas sensibles a la luz, no lo hagasexponer las secciones de tejido a la luz hasta su análisis. Para IIF en secciones de tejido, favorecer anticuerpos secundarios acoplados a un fluoróforo rojo desde las membranas celulares tienden a generar una auto-fluorescencia verde que puede interferir con el etiquetado baja. Por otra parte, la elección de un anticuerpo secundario acoplado a fluoróforo rojo permite el etiquetado concomitante de lípidos neutros (véase más adelante).
- Lave bien las secciones de tejido con PBS por lo menos cuatro veces durante 10 minutos a temperatura ambiente.
- Para algunos experimentos, realice post-fijación mediante la incubación de las muestras con 2% PFA diluidas en PBS durante 10 min a RT a fin de estabilizar los andamios de antígeno / anticuerpo. Sin embargo, este paso se puede prescindir en la mayoría de los casos.
- Para visualizar CLD y MFG, color lípidos neutros mediante la incubación de las secciones de tejido en el 30-50 l de una solución de PBS que contenía 3 mg / ml de bodipy 493/ 503 durante 10 min a RT. Rápidamente enjuague secciones de tejido dos veces con PBS.
- Contratinción ADN nuclear con un 30-50 l de una solución de PBS que contenía 3 mM de DAPI (4-6-diamidino-2-fenilindol, 5 mg solución madre / ml) durante 10 min a TA. Lávese las secciones de tejido dos veces con PBS antes de montar las diapositivas para la observación.
- Retire PBS y colocar una gota de medio de montaje en cada sección de tejido.
- Coloque un lado de la hoja de la cubierta en un ángulo contra la diapositiva, haciendo contacto con el borde exterior de la gota líquida y luego baje la cubierta lentamente, evitar las burbujas de aire. Deje que el líquido se extendió entre el portaobjetos y el cubreobjetos durante unos minutos y luego retirar el exceso de medio de montaje con una toalla de papel.
- Sellar el cubreobjetos al portaobjetos de vidrio con secciones esmalte de uñas y tejido se almacena a 4 ° C para evitar su exposición a la luz hasta la observación.
4. Fluorescencia Observación y adquisición de imágenes
Nota: un microscopio de fluorescencia equipado con una cámara se requiere controlado por el software de adquisición de imágenes para observar los resultados de IHC.
- Antes de la adquisición de imágenes, comprobar la intensidad del etiquetado y evaluar el fondo del experimento observando los controles negativos. Adquirir imágenes de cada marcador fluorescente (canal de color) de forma individual.
- Adquirir todas las imágenes, incluidas las de los controles correspondientes, en las mismas condiciones (exposición y los ajustes generales) para cada canal de color.
- Microscopía convencional
- Realizar la microscopía de epifluorescencia con un microscopio equipado con filtros estándar de isotiocianato de fluoresceína (FITC, verde), rodamina (rojo) y DAPI (azul) emisiones, × 20 y × 63 (de inmersión en aceite, NA 1.3) objetivos y una cámara de imagen DP50.
- Microscopía confocal
- Realizar microscopía confocal con unos microscope equipado con el software ZEN, utilizando × 20 × 63 a (aceite de inmersión, NA 1.4) los objetivos y las longitudes de onda de excitación 488- y 568 nm del láser.
Tratamiento 5. Imagen
Nota: Todas las imágenes post-tratamientos se realizaron con el software libre ImageJ (http://imagej.nih.gov/ij/).
- Superponer imagen (fusionar)
- Abra las imágenes adquiridas en cada canal que se combinará (Archivo / Abrir). Si se trabaja con 8 bits escala de grises, color artificial atribuir a cada canal utilizando la tabla de búsqueda (Tablas de imagen / de búsqueda).
- Generar la imagen compuesta a partir de imágenes en escala de grises o de color mediante el comando "Combinar canales" (Imagen / Color / Combinar Canales) y luego atribuir un color a cada canal.
- Realice imagen apila superposición de la misma manera con la apertura de pilas adquiridos en cada canal que se combina (Archivo / Abrir) y usando el comando "Combinar canales y# 8221; (Imagen / Color / Combinar Canales) atribuir un color a cada canal. Guarde la pila compuesta como una secuencia de imágenes o como una película (véase la sección 5.4).
- Imagen pila Z proyección
- Utilice la función Z proyección (Imagen / pila / Zproject, Intensidad máxima) para proporcionar una vista bidimensional de todas las imágenes de una pila de imágenes mediante la proyección a lo largo del eje perpendicular al plano de la imagen (eje z). La opción "Intensidad máxima" crea una imagen en la que cada píxel contiene el valor máximo sobre todas las imágenes de la pila. Esto genera una sola imagen que permite la visualización de toda la tinción observada a través de toda la pila imagen para un canal en particular o después de la superposición de varios canales.
- Imagen proyección pila 3D
- Utilice el comando de proyección en 3D (proyecto de imagen / Stack / 3D, más brillante Point, eje y) para generar una secuencia de proyecciones de un volumen de rotación sobre un plano. La representación visual de susus superficies de las estructuras internas y depende tanto del método de proyección (punto más cercano, punto más brillante (utilizado aquí), o media-valor) y los parámetros de visualización seleccionados. Cada fotograma de la secuencia de animación es el resultado de que se proyecta desde un ángulo de visión diferente.
- Rotar la imagen 3D creada alrededor de cada uno de los tres ejes ortogonales (el eje y se ha seleccionado aquí). Guarde la secuencia producida como una sola imagen o una película.
- Pila de imagen para la conversión de la película
- Abra una pila de imágenes (Archivo / Abrir) y guardarlo como una película en formato .AVI con el comando "AVI" (Archivo / Guardar como / AVI).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
La glándula mamaria es una glándula subcutánea situado a lo largo de la estructura ventral tanto del tórax y el abdomen en los roedores. La ubicación de los cinco pares de glándulas del ratón durante la gestación se muestra en la Figura 4. La morfología de la glándula mamaria cambia dramáticamente durante su desarrollo, lo que refleja modificaciones funcionales necesarias para prepararse para la plena lactancia (Figura 1B). En los animales vírgenes o nulíparas, la glándula mamaria consiste en un epitelio ductal escasamente ramificado incrustado dentro de un estroma grasos delgada que puede ser difícil de ver. Desde el inicio del embarazo, los prolifera epitelio mamario y se expande, lo que resulta en glándulas mamarias más grandes que se vuelven más fáciles de ver y quitar (Figura 4). Durante la lactancia, el tejido mamario es más gruesa y más blanca aparece debido a la presencia de leche. Sólo glándulas mamarias abdominales e inguinales se recogen porque Glan mamaria cervical y torácicads se eliminan con menos facilidad debido a su estrecha relación con los músculos. Para algunos experimentos, las crías se pueden separar de la lactación hembra 4-6 horas antes del sacrificio a fin de limitar la secreción de leche por mESCs 46,47.
Identificación de mioepitelial mamaria y células epiteliales
Células mioepiteliales contráctiles que rodean los alvéolos se pueden distinguir de mESCs luminal a través del uso de anticuerpos dirigidos contra marcadores expresados específicamente por cada uno de estos tipos de células. En la glándula mamaria, los marcadores actuales utilizados son citoqueratinas (CKs). CKs son una gran familia de proteínas citoplásmicas que polimerizan para formar filamentos intermedios del citoesqueleto (10 nanómetros de diámetro en promedio) que se encuentran en los tejidos epiteliales. Los filamentos intermedios son extremadamente estables y proporcionan un soporte mecánico para la arquitectura celular, y organizan los tejidos, contribuyendo a la adhesión célula-célula y célula-conectivo basalinteracciones tisulares. Los subconjuntos de CKs expresada por las células epiteliales dependen principalmente del tipo de epitelio, su etapa de desarrollo y su estado de diferenciación. Por otra parte, esto también se aplica a las contrapartes malignas del epitelio. Por lo tanto, estos marcadores son herramientas simples y valiosas para caracterizar las poblaciones de células en un tejido en condiciones fisiológicas y se utilizan para el diagnóstico de tumores y caracterización de la patología quirúrgica 48.
