

Introduction
Cyanobakterier är en evolutionärt gamla och skiftande stammen av bakterier som finns i nästan varje fysisk miljö på jorden. I marina ekosystem de är särskilt riklig och spelar en viktig roll i många näringsämnenas kretslopp, som står för ungefär hälften av koldioxidfixering 1, de flesta av kvävefixering 2 och hundratals miljoner ton kolväten produktion 3 i haven årligen. Kloroplasterna, den organell som ansvarar för fotosyntesen i eukaryota alger och växter, kommer sannolikt att ha utvecklats från en cyanobakterie som drabbades av en värdorganism 4. Cyanobakterier har visat sig användbara modellorganismer för studier av fotosyntes, elektrontransport 5 och biokemiska vägar, av vilka många är bevarade i växter. Dessutom cyanobakterier används alltmer för produktion av livsmedel, biobränslen 6, el 7 och industriella föreningar 8, på grund av sin highly effektiv omvandling av vatten och CO2 till biomassa med hjälp av solenergi 9. Många arter kan odlas på icke-åkermark med minimala näringsämnen och havsvatten, vilket tyder på att cyanobakterier skulle kunna odlas i stor skala utan att påverka jordbruksproduktionen. Vissa arter är också källor av naturprodukter, inklusive svampdödande, antibakteriella och anti-cancerföreningar 10,11.
Förmågan att generera mutanter är nyckeln till att förstå cyanobakterier fotosyntes, biokemi och fysiologi, och en förutsättning för utveckling av stammar för industriella ändamål. Majoriteten av publicerade studier generera genetiskt modifierade stammar genom insättning av en antibiotikaresistent kassett i stället av intresse. Detta begränsar antalet mutationer som kan införas i en stam, som endast ett fåtal antibiotikaresistens kassetter är tillgängliga för användning i cyanobakterier. Stammar innehållande gener som ger antibiotisk reresistansen kan inte användas för industriell produktion i öppna dammar, som sannolikt kommer att vara den enda kostnadseffektiva sättet att producera biobränslen och andra lågt värde produkter 12. Genereringen av omärkta mutanter övervinner dessa begränsningar. Omärkta mutanter innehåller inga främmande DNA, om inte avsiktligt har inkluderats, och kan manipuleras flera gånger. Därför är det möjligt att generera så många förändringar i en stam som önskas. Dessutom kan polära effekter på gener nedströms om modifiering plats minimeras, vilket gör att mer exakt modifiering av organismen 13.
För att generera muterade stammar, självmords plasmider innehållande två DNA-fragment identiskt med regioner i cyanobakterier kromosomen som flankerar genen som ska tas bort (kallas 5 "och 3 'flankerande regioner) först konstrueras. Två gener sätts sedan in mellan dessa flankerande regioner. En av dessa kodar för en antibiotisk resistensprotein; den andra kodar sacB, som produces levansackaras, en förening som ger känslighet för sackaros. I det första steget i processen, märkta mutanter, dvs stammar innehållande några främmande DNA, genereras. Plasmidkonstruktionen blandas med de blågröna celler och DNA tas upp naturligt av organismen. Transformanter selekteras genom tillväxt på agarplattor innehållande det lämpliga antibiotikumet och den mutanta genotypen verifierades genom PCR. Självmords plasmider kan inte replikera i stammen av intresse. några antibiotikaresistenta kolonier kommer därför att leda från en rekombinationshändelse varvid genen av intresse i införas i kromosomen. För att generera omärkta mutanter, är den markerade mutanten blandas sedan med en andra självmord plasmid innehållande bara de 5 'och 3' flankerande regioner. Men om det krävs insättning av främmande DNA, en plasmid som består av de 5 'och 3' flankerande regioner med en kassett innehållande generna av intresse införas mellan dessa DNA-fragment, kan användas. SeleInsatser är via tillväxt på agarplattor innehållande sackaros. Som sackaros är dödlig för celler när sacB-genprodukten uttrycks, är de enda celler som överlever är de i vilka en andra rekombinationshändelse har skett, varigenom sackaros känslighet genen, förutom att den antibiotiska resistensgenen, har rekombinerats ut ur kromosomen och på plasmiden. Som en konsekvens av den rekombinatoriska utbyte, är de flankerande regionerna eller varje DNA mellan dessa införas i kromosomen.