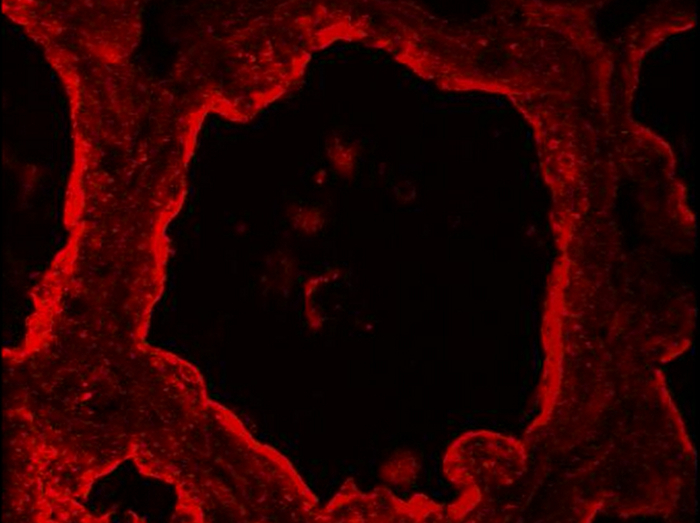
En la glándula mamaria normal, las células mioepiteliales y mESCs luminales pueden distinguirse en base a su expresión diferencial de CK14 y CK8, respectivamente (Figura 5). Estos marcadores citoplásmicos se detectan en las secciones mamarias de ratones lactantes después de PFA fijación y AR. Las imágenes fueron adquiridas con un microscopio de epifluorescencia convencional. CK8 parece estar distribuidos por todo el citoplasma de las mESCs luminal (Figura 5, Ck8). Tenga en cuenta que el fondo rojo observard para el control negativo sin anticuerpo primario (Figura 5, -Ig1) se debe principalmente a la sección de tejido plegado, según lo sugerido por el marcaje de ADN azul, que muestra varias capas de núcleos (Figura 5, -Ig1, núcleos). CK14 se observa específicamente en las células mioepiteliales planas y alargadas situadas en la base de los alvéolos (Figura 5, CK14). Otra forma común de identificar las células mioepiteliales es detectar actina de músculo liso alfa (a - SMA) presente en estas células contráctiles (véase la figura 4 en 49).
La detección de los productos lácteos ratón
Después del parto, los mESCs completamente diferenciadas comienzan a producir abundantes cantidades de leche. Componentes de la leche son secretadas por vías distintas 40,50. Micelas de caseína son secretadas por exocitosis de SV derivados de Golgi, mientras que los lípidos son liberados como MFG por el florecimiento de la pl apicalasma membrana de mESCs (Figura 2, célula secretora epitelial mamaria). Para algunos experimentos, las crías se separan de la hembra 4-6 horas antes de recoger las glándulas mamarias, con el fin de frenar la secreción de leche 46,47. En estas condiciones, la membrana plasmática apical de mESCs y el contenido del lumen se pueden observar fácilmente, lo que no es el caso durante la lactancia desde alvéolos se contraen y el lumen están cerrados. Por otra parte, lo que frena la secreción también es esencial en el estudio de las proteínas implicadas en el tráfico de membrana tales como trampas. De hecho, trampas ciclo entre el donante y aceptor compartimentos y su localización subcelular es difícil de determinar ya que el etiquetado es a menudo difunde cuando el volumen de negocios de la membrana es alto, es decir., Durante la succión. Por lo tanto, lo que frena la secreción de leche mediante la eliminación de las crías proporciona las condiciones adecuadas para estudiar la localización intracelular de trampas cuando el t- y v-SNARE preferentemente residen en el donantey el compartimiento aceptor, respectivamente (ver más abajo).
La Figura 6 muestra la localización de las caseínas en la glándula mamaria lactante ratón en día 10 de lactancia, en presencia (Figura 6, + p) o en ausencia (Figura 6, -p) de las crías. Las secciones de tejido fueron observados tanto por microscopía de epifluorescencia convencional (las tres columnas de la derecha) y microscopía confocal (Figura 6, columna izquierda). Durante la succión, las caseínas parecen estar acumulado en su mayoría en la región apical (Figura 6, + p, puntas de flecha). La microscopía confocal revela que las caseínas también están presentes, aunque en menor medida, en el lado basal de mESCs en la presencia de las crías (Figura 6, + p, flechas), que no se pueden observar claramente en la microscopía convencional (Figura 6, caseínas, comparar los paneles izquierdo y derecho). De hecho, en campo amplio de epifluorescencia, la fluorescencia emitida por la muestra (fondo fluorescence) pasa a través del volumen excitado y altera la resolución de los objetos observados en el plano focal objetivo (fuera de foco de fluorescencia). Esto es especialmente cierto para los especímenes de espesor (más gruesa que 2 M). La microscopía confocal permite obtener imágenes de alta calidad a partir de muestras preparadas para epifluorescencia, como la profundidad del campo puede ser controlada y la fluorescencia de fondo excluido desde el plano focal. Además, en la presencia de las crías (Figura 6, + p), el lumen de los alvéolos son bastante cerrada y la parte apical de mESCs se observa mejor en la ausencia de las crías (Figura 6, -P), cuando el lumen de la alvéolos se dilata debido a la acumulación de productos lácteos. Cuando la secreción de leche se ralentiza, caseínas también aparecen acumulan debajo de la membrana plasmática apical (Figura 6, -p, puntas de flecha), y se observan claramente en el lado basal de mESCs (Figura 6, -p, flechas). Los controles negativos sin antib primariaody no mostró ningún etiquetado (Figura 6, -Ig1).
Los productos lácteos pueden ser co-detectan fácilmente mediante la combinación de IHC para caseínas y neutral counterstaining lipídica de CLD y MFG (Figura 7). Las secciones de tejido se obtuvieron imágenes como Z-pilas por microscopía confocal, que eran post-tratadas con ImageJ para producir proyecciones Z o proyecciones en 3D para cada uno (Figura 7, caseínas, lípidos) o todos los canales de color (Figura 7, Merge). Las secuencias de imágenes producidas se han guardado como imágenes individuales (Figuras 7 y 8) o películas (ver Películas complementarias).
Aunque algunos de marcaje se observó en su lado basal, las caseínas se acumularon sobre todo en la parte apical de mESCs (Figura 7, + p), como ya se ha descrito cuando las hembras no estaban separados previamente de cachorros (Figura 6, + P). CLD también se localizan principalmente en la región apical de mESCs, mientras más grande secretoMFG ed están presentes en el lumen de los alvéolos. Tenga en cuenta que las caseínas y MFG son fácilmente visualizados en el lumen de los alvéolos en la ausencia de las crías (Figura 7, comparar + p y -p). Las caseínas no co-localizar con CLD o MFG en cualquiera de estas enfermedades ya que la superposición de los dos canales de color no produce etiquetado amarilla (Figura 7, combinar fotos). Sin embargo, pila de imágenes post-tratamientos muestran que rodean las caseínas MFG secretadas en el lumen de los alvéolos, lo que sugiere que estas proteínas pueden interactuar con el MFG (Figura 7, fusionar imágenes). Nótese la diferencia de las imágenes producidas por cada post-tratamiento utilizado (Figura 7, comparar Zproj y 3D proj para cada canal de color).
La detección de butyrophilin, un marcador de la proteína de MFG.
Btn1 es una de las principales proteínas asociadas con MFG en la leche 51. Esta proteína transmembrana es pppgy localizada en la membrana plasmática apical de las mESCs y por consiguiente se encuentra en la superficie de la MFG después de su lanzamiento por gemación 52. Figura 8 muestra que al día 10 de lactancia, btn1 se localiza principalmente en la membrana plasmática apical y, a una en menor medida, en la región apical de mESCs. Btn1 también rodea los MFG presentes en el lumen de los alvéolos, así como algunos de los CLD apical (Figura 8, proj de combinación de 3D, puntas de flecha). Los resultados se muestran como una sola imagen extraído de la imagen Z-pila adquirida (Figura 8, imagen) o como una vista 3D generado con el comando de proyección en 3D de ImageJ, como se describió anteriormente (Figura 8, proj 3D). Tenga en cuenta que una sola imagen puede ser suficiente para observar la distribución apical de la proteína, pero la asociación espacial de btn1 con MFG secretadas o apical CLD sólo se observa después de la reconstrucción 3D de la Z-pila (Figura 8 compara la imagen btn1 y de combinación de 3D proj pictoUres). El Z-pila también puede ser reconstruida como una película para dar una mejor visión espacial de la distribución de la proteína. La imagen de la Z-pila adquirido para btn1 solo (películas de apoyo 1 y 3) o superpuesta con los otros dos canales de color (Merge, películas complementarias 2 y 4) se muestran como ejemplos. El Z-pila puede ser leidos imagen por imagen desde la parte superior a la parte inferior (películas de apoyo 1 y 2) o como una vista de rotación (eje y) de la proyección en 3D de toda la pila de imágenes (películas complementarias 3 y 4 ).