Vi har framgångsrikt använt dessa metoder för att generera flera kromosom mutationer i samma stam av Synechocystis sp. PCC6803 (hädanefter kallad Synechocystis) 13,14, för att introducera enstaka punktmutationer i en gen av intresse 13 och för uttryck av genkassetterna. Medan generering av omärkta knockouts har visats innan vårt arbete i Synechocystis 15,16, en detaljerad metod, med hjälp aven visuell presentation av de kritiska stegen är inte tillgängliga för allmänheten. Vi har också tillämpat samma metod för generering av markerade knockouts i en annan modell cyanobakterien, Synechococcus sp. PCC7002 (nedan kallat Synechococcus). Detta protokoll ger en tydlig, enkel metod för att generera mutanter och en snabb protokoll för validering och lagring av dessa stammar.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Beredning av kultur Media
- Förbereda BG11 medium enligt Castenholz, 1988 17.
- Framställa stamlösningar av 100x BG11, spårämnen och järn lager (tabell 1).
- Bered separata lösningar av fosfatstam, Na 2 CO 3 lager, N - [tris (hydroximetyl) metyl] -2-aminoetansulfonsyra (TES) buffert och NaHCOs 3 (tabell 1).
- Autoklavera fosfat och Na 2 CO 3 lager. Filter-sterilisera TES buffert och NaHCO 3 med 0,2 um filter.
- Förbereda BG11 genom att kombinera 976 ml vatten, 10 ml av 100x BG11, en ml spårelement och 1 ml järn lager och autoklavera lösningen. Sedan denna lösning kylts till rumstemperatur, tillsätt 1 ml av fosfatstam, 1 ml Na 2 CO 3 lager och 10 ml NaHCOs 3.
- För BG11 fast medium, tillsätt 15 g agar och 700 ml vatten till en flask. Till den andra flaskan, tillsätt 3 g Na2S 2 O 3, 226 ml vatten, 10 ml av 100x BG11, en ml av spårämnen och 1 ml järn lager. Autoklav båda lösningarna. Efter dessa lösningar har svalnat till rumstemperatur, kombinera dem och tillsätt 1 ml fosfat lager, 1 ml av Na 2 CO 3 lager, 10 ml TES-buffert och 10 ml NaHCOs 3.
Note: Lösningar framställes separat för att undvika utfällning av vissa salter.
- För selektion på sackaros, förbereda en 50% (vikt / volym) sackaroslösning. Filtersterilisera lösningen med 0,2 | j, m filter och lägg till BG11 (100 ml av 50% sackaros till 900 ml BG11) för att producera BG11 / 5% sackaros plattor.
Obs: Lägg inte till NaHCO 3 till BG11 / 5% sackaros agarplattor. Lägg Na 2 CO 3 som vanligt. - För odling av Synechococcus tillsätt 10 ml 1 M 4- (2-hydroxietyl) piperazin-1-etansulfonsyra, N - (2-hydroxietyl) piperazin- NR42, - (2-etansulfonsyra) (HEPES) och 1 ml av vitamin B 12 (tabell 1) till ett L av BG11 medium.
Obs: Transformation av stammar odlade i kommersiellt tillgängliga BG11 media är betydligt mindre effektivt än i BG11 medie recept som beskrivs här och rekommenderas därför inte.
2. Tillväxt av Blågrön Stammar
- Kultur stammar i 100 ml koniska kolvar med en maximal volym av 50 ml och skaka vid 120 rpm. Försegla BG11 plattor med Parafilm och punkterings tre små hål i sidan av plattan för att möjliggöra gasväxling. Inkubera alla stammar vid 30 ° C under lysrör i en fotobioreaktor vid en ljusintensitet mellan 20-40 fimol fotoner m -2 s -1.
- Använd bästa sterila tekniker. Hantera alla blågrön stammar i ett laminärt flöde huva.
Notera: Detta är särskilt viktigt när stammar odlas med medium innehållande sackaros, som lätt kan contaminpade.