La detección de dos proteínas SNARE: Stx6 y VAMP4
Como se mencionó anteriormente, trampas son proteínas de membrana que desplazarse entre las membranas de donante y aceptor. Por tanto, es mejor reducir la velocidad rotación de membrana asociada a la alta actividad secretora de mESCs separando las hembras de las crías antes de recoger la glándula mamaria en el estudio deestas proteínas. Stx6 y VAMP4 ambos se han descrito como asociadas con la red trans-Golgi 53,54. Sin embargo, estas proteínas SNARE también pueden desempeñar un papel importante en el nivel de otros compartimentos celulares, tales como los gránulos secretores (Stx6 55,56) y el aparato de Golgi (VAMP4) 57. Estudios previos sugieren que las proteínas SNARE juegan un papel en la secreción de la caseína 35,36. Durante la lactancia, Stx6 y VAMP4 se encuentran en la región sub-apical de mESCs. Se observa Stx6 entre el núcleo y la membrana apical de MECs, correspondiente a la Golgi y la red trans-Golgi (Figura 9, Stx6), y también está presente, aunque en menor medida, en SVs 36 que contiene caseína. VAMP4 también se localiza en la región sub-apical de mESCs, pero el etiquetado parece ser más punteada y se acumula debajo de la membrana plasmática apical (Figura 9, VAMP4) debido a su asociación con ambas CLD y la caseína-contaSV INING 36. Control negativo sin anticuerpo primario no dio lugar a ninguna etiqueta.

Figura 1. Ratón desarrollo de la glándula mamaria durante la vida embrionaria y adulta. (A) Las glándulas mamarias de ratones comienzan a desarrollarse alrededor de día embrionario 10 (E10) de los ectodérmica (azul claro) líneas de leche (de color rosa). En E11.5, placodas forman simétricamente a lo largo de la línea de leche mamaria y el mesénquima circundante (azul oscuro) comienza a condensarse. Los placodas invaginan para formar brotes (E12.5-E14.5) y, en E15.5, el epitelio mamario (rosa), proliferan y alargado para formar el brote principal que empuja a través del mesénquima mamario hacia la almohadilla de grasa (verde claro ). Se forma un lumen huecos y se abre para dar lugar a la boquilla (morado). En E18.5, el epitelio mamario forma una Rudimentaestructura ramificada ry conectado al exterior. Adaptado de 6 con permiso de Macmillan Publishers Ltd: Nature Reviews Genetics, derechos reservados 2007. (B) Durante la pubertad, el epitelio mamario (púrpura) entra en una fase significativa del crecimiento (extensa elongación, bifurcación y ramificación lateral). En el inicio de la gestación, extensa y rápida proliferación, así como ramificación lateral ocurrir, llevando a la considerable expansión del epitelio mamario, que invade totalmente toda la almohadilla de grasa mamaria. El epitelio mamario alcanza un estado funcional altamente diferenciado durante la lactancia cuando mESCs luminales secretan grandes cantidades de leche. Cuando la lactancia cesa después del destete, los involuciona glándula mamaria. MESCs se eliminan mediante apoptosis y la fagocitosis, lo que lleva a la desaparición de las estructuras lóbulo-alveolar que se sustituye por el tejido adiposo. Adaptado del esquema 1 de http://brisken-lab.epfl.ch/research y el capítulo 2.2. http://tvmouse.ucdavis.edu/bcancercd/22/index.html. Haga clic aquí para ver una versión más grande de esta figura.
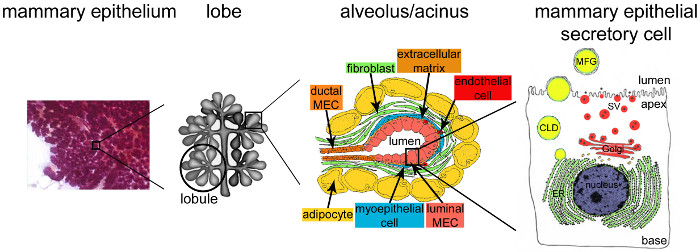
Figura 2. Arquitectura de la glándula mamaria durante la lactancia. Durante la lactancia, el completamente desarrollado y el epitelio altamente ramificada (púrpura) da cuenta de la gran mayoría del tejido mamario. El tejido epitelial está formado por estructuras túbulo-alveolar embebidos en un estroma que contiene diferentes tipos de células (fibroblastos, adipocitos, células musculares lisas, la sangre y los vasos linfáticos y terminaciones nerviosas). MESCs se organizan en estructuras o alvéolos acinares, reunidos en lóbulos que forman lóbulos. Cada alvéolo es una unidad productora de leche funcional que está conectado a una red muy ramificada de canales lobular y interlobulares, permitiendo así que la leche a ser dllovido hacia el exterior. Cada alvéolo está delimitado por una monocapa de mESCs polarizados, la parte apical de que limita con un lumen central. El lado basal de la mESCs está en estrecho contacto con una matriz extracelular y células mioepiteliales contráctiles. Los productos lácteos son liberados en la parte apical de mESCs. Major leche (caseínas) son secretadas como micelas de caseína (puntos negros) por exocitosis de vesículas secretoras derivadas de Golgi (SVS), mientras que los lípidos son liberados en forma de glóbulos de grasa de leche (MFG) por gemación de la membrana plasmática apical de mESCs. EPC: gotas de lípidos citoplasmática; ER: retículo endoplasmático; MEC: células epiteliales mamarias. Adaptado del capítulo 2.2. http://tvmouse.ucdavis.edu/bcancercd/22/index.html., Fig. 02 www.cellbiol.net/ste/alpHERCEPTIN1.php, Fig. 26-02 en el 58, y de 50. Haga clic aquí para ver una versión más grande de esta figura.
.Dentro-page = "always"> 

Figura 3. Procedimiento experimental para realizar inmunofluorescencia indirecta en secciones congeladas de glándula mamaria de ratón. La glándula mamaria se recoge de un ratón hembra CD1 en el día 10 de la lactancia. El tejido mamario se corta en pequeños fragmentos que se fijan con paraformaldehído y infunden en sacarosa antes de ser incrustado en compuesto OCT y snap-congelado. Las muestras de glándula mamaria se cortan en secciones congeladas finas y se procesaron para IIF por incubación sucesiva con anticuerpos primarios y secundarios conjugados con fluorocromo, respectivamente. Después del montaje, las muestras se analizaron con un microscopio de fluorescencia, lo que permite la adquisición de imágenes que posteriormente pueden ser post-tratado./53179/53179fig3large.jpg "Target =" _ blank "> Haga clic aquí para ver una versión más grande de esta figura.

. Figura 4. Anatómico localización de las glándulas mamarias de ratón izquierdo: vista ventral del sistema mamario del ratón en la etapa de gestación tardía. Derecha: localización y aspecto de la glándula mamaria en la etapa de gestación tardía en el ratón. Tenga en cuenta que durante la lactancia, las glándulas mamarias son más gruesas y aparecen más blanca debido a la presencia de leche en los alvéolos. Adaptado de http://ctrgenpath.net/static/atlas/mousehistology/Windows/femaleu/mousemammgldiagram.html y http://www.pathbase.net/Necropsy_of_the_Mouse/index.php?file=Chapter_3.html. Haga clic aquí para ver una versión más grande de esta figura.
-together.within-page = "always"> 
Figura 5. Identificación de células epiteliales luminales y células mioepiteliales basales en la glándula mamaria del ratón. MESCs luminal y células mioepiteliales son identificados por IIF en la glándula mamaria del ratón en el día 10 de lactancia, en base a su expresión de CK-8 y CK-14 , respectivamente. El ADN nuclear fue teñido con DAPI (azul). Las imágenes fueron adquiridas con un microscopio de epifluorescencia convencional. La imagen compuesta (fusionar) muestra la superposición del etiquetado correspondiente a las caseínas (rojo) y núcleos (azul), respectivamente. -Ig1, Control negativo sin anticuerpo primario. Los asteriscos indican lúmenes. Barra de escala = 100 micras. Haga clic aquí para ver una versión más grande de esta figura.
e 6 "src =" / files / ftp_upload / 53179 / 53179fig6.jpg "/>
Figura 6. localización celular de las caseínas en la glándula mamaria del ratón. Las caseínas son detectados por IIF en la glándula mamaria del ratón en el día 10 de lactancia. La glándula mamaria se recogió a partir de hembras en presencia (+ p) o en ausencia (-p) de las crías. Las imágenes fueron adquiridas con convencionales (derecha del panel, caseínas, núcleos y se fusionan) o una confocal (caseínas (rojo), izquierda panel) microscopio de fluorescencia. En ambas condiciones, caseínas (rojo) se detectan en la región apical (puntas de flecha) y más o menos al basal de mESCs (flechas). Los controles negativos sin anticuerpos primarios no muestran ningún etiquetado (-Ig1). El ADN nuclear se tiñe con DAPI (azul). La imagen compuesta (fusionar) muestra la superposición del etiquetado correspondiente a las caseínas (rojo) y núcleos (azul), respectivamente. Los asteriscos indican lúmenes. Barra de escala = 100 micras para las imágenes de epifluorescencia (panel de la derecha, caseínas, núcleos, fusión) y = 10 y# 181;. M para imágenes confocal (columna izquierda) Haga clic aquí para ver una versión más grande de esta figura.
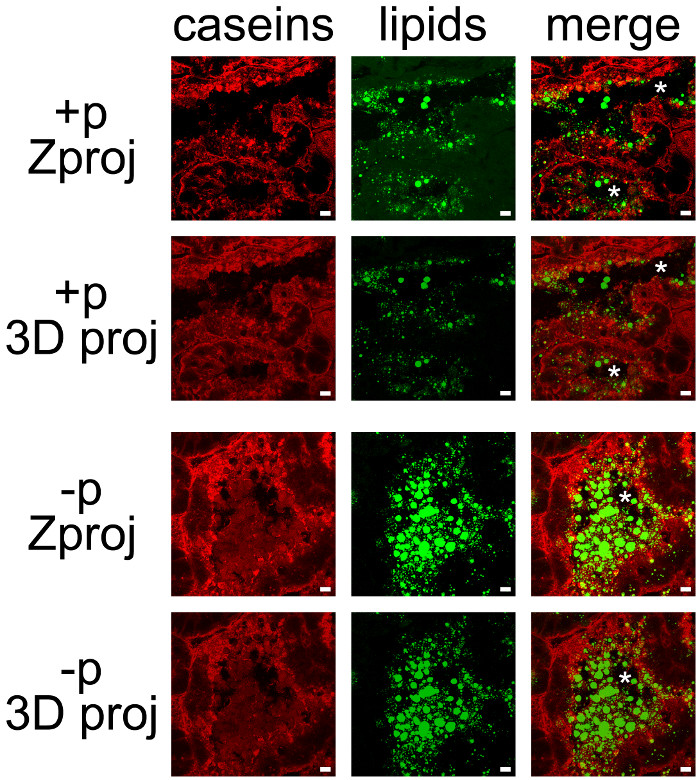
Figura 7. localización celular de los productos lácteos en la glándula mamaria del ratón. Las caseínas (rojo) son detectados por IIF en la glándula mamaria del ratón en el día 10 de lactancia en presencia (+ p) o en ausencia (-p) de las crías. Lípidos neutros (CLD y MFG) se counterstained con bodipy 493/503 (verde). Las fotografías compuestas (Merge) muestran la superposición de los dos marcajes. Las imágenes fueron adquiridas como Z-pilas con un microscopio confocal. Z-pilas fueron post-tratados con ImageJ para generar proyecciones Z (Zproj) o proyecciones en 3D (eje y) (proy 3D) de toda la pilas en cada canal para ambos (fusionar). Los asteriscos indican lúmenes. Barra de escala= 10 micras. Haga clic aquí para ver una versión más grande de esta figura.

Figura 8. localización celular de butyrophilin y los lípidos en la glándula mamaria del ratón. Btn1 (rojo) es detectado por IIF en la glándula mamaria del ratón en el día 10 de lactancia en la ausencia de los cachorros. Lípidos neutros (CLD y MFG) y ADN nuclear se counterstained con bodipy 493/503 (verde) y DAPI (azul), respectivamente. Las imágenes fueron adquiridas con un microscopio confocal como imagen Z-pilas. Los resultados se muestran como una sola imagen extraída de la pila de imágenes (imagen, btn1, lípidos, núcleos y se fusionan) o después de post-tratamiento con ImageJ para generar una vista 3D (eje y) de toda la pila de imágenes (proy 3D, btn1 , los lípidos, los núcleos, se fusionan). Las fotografías compuestas (Merge) muestran lasuperposición de los tres canales de color. -Ig1, Control negativo sin anticuerpo primario. Los asteriscos indican lúmenes. La barra de escala = 10 micras. Haga clic aquí para ver una versión más grande de esta figura.

Figura 9. localización celular de dos proteínas SNARE en la glándula mamaria de ratón. Sintaxina 6 (Stx6) y VAMP4 (V4) son detectados por IIF en la glándula mamaria del ratón en el día 10 de lactancia. Las imágenes fueron adquiridas con un (conv) epifluorescencia convencional o un confocal (LSM) microscopio. Las imágenes compuestas (Merge) muestran la superposición de la etiqueta observada para cada proteína SNARE (rojo) y para el ADN nuclear a contratinción con DAPI (color verde falsa), respectivamente. -Ig1, Control negativo sin anticuerpo primario. Asteriscos indican lúmenes. Barra de escala = 10 micras de imágenes confocal y = 100 m para epifluorescencia imágenes. Por favor, haga clic aquí para ver una versión más grande de esta figura.


Tabla 1. Guía de solución de problemas La inmunohistoquímica.

Película suplementario 1. La localización de butyrophilin en la glándula mamaria del ratón. Btn1 (rojo) es detectado por IIF en la glándula mamaria del ratón en el día 10 de lactancia. Las imágenes fueron adquiridas con un compañero microscopio nfocal como Z-pila y post-tratada con ImageJ para generar una película. El Z-pila se lee desde la parte superior a la parte inferior. Por favor haga clic aquí para ver el vídeo.

Película complementario 2. La localización de butyrophilin y lípidos neutros en la glándula mamaria del ratón. Btn1 (rojo) es detectado por IIF en la glándula mamaria del ratón en el día 10 de lactancia. Lípidos neutros (CLD y MFG) y ADN nuclear se counterstained con bodipy 493/503 (verde) y DAPI (azul), respectivamente. Las imágenes fueron adquiridas con un microscopio confocal como Z-pila para cada canal de color y fueron post-tratados con ImageJ para generar un Z-pila compuesta que superpone los tres canales de color. El Z-pila de material compuesto resultante se lee desde la parte superior a la parte inferior.https://www.jove.com/files/ftp_upload/53179/supvid2.mp4 "target =" _ blank "> Haga clic aquí para ver el vídeo.
Película suplementario 3. La localización de butyrophilin en la glándula mamaria del ratón. Btn1 (rojo) es detectado por IIF en la glándula mamaria del ratón en el día 10 de lactancia. Las imágenes fueron adquiridas con un microscopio confocal como Z-pila y post-tratado con ImageJ (proyección 3D) para generar un (eje y) que gira vista espacial del etiquetado btn1. Haga clic aquí para ver el vídeo.