3. Framställning av plasmidkonstrukt
- Design uppsättningar av primers, inklusive de nödvändiga restriktionsenzymställen, med hjälp av primer design mjukvara som Primer3 (http://frodo.wi.mit.edu/primer3/), för att förstärka två ~ 1 kb regioner 5 'och 3' om genen av intresse. Konsultera genomsekvensen av blågrön arter via Cyanobase (http://genome.kazusa.or.jp/cyanobase). Se tabell 2 för alla primrar som används här. Vid utformningen primers överväga följande faktorer:
- Säkerställa att amplifierade regioner innefattar 5'- och 3 'regionerna av genen som skall muteras, t ex Figur 1.
- Inte mutera intergena regioner för att undvika oavsiktlig mutation av antisens och icke-kodande RNA. För generering av mutanter i Synechocystis, se listan över transkriptionsstartställena dokumenterade i Mitschke et al., 2011 18, i syfte att undvika mutation av antisenseller icke-kodande RNA.
- När du väljer flankerande regionerna inte omfattar hela den öppna läsramen av intilliggande gener som uttryck av dessa gener i Escherichia coli kan störa kloning.
- Förstärk produkter av PCR med användning av high fidelity DNA-polymeras enligt tillverkarens instruktioner.
Obs: Enligt vår erfarenhet ger detta enzym några fel.- Inrättat 50 pl PCR-reaktioner innehållande HF buffert och antingen 0, 1,5 eller 3 pl DMSO. Använd 100 ng av genomiskt DNA per reaktion. Använda ett program som består av ett initialt denatureringssteg av 98 ° C under 30 sek, 35 omgångar 98 ° C under 10 sek, 67 ° C under 30 sek, 72 ° C under 30 sekunder, följt av en slutlig förlängningssteg av 72 ° C i 5 min. Detta ger vanligtvis konsekvent produkter.
- Kontrollera PCR-produkter och prover smälta med endonukleas enzymer för rätt storlek via gelelektrofores. Köra en% (vikt / volym) agarosgeler innehållande 0,02%(Volym / volym) etidiumbromid under 45 min vid 100 V.
VARNING: Etidiumbromid är en potentiell mutagen och bör hanteras med lämpligt skydd. - Rena PCR-produkter med användning av en DNA-reningskit enligt tillverkarens instruktioner. Också använda detta kit för rening av plasmid-fragment, inklusive bitar skärs från agarosgeler. Eluera renade DNA i 14 pl vatten.
- För kloningssteg, inkubera restriktionsendonukleas reaktionsblandningarna vid 37 ° C under> 1 timme i en total volym av 30 | il enligt tillverkarens instruktioner.
- För ligering steg, Ligera DNA-fragment vid rumstemperatur för> 1 timme i en total volym av 20 | j, l, innehållande 5 | j, l av renad digererad plasmid, 12 | j, l av renat diger insert, 2 | j, l av buffert och 1 yl ligas.
- Förbered Escherichia coli DH5a transformanta cellerna enligt följande metod.
- Odla en övernattning E. coli
- Inokulera 400 ml LB i en 1 L konisk kolv innehållande 6 ml 1 M MgCl2 (tabell 1) med 1 ml av övernattskultur.
- Odla kulturen vid 37 ° C vid 220 varv per minut under ca 4 h eller tills OD 600 nm når 0,4-0,6.
- Placera cellerna på is under 1 h.
- Centrifugera vid 2800 xg under 10 min för att pelletera celler vid 4 ° C.
- Avlägsna supernatanten och återsuspendera i 160 ml lösning A (tabell 1) och inkubera på is under 20 min.
- Centrifugera vid 2800 xg under 10 min för att pelletera celler vid 4 ° C.
- Avlägsna supernatanten och återsuspendera i 4 ml Lösning A + glycerol (tabell 1).
- Förbered 50 | il alikvoter, frysa i flytande N2, förvara vid -80 ° C.
- Blanda 5 | j, l av ligeringsblandning med 50 pl av kompetenta celler och inkubera under 1 timme på is.
- Värmechock cellerna vid 42 ° C under 90 sek, followed av inkubering på is under 2 min.
- Lägg 950 | il LB-medium (tabell 1) och inkubera vid 37 ° C under 1 timme.