Película suplementario 4. La localización de butyrophilin y neutrales lípidos en la ma ratónglándula mmary. btn1 (rojo) es detectado por IIF en la glándula mamaria del ratón en el día 10 de lactancia. Lípidos neutros (CLD y MFG) y ADN nuclear se counterstained con bodipy 493/503 (verde) y DAPI (azul), respectivamente. Las imágenes fueron adquiridas con un microscopio confocal como Z-pila para cada canal de color y fueron post-tratados con ImageJ para generar un Z-pila compuesta que superpone los tres canales de color. ImageJ (proyección 3D) se utilizó además para generar un (eje y) que gira vista espacial de la Z-pila compuesto. Haga clic aquí para ver el vídeo.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
IHC es un método experimental relativamente simple y directo para localizar el antígeno en secciones de tejido, que depende principalmente de las interacciones específicas de epítopo de anticuerpos. Aunque un gran número de protocolos se utilizan para localizar una proteína por IIF, el núcleo de estos procedimientos es casi siempre el mismo. Sin embargo, hay algunos aspectos críticos que pueden influir fuertemente el resultado y por lo tanto deben ser optimizados para cada estudio individual IHC. El aspecto más difícil de este enfoque es determinar las mejores condiciones experimentales, es decir., Los que generan una señal fuerte y específico para el antígeno de interés. Las variables que se deben considerar para el diseño experimental y optimización son: (1) el tipo de antígeno (especies, los niveles de expresión, localización subcelular); (2) el tipo epítopo (secuencia, conformación, modificaciones post-traduccionales putativo); (3) preparación de la muestra (la incrustación en parafina o para secciones congeladas); (4) la fijación metod (la perfusión o inmersión); (5) el fijador utilizado (formaldehído, alcohol o acetona); (6) el reactivo de bloqueo utilizado (suero normal, BSA o leche sin grasa); (7) la etapa de AR; (8) el método de detección (directa o indirecta); (8) el tipo de anticuerpo primario (policlonal o monoclonal); (9) El anticuerpo secundario (especies y etiqueta); (10) contratinciones (nuclear y / u otro compartimento celular etiquetado); y (11) del medio de montaje (véase la Tabla 1 para más detalles). La fijación y las etapas de bloqueo, al menos, requieren la optimización de factores adicionales, tales como la concentración, pH, temperatura, tiempo de incubación y diluyente.
El primer aspecto crucial se refiere a la preparación de muestras de tejido, que está estrechamente vinculado al método de fijación, que a su vez influye en la calidad de los resultados. Por ejemplo, trozos de tejido pueden ser fijas o no con anterioridad a la incrustación. Este paso también puede depender del método de incorporación elegido, es decir, un compuesto OCT vs. inclusión en parafina, Que a su vez depende a veces en el anticuerpo primario utilizado. La fijación del tejido se puede realizar in vivo por perfusión un animal anestesiado con una solución de fijación. Este método es útil para preservar antígenos cuando se estudia tejidos intactos pero puede no ser suficiente para fijar el tejido de interés. En este caso, las pequeñas piezas de tejido (no más grueso que 10 mm) se puede sumergir en la solución de fijación. El tejido congelado se puede preparar mediante la inmersión del tejido en nitrógeno líquido o isopentano, y complemento de congelación es altamente recomendable para la posterior detección de modificaciones post-traduccionales tales como fosforilación. Sin embargo, a diferencia de tejido incluido en parafina, la congelación no es adecuada para la preservación a largo plazo de los tejidos debido a la formación de cristales de hielo dentro de las células que pueden alterar la morfología subcelular. Una vez cortadas, las secciones de tejidos congelados se pueden almacenar a -80 ° C por hasta 1 año. En cualquier caso, la preparación de las muestras de tejido es un compromiso entre la preservación del tejido/ arquitectura celular y la preservación de la integridad epítopo.
Ya que altera la composición química de los tejidos, es fundamental para optimizar las condiciones de fijación para evitar tanto incompleta (menos de fijación) y excesiva (overfixation) fijación.
De hecho, underfixation puede reducir la señal específica mediante la promoción de la degradación proteolítica de ciertos antígenos. Por otro lado, overfixation puede alterar el etiquetado específico enmascarando el epítope o la generación de un fuerte fondo no específica. Por lo tanto, además de la elección de la solución de fijación, otros parámetros tales como el tiempo de incubación, la temperatura y el pH afectan a la fijación del tejido. Aunque PFA es el fijador más común usado para IHC, no puede ser considerado como un fijador "universal". PFA induce proteína-proteína y ácidos enlaces cruzados de proteínas nucleico y por lo tanto puede modificar artefactually el epítopo (overfixation) y luego impedir su recognition por el anticuerpo primario. Sin embargo, el epítopo puede ser desenmascarado adicionalmente mediante técnicas AR (ver más abajo). PFA también puede ser inadecuado para la detección de ciertos antígenos, ya que se ha demostrado que induce la translocación de algunas proteínas fosforiladas desde la membrana hasta el citoplasma. En tales casos, PFA debe ser reemplazado por fijadores alternativos apropiados, tales como alcohol. A diferencia de PFA, alcoholes tales como metanol o etanol no enmascaran epítopos ya que permiten la fijación del tejido mediante la sustitución de las moléculas de agua en los tejidos. Esto puede conducir a la precipitación de las proteínas y luego evitar la interacción anticuerpo / epítopo debido a los cambios conformacionales. En general se piensa que los alcoholes no penetran y por lo tanto no conservan la morfología del tejido, así como PFA. La acetona es otro fijador alternativa, que se utiliza comúnmente cuando se trabaja con, secciones de tejido congeladas complemento no fijadas. Sin embargo, la acetona es un agente deshidratante fuerte y puede conducir a la precipitación irreversible de las proteínas del tejido.
Para algunos antígenos, una etapa adicional de AR puede ser necesaria para obtener una buena señal, sobre todo si el fijador induce el cambio conformacional o altera la carga electrostática del epítopo (enmascaramiento del epítopo). Métodos AR tienen como objetivo revertir estos procesos para restaurar la inmunorreactividad del epítopo y su posterior interacción con el anticuerpo primario. Métodos AR se basan principalmente en dos enfoques: (1) recuperación del epítopo inducida por la proteasa, es decir, con enzimas tales como la proteinasa K, la tripsina o pepsina, que escinden péptidos que enmascaran el epítopo; y (2) la recuperación de epítopo inducida por calor, es decir, el uso de un horno de microondas, ollas a presión, vapores vegetales, autoclaves o baños de agua. Este último enfoque es especialmente tiempo-, con temperatura,-tapones, y sensible al pH, y las condiciones óptimas se debe determinar empíricamente (un ejemplo se proporciona en la sección Protocolo). Alternativamente, la afinidad de un anticuerpo por su antígeno se puede mejorarcambiando el pH o la concentración de cationes del diluyente de anticuerpo.
Un paso de permeabilización a veces se requiere para obtener una buena señal para un epítopo intracelular en secciones de tejidos gruesos, en particular para la tinción de antígeno nuclear. Esto puede lograrse de diversas maneras mediante el uso de: (1) alcoholes o acetona como fijadores; o (2) detergentes tales como Triton, NP-40 (0,1 hasta 0,2% en PBS, 10 min), digitonina, saponina o Tween 20 (0,2 a 0,5% durante 10 a 30 min) después de PFA fijación. Sin embargo, la elección del detergente depende de la localización celular del epítopo detectado. De hecho, detergentes fuertes, tales como Triton-X100, que solubilizan las membranas celulares, son adecuados para la detección epítopo nuclear, pero pueden llevar a indicar alteración en la extracción de algunas proteínas membranosas. El uso de detergentes más suaves (de saponina y Tween 20) son más adecuados para la detección de epítopos citoplasmáticos.
El segundo paso crítico es el blocking de la tinción no específica. La unión de un anticuerpo a su epítopo diana se rige por las fuerzas intermoleculares (interacciones por ejemplo, hidrófobos e iónicos, enlaces de hidrógeno). Así, las interacciones de anticuerpos primarios y / o secundarios con otras proteínas que sus antígenos diana pueden resultar en la tinción no específica. Esto genera fondo de alta fluorescencia, lo que impide la visualización de la proteína de interés (baja relación señal / ruido). El bloqueo de reactivos reduce las interacciones no específicas, sin perjudicar interacción específica de anticuerpo / epítopo. Un procedimiento común consiste en incubar las secciones de tejido con suero normal inactivado por calor o BSA. Cuando se utiliza un suero normal, debe ser de la misma especie que la del animal huésped del anticuerpo secundario o de una especies no relacionadas. En todos los casos, el reactivo de bloqueo seleccionado también debe ser añadido a los diluyentes para los anticuerpos primarios y secundarios. Además, el uso de detergentes no iónicos tales como Triton X-100, Tween 20 o saponina ayuda a reducir las interacciones no específicas.
La tercera y probablemente más importante parámetro es la selección anticuerpo primario y optimización. Obviamente, la mejor elección es un anticuerpo de alta calidad con un mínimo de reactividad cruzada. Como anticuerpos monoclonales usualmente exhiben alta afinidad y especificidad para un único epítopo, son las mejores herramientas para discriminar un miembro particular de una familia de proteínas con alta identidad de secuencia. Sin embargo, la interacción anticuerpo / epítopo puede verse comprometida si el epítopo diana ha perdido su estado conformacional nativo o cuando el acceso al epítopo se evita mediante interacciones con otras proteínas, modificaciones post-traduccionales, temperatura, pH, la fijación y la concentración de sal. En tales casos, los anticuerpos policlonales son más adecuados ya que reconocen múltiples epítopos de la misma proteína. Además, a menudo son más estables que los anticuerpos monoclonales en un amplio rango de pH y concentración de sal.Los estudios preliminares tienen que definir las condiciones de incubación apropiados, es decir, trabajando de dilución (anticuerpo monoclonal: 5-25 mg / ml, anticuerpo policlonal: 1,7-15 mg / ml), tiempo de incubación, diluyente y de temperatura, que tiene que ser determinado empíricamente para cada anticuerpo primario. Estos parámetros tienen que ser optimizado para determinar las condiciones que producen la señal óptima con bajo nivel de ruido de fondo. La especificidad del etiquetado es favorecido por tiempos de incubación más largos a temperaturas más bajas (es decir., 4 ° C vs. RT).
La elección para realizar la detección directa o indirecta a menudo depende del nivel de expresión del antígeno. Por ejemplo, un epítopo altamente expresado simplemente se puede detectar con un anticuerpo primario conjugado a un fluorocromo, permitiendo así una tinción multicolor rápida y sencilla posible evitando al mismo tiempo no específica de fondo debido a la utilización de un anticuerpo secundario. Sin embargo, dirigir SI mayo genera una señal de baja a un costo más alto, y puede slgunas veces ser difícil, anticuerpo marcado cuando no están disponibles comercialmente. A la inversa, IIF es más sensible para detectar epítopos inferiores expresadas como la señal generada es más intensa debido a la interacción de al menos dos anticuerpos secundarios marcados (generado contra la especie huésped anticuerpo primario) con el anticuerpo primario (amplificación). Además, una amplia gama de anticuerpos secundarios conjugados con diferentes fluoróforos están disponibles comercialmente, relativamente barato, y calidad controlada. Sin embargo, este enfoque puede inducir la reactividad cruzada y por lo tanto requiere que elegir cuidadosamente anticuerpos primarios que no se producen en las mismas especies o de diferentes isotipos cuando se realizan experimentos de etiquetado múltiple. IIF también a veces requiere pasos adicionales de bloqueo y debe incluir controles negativos sistemáticas (ver más abajo). La amplificación se puede lograr aún más mediante el uso de un anticuerpo secundario conjugado con biotina y marcado con fluorescencia avidina o estreptavidina (cuatro biotinas unidas por molecule). Sin embargo, este método de amplificación requiere pasos adicionales para evitar la unión no específica y no puede ser adaptado para la tinción de algunos tejidos (hígado, riñón, corazón, cerebro, pulmón y glándula mamaria lactante) debido a la presencia de altos niveles de biotina endógena . Sin embargo, la biotina endógena puede ser bloqueada por pre-incubación de la muestra con avidina y, posteriormente, con biotina antes de la incubación con el anticuerpo primario. La elección de los fluorocromos conjugados, que son pequeñas moléculas químicas con la propiedad de emitir luz cuando es excitado por la luz de una longitud de onda más corta, depende principalmente del tipo de equipo microscopio disponible.
Cuando está correctamente diseñado para limitar tanto la reactividad cruzada entre los anticuerpos y de cruce entre las propiedades espectrales de los fluorocromos utilizados, IHC basado inmunofluorescencia-permite la visualización simultánea de múltiples dianas celulares.
La última críticapunto con respecto a experimentos de IHC se refiere a los controles positivos y negativos que se deben realizar para apoyar la validez de la tinción, identificar artefactos experimentales y para la interpretación precisa de los resultados. Algunos tejidos presentan alto fondo fluorescente (denominado autofluorescencia) que podría conducir a una mala interpretación de los resultados. Por lo tanto, las secciones de tejido tienen que ser observado tanto bajo la iluminación de fluorescencia y de campo brillante antes de comenzar el experimento IHC. Un control negativo que omite el anticuerpo primario sistemáticamente debe ser incluido en cada experimento IHC con el fin de asegurar que una no específica potencial de unión del anticuerpo secundario es insignificante y no oculte ni parecerse al patrón de tinción específica. Un control de isotipo se puede realizar cuando se trabaja con un anticuerpo primario monoclonal mediante su sustitución con un anticuerpo no inmune del mismo isotipo (por ejemplo, IgG1, IgG2A, IgG2b, IgM) a la misma concentración. Este control ayuda a estimate la tinción no específica, que puede ser debido a las interacciones de anticuerpos con la muestra. Para demostrar la unión de un anticuerpo a su antígeno específico, un control de la absorción se puede lograr de dos maneras por pre-incubación del anticuerpo primario: (1) con su inmunógeno soluble (10: 1 relación molar) O / N a 4 ° C ; y (2) con células o secciones de tejido que expresan el epítopo de interés, pero que difieren de los tejidos estudiados (por ejemplo, véase la Figura 4B en 59). En ambos casos, el consiguiente agotamiento del anticuerpo primario debe dar lugar a poca o ninguna tinción. Otro tipo de control se puede hacer usando un anticuerpo primario irrelevante, es decir., Dirigido contra un epítopo que presenta una localización celular que es diferente de la epítopo de interés (nuclear vs. citoplasmática). El anticuerpo irrelevante debe ser del mismo isotipo y las especies como el anticuerpo primario de interés. Controles adicionales para los experimentos de IHC pueden incluir el uso de muestras de TISSues se sabe que expresan (animales transgénicos) o no (los animales knock-out), el epítopo de interés. Esto puede proporcionar una referencia útil y ayudar a optimizar el procedimiento de IHC.
Una limitación principal de las técnicas de IF es que sólo se pueden aplicar a fijo (muerto) y / o células permeabilizadas, tanto procedimiento potencialmente inducir artefactos. Otras limitaciones de este enfoque son debido al uso de un microscopio para la observación de las muestras. En primer lugar, ya que la resolución óptica de epifluorescencia y confocal microscopios es limitado, la ubicación o la co-localización de las proteínas detectadas no debe ser sobre-interpretado. En segundo lugar, photobleaching, es decir. la decoloración de la intensidad de fluorescencia con el tiempo cuando se exponen a la luz, se debe esencialmente a la generación de especies reactivas del oxígeno en la muestra tras la excitación de fluorescencia, que, a su vez, conduce a la destrucción fotoquímica del fluoróforo. Photobleaching se puede reducir por: a) mantener las muestras protegidas dela luz durante el experimento SI y almacenamiento hasta su observación; b) usando un agente antifade (eliminadores de especies reactivas de oxígeno) en el medio de montaje; c) la reducción de la intensidad y / o duración de la luz de excitación; d) el aumento de la concentración de fluoróforos o el uso de una baja concentración de un fluorocromo con alta eficiencia cuántica; y e) el uso de fluoróforos robustos que son menos propensos a photobleaching (es decir. Alexa Fluors, Seta Fluors, o DyLightFluors). En tercer lugar, la autofluorescencia es a menudo debido a la presencia de coenzimas flavina (FMN y FAD: absorción, 450 nm; emisión, 515 nm) y nucleótidos de piridina reducidos (NADH: absorción, 340 nm; emisión, 460 nm) en células de mamífero. Además, el uso de aldehídos, especialmente glutaraldehído, para fijar las muestras, puede resultar en altos niveles de autofluorescencia. Esto puede ser minimizado mediante el lavado de las muestras con 0,1% en PBS borohidruro de sodio antes de la incubación del anticuerpo y / o mediante la selección de sondas y filtros ópticos que Maximize la señal de fluorescencia con respecto a la autofluorescencia. En cuarto lugar, la superposición de fluorescencia (también denominado derrame, cruzado o crosstalk) es principalmente debido a las propiedades espectrales de emisión de los fluoróforos ya que a menudo presentan anchos de banda muy amplio, diferentes perfiles espectrales, asimétricas, así como varias longitudes de onda pico de emisión y el número de maxima. Solapamiento de la fluorescencia se produce cuando se trabaja con varios fluoróforos (etiquetado multicolor) y se caracteriza por la emisión de un fluoróforo en el canal (filtro) de otro fluoróforo. Artefactos derrame deben minimizarse ya que a menudo complican la interpretación de los resultados de SI, en particular en el caso de co-localización o estudios cuantitativos. Como balance de la emisión fluoróforos sólo puede mejorarse ligeramente por el procedimiento de IF, sangrar a través sobre todo puede ser reducido en el momento de la adquisición de imágenes mediante el uso de un filtro de fluorescencia optimizados conjuntos y / o detector fotomultiplicador para poder adecuadamente separcomieron los perfiles espectrales de los fluoróforos. En este sentido, la microscopía confocal es muy adecuado para formación de imágenes multicolor, ya que permite la diferenciación espectros de emisión de fluorescencia de los fluoróforos individuales, dirigiendo cada señal a un canal de detección particular. Por otra parte, la microscopía confocal permite ajustar la ganancia, voltaje fotomultiplicador, o la potencia del láser para los canales de detección individuales para la adquisición secuencial (sólo un fluoróforo a la vez) de la etiqueta. Idealmente, los controles de etiqueta única se deben realizar para cuantificar el derrame y finalmente eliminarlo computacionalmente. Un control sin anticuerpos secundarios (control de fondo) se puede preparar para establecer los límites de la ganancia de la señal y el desplazamiento de cada canal para la adquisición de imagen óptima. También se puede utilizar para el procesamiento posterior a la adquisición de fondo de la imagen correcta (autofluorescencia).
En conclusión, el método descrito proporciona un protocolo estándar simple para realizati fácilesen inmunotinción de secciones en la glándula mamaria. Sin embargo, las principales etapas de un experimento IHC deben ser optimizados para cada antígeno / par de anticuerpos con el fin de visualizar la tinción específica y para minimizar las señales de fondo no específicas. El método descrito también incluye varios métodos básicos para el post-tratamiento de la mayoría de las imágenes obtenidas. Inmunodetección basada en fluorescencia es un método potente con una amplia gama de aplicaciones, desde la localización celular de un antígeno para el diagnóstico. Los nuevos avances en estos enfoques se lograrán con el futuro desarrollo de nuevos fluoróforos, dispositivos de adquisición y técnicas de microscopía, a la imagen previamente detalles no observadas de estructuras y procesos biológicos.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Los autores declaran que no tienen intereses financieros en competencia.
Acknowledgments
Los autores agradecen a la facilidad de la base de imágenes INRA MIMA2 (INRA, UMR1198, Jouy-en-Josas) y para el personal de la unidad IERP (UE 0907, el INRA, Jouy-en-Josas) para el cuidado de los animales y las instalaciones. También nos gustaría dar las gracias a IH Mather, MC Neville y S. Tooze por darnos antibodie muy útil.
Materials
| Name | Company | Catalog Number | Comments |
| Dissection | |||
| Pins | |||
| Ethanol | |||
| Scissors | |||
| Scalpel and adapted blades | |||
| Ice | |||
| Towel paper | |||
| Tissue sample preparation | Company | Catalog Number | Comments/Description |
| Phosphate Buffered Saline (pH7.4) | Sigma | P-3813 | |
| Paraformaldehyde (PFA, 32% EM grade, 100 ml) | Electron Microscopy Sciences | 15714-S | personnal protection equipment required WARNING: this product will expose you to Formaldehyde Gas, a chemical known to cause cancer |
| OCT compound/Tissue Tek | Sakura | 4583 | |
| Sucrose (D-saccharose) | VWR | 27480.294 | |
| Plastic molds | Dominique Dutscher | 39910 | |
| Liquid nitrogen | |||
| Cryostat/sample support | Leica | CM3050S | |
| Razor blades (SEC35) | Thermo Scientific | 152200 | |
| Slide box | |||
| Glass slides Superfrost/Superfrost Ultra Plus | Thermo Scientific | 10143560W90/1014356190 | |
| Brushes | |||
| IHC | Company | Catalog Number | Comments/Description |
| Super Pap Pen | Sigma | Z377821-1EA | |
| Permanent marker (black) | |||
| 50 mM NH4Cl in PBS | Sigma | A-0171 | |
| 0.1 M glycine in PBS | VWR | 24403.367 | |
| Antigen Retrieval solution: Tris 100 mM 5% urea pH9.6 | |||
| Heater (up to 100°C) | |||
| Bovine Serum Albumin (BSA) | Sigma | A7906-100G | |
| Vectashield (anti-fading mounting medium) without DAPI/with DAPI | Vector Laboratories | H-1000/H-1200 | |
| Glass coverslips 22x50mm (microscopy grade) | VWR | CORN2980-225 | |
| Nail polish | |||
| Primary antibodies | Company | Catalog Number | Comments/Description |
| Rabbit anti-mouse caseins (#7781; 1:50 dilution) | generously gifted by M.C. Neville (University of Colorado Health Sciences Center, USA) |
||
| Mouse anti-cytokeratin 8 (CK8, clone 1E8, 1:50 dilution) | Biolegend (Covance) | MMS-162P | |
| Mouse anti-cytokeratin 14 (CK14, cloneLL002, 1:50 dilution) | Thermo Scientific | MS-115-P0/P1 | |
| Rabbit anti-butyrophilin (1:300 dilution) | generously gifted by I.H. Mather (Department of Animal and Avian Sciences University of Maryland College Park, USA) | ||
| Rabbit anti-Stx6 (1:50 dilution) | generously gifted S. Tooze (Cancer Research UK, London Research Institute, London, UK) |
||
| Rabbit anti-VAMP4 (1:50 dilution) | Abcam | ab3348 | |
| Secondary antibodies | Company | Catalog Number | Comments/Description |
| Rhodamine-conjugated goat anti-rabbit IgG (H + L) (1:300 dilution) | Jackson ImmunoResearch Laboratories | 111-025-003 | |
| Counterstains | Company | Catalog Number | Comments/Description |
| Bodipy 493/503 | Life Technologies (Molecular Probes) | D-3922 | |
| DAPI (4-6-diamidino-2-phenylindole) | Life Technologies (Molecular Probes) | D-1306 | |
| Observation/Image capture | Company | Catalog Number | Comments/Description |
| conventional fluorescence microscope | Leica Leitz DMRB microscope |
Standard filters for FITC, Rhodamine and DAPI emissions, ×63 oil-immersion objective (NA 1.3), DP50 imaging camera (Olympus), CellˆF software (Olympus) |
|
| Laser Scanning Microscope (confocal microscopy) | Zeiss LSM 510 microscope |
Plan-Apochromat ×63 oil-immersion objective (NA 1.4), CLSM 510 software, Confocal facilities, MIMA2 Platform, INRA Jouy-en-Josas, France, http://mima2.jouy.inra.fr/mima2) | |
| Image treatment | Company | Catalog Number | Comments/Description |
| ImageJ 1.49k software | Free software |
References
- Watson, C. J., Khaled, W. T. Mammary development in the embryo and adult: a journey of morphogenesis and commitment. Development. , 135-995 (2008).
- Smith, G. H. Experimental mammary epithelial morphogenesis in an in vivo model: evidence for distinct cellular progenitors of the ductal and lobular phenotype. Breast Cancer Res Treat. 39, 21-31 (1996).
- Van Keymeulen, A., et al. Distinct stem cells contribute to mammary gland development and maintenance. Nature. 479, 189-193 (2011).
- Oakes, S. R., Gallego-Ortega, D., Ormandy, C. J. The mammary cellular hierarchy and breast cancer. Cell Mol Life Sci. 71, 4301-4324 (2014).
- Visvader, J. E., Stingl, J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes & development. 28, 1143-1158 (2014).
- Robinson, G. W. Cooperation of signalling pathways in embryonic mammary gland development. Nat Rev Genet. 8, 963-972 (2007).
- Cowin, P., Wysolmerski, J. Molecular mechanisms guiding embryonic mammary gland development. Cold Spring Harb Perspect Biol. 2, a003251 (2010).
- Brisken, C., O'Malley, B. Hormone action in the mammary gland. Cold Spring Harb Perspect Biol. 2, a003178 (2010).
- Gjorevski, N., Nelson, C. M. Integrated morphodynamic signalling of the mammary gland. Nat Rev Mol Cell Biol. 12, 581-593 (2011).
- Daniel, C. W., Smith, G. H. The mammary gland: a model for development. J Mammary Gland Biol Neoplasia. 4, 3-8 (1999).
- Howlett, A. R., Bissell, M. J. The influence of tissue microenvironment (stroma and extracellular matrix) on the development and function of mammary epithelium. Epithelial Cell Biol. 2, 79-89 (1993).
- Edwards, G., Streuli, C. Signalling in extracellular-matrix-mediated control of epithelial cell phenotype. Biochem Soc Trans. 23, 464-468 (1995).
- Hennighausen, L., Robinson, G. W. Think globally, act locally: the making of a mouse mammary gland. Genes & development. 12, 449-455 (1998).
- Topper, Y. J., Freeman, C. S. Multiple hormone interactions in the developmental biology of the mammary gland. Physiol Rev. 60, 1049-1106 (1980).
- Brisken, C., et al. A paracrine role for the epithelial progesterone receptor in mammary gland development. Proc Natl Acad Sci U S A. 95, 5076-5081 (1998).
- Ormandy, C. J., Binart, N., Kelly, P. A. Mammary gland development in prolactin receptor knockout mice. J Mammary Gland Biol Neoplasia. 2, 355-364 (1997).
- Oakes, S. R., Rogers, R. L., Naylor, M. J., Ormandy, C. J. Prolactin regulation of mammary gland development. J Mammary Gland Biol Neoplasia. 13, 13-28 (2008).
- Hennighausen, L., Robinson, G. W. Information networks in the mammary gland. Nat Rev Mol Cell Biol. 6, 715-725 (2005).
- Kouros-Mehr, H., Werb, Z. Candidate regulators of mammary branching morphogenesis identified by genome-wide transcript analysis. Dev Dyn. 235, 3404-3412 (2006).
- Khaled, W. T., et al. The IL-4/IL-13/Stat6 signalling pathway promotes luminal mammary epithelial cell development. Development. 134, 2739-2750 (2007).
- Asselin-Labat, M. L., et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat Cell Biol. 9, 201-209 (2007).
- Barcellos-Hoff, M. H., Aggeler, J., Ram, T. G., Bissell, M. J. Functional differentiation and alveolar morphogenesis of primary mammary cultures on reconstituted basement membrane. Development. 105, 223-235 (1989).
- Robinson, G. W., McKnight, R. A., Smith, G. H., Hennighausen, L. Mammary epithelial cells undergo secretory differentiation in cycling virgins but require pregnancy for the establishment of terminal differentiation. Development. 121, 2079-2090 (1995).
- Streuli, C. H., Bissell, M. J. Mammary epithelial cells, extracellular matrix, and gene expression. Cancer Treat Res. 53, 365-381 (1991).
- Streuli, C. H., et al. Laminin mediates tissue-specific gene expression in mammary epithelia. J Cell Biol. 129, 591-603 (1995).
- Boudreau, N., Sympson, C. J., Werb, Z., Bissell, M. J. Suppression of ICE and apoptosis in mammary epithelial cells by extracellular matrix. Science. 267, 891-893 (1995).
- Pullan, S., et al. Requirement of basement membrane for the suppression of programmed cell death in mammary epithelium. J Cell Sci. 109 (Pt 3), 631-642 (1996).
- Schmidhauser, C., Bissell, M. J., Myers, C. A., Casperson, G. F. Extracellular matrix and hormones transcriptionally regulate bovine beta-casein 5' sequences in stably transfected mouse mammary cells. Proc Natl Acad Sci U S A. 87, 9118-9122 (1990).
- Streuli, C. H., et al. Stat5 as a target for regulation by extracellular matrix. J Biol Chem. 270, 21639-21644 (1995).
- Sollner, T., et al. SNAP receptors implicated in vesicle targeting and fusion. Nature. 362, 318-324 (1993).
- Jahn, R., Scheller, R. H. SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol. 7, 631-643 (2006).
- Weber, T., et al. SNAREpins: minimal machinery for membrane fusion. Cell. 92, 759-772 (1998).
- Sollner, T., Bennett, M. K., Whiteheart, S. W., Scheller, R. H., Rothman, J. E. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 75, 409-418 (1993).
- McNew, J. A. Regulation of SNARE-mediated membrane fusion during exocytosis. Chem Rev. 108, 1669-1686 (2008).
- Wang, C. C., et al. VAMP8/endobrevin as a general vesicular SNARE for regulated exocytosis of the exocrine system. Mol Biol Cell. 18, 1056-1063 (2007).
- Chat, S., et al. Characterisation of the potential SNARE proteins relevant to milk product release by mouse mammary epithelial cells. Eur J Cell Biol. 90, 401-413 (2011).
- Reinhardt, T. A., Lippolis, J. D. Bovine milk fat globule membrane proteome. J Dairy Res. 73, 406-416 (2006).
- Robenek, H., et al. Butyrophilin controls milk fat globule secretion. Proc Natl Acad Sci U S A. 103, 10385-10390 (2006).
- Fujimoto, T., Ohsaki, Y., Cheng, J., Suzuki, M., Shinohara, Y. Lipid droplets: a classic organelle with new outfits. Histochem Cell Biol. 130, 263-279 (2008).
- Mather, I. H., Keenan, T. W. Origin and secretion of milk lipids. J Mammary Gland Biol Neoplasia. 3, 259-273 (1998).
- Heid, H. W., Keenan, T. W. Intracellular origin and secretion of milk fat globules. Eur J Cell Biol. 84, 245-258 (2005).
- McManaman, J. L., Russell, T. D., Schaack, J., Orlicky, D. J., Robenek, H. Molecular determinants of milk lipid secretion. J Mammary Gland Biol Neoplasia. 12, 259-268 (2007).
- de Assis, S., Warri, A., Cruz, M. I., Hilakivi-Clarke, L. Changes in mammary gland morphology and breast cancer risk in rats. Journal of visualized experiments : JoVE. , (2010).
- Plante, I., Stewart, M. K., Laird, D. W. Evaluation of mammary gland development and function in mouse models. Journal of visualized experiments : JoVE. , (2011).
- Galio, L., et al. MicroRNA in the ovine mammary gland during early pregnancy: spatial and temporal expression of miR-21, miR-205, and miR-200. Physiol Genomics. 45, 151-161 (2013).
- Linzell, J. L., Peaker, M. The effects of oxytocin and milk removal on milk secretion in the goat. J Physiol. 216, 717-734 (1971).
- Knight, C. H., Peaker, M., Wilde, C. J. Local control of mammary development and function. Rev Reprod. 3, 104-112 (1998).
- Walid, M. S., Osborne, T. J., Robinson, J. S. Primary brain sarcoma or metastatic carcinoma? Indian J Cancer. 46, 174-175 (2009).
- Hue-Beauvais, C., et al. Localisation of caveolin in mammary tissue depends on cell type. Cell Tissue Res. 328, 521-536 (2007).
- McManaman, J. L., Neville, M. C. Mammary physiology and milk secretion. Adv Drug Deliv Rev. 55, 629-641 (2003).
- Mather, I. H., Jack, L. J. A review of the molecular and cellular biology of butyrophilin, the major protein of bovine milk fat globule membrane. J Dairy Sci. 76, 3832-3850 (1993).
- Heid, H. W., Winter, S., Bruder, G., Keenan, T. W., Jarasch, E. D. Butyrophilin, an apical plasma membrane-associated glycoprotein characteristic of lactating mammary glands of diverse species. Biochim Biophys Acta. 728, 228-238 (1983).
- Bock, J. B., Klumperman, J., Davanger, S., Scheller, R. H. Syntaxin 6 functions in trans-Golgi network vesicle trafficking. Mol Biol Cell. 8, 1261-1271 (1997).
- Tran, T. H., Zeng, Q., Hong, W. VAMP4 cycles from the cell surface to the trans-Golgi network via sorting and recycling endosomes. J Cell Sci. 120, 1028-1041 (2007).
- Wendler, F., Page, L., Urbe, S., Tooze, S. A. Homotypic fusion of immature secretory granules during maturation requires syntaxin 6. Mol Biol Cell. 12, 1699-1709 (2001).
- Wendler, F., Tooze, S. Syntaxin 6: the promiscuous behaviour of a SNARE protein. Traffic. 2, 606-611 (2001).
- Shitara, A., et al. VAMP4 is required to maintain the ribbon structure of the Golgi apparatus. Mol Cell Biochem. 380, 11-21 (2013).
- Thibault, C., Levasseur, M. C. La reproduction chez les mammifères et l'homme. , INRA Editions. 928 (2001).
- Truchet, S., Wietzerbin, J., Debey, P. Mouse oocytes and preimplantation embryos bear the two sub-units of interferon-gamma receptor. Mol Reprod Dev. 60, 319-330 (2001).