- Alikvot 50 och 200 | j, l på plattor med lämpligt antibiotikum, antingen ampicillin (100 | ig / ml) och / eller kanamycin (30 | ig / ml).
VARNING: Både kanamycin och ampicillin är giftiga och bör hanteras med lämpligt skydd. - Plocka och inkubera enskilda kolonier i 2 ml LB-medium som inokulerats med det lämpliga antibiotikumet.
- Rena alla plasmider med användning av en miniprep-plasmid-reningskit enligt tillverkarens instruktioner.
- Generera plasmider, i detta specifika exempel för att slå ut de cpcC1C2 generna, enligt följande steg.
- Amplifiera 1012 bp 5 'flankerande regionen (vänster-fragment) med användning av primrar cpcC1C2leftfor och cpcC1C2leftrev (Se steg 3,2, tabell 2). Avlägsna en liten mängd av PCR-reaktionen och bekräfta huruvida denrätt storlek produkt har förstärkt via gelelektrofores (steg 3,3). Smälta detta fragment och pUC19 med Xbal och BamHI (steg 3,5).
- Rena båda preparaten (steg 3,4), ligera (steg 3,6), transform (steg 3,7) och inrätta fyra 2 ml flytande LB-kulturer med ampicillin (100 mikrogram / ml) från separata kolonier för plasmidrening via minipreps (steg 3,8).
- Kontrollera för insättning av fragmentet in i pUC19 via Xbal / BamHI-digerering och gelelektrofores (steg 3,3). Band av 2660 bp och 1012 bp indikerar korrekt införande av insatsen i plasmiden.
- Amplifiera 1016 bp 3 'flankerande regionen (höger-fragment) med användning av primrar cpcC1C2rightfor och cpcC1C2rightrev (Se steg 3,2, tabell 2). Avlägsna en liten mängd av PCR-reaktionen och bekräfta om korrekt storlek produkten har amplifierats via gelelektrofores (steg 3,3). Smälta detta fragment och pUC19 med SacI och EcoRI (step 3,5).
- Rena båda preparaten (steg 3,4), ligera (steg 3,6), transform (steg 3,7) och inrätta fyra 2 ml flytande LB-kulturer med ampicillin (100 mikrogram / ml) från separata kolonier för plasmidrening via minipreps (steg 3,8).
- Kontrollera för insättning av fragmentet in i pUC19 via SacI / EcoRI-digestion (steg 3.5) och gelelektrofores (steg 3,3). Band av 2660 bp och 1016 bp indikerar korrekt införande av insatsen i plasmiden.
Notera: Xbal / BamHI-ställena för kloning av 5'-regionen och SacI / EcoRI för kloning av 3'-regionen i pUC19 används där så är möjligt. Om möjligt, alltid inkludera ett BamHI-ställe på den omvända primern för 5'-regionen eller den framåtriktade primern för 3'-regionen för att säkerställa att senare kloningssteg är enklare att utföra. - Sekvens båda skären för att bestämma om sekvensen är korrekt med användning av primers som spänner över insättningsstället, t.ex. M13 framåt och M13 omvänd (tabell 2). Sekvensen måste vara korrekt för att garantera att inga fel införs i flankerande regioner.
- Skära den vänstra fragmentet från pUC19 via Xbal / BamHI digestion. Smälta pUC19 + höger fragment med Xbal / BamHI (steg 3,5).
- Rena den 1012 bp vänstra fragmentet och 3676 bp pUC19 + högra fragmentet från en agarosgel (steg 3,3) via excision av DNA med användning av ett skalpellblad.
- Rena båda preparaten (steg 3,4), ligera (steg 3,6), transform (steg 3,7) och inrätta fyra 2 ml flytande LB-kulturer med ampicillin (100 mikrogram / ml) från separata kolonier för plasmidrening via minipreps (steg 3,8).
- Kontrollera för insättning av fragmentet in i pUC19 + högra fragmentet via Xbal / BamHI-digerering (steg 3,5) och gelelektrofores (steg 3,3). Band av 3676 bp och 1012 bp indikerar korrekt insättning av insatsen i plasmiden (se detta som plasmid B).
Obs: NPT1 / sacB kassett behöver inte renas från agarosgeler sedan pUM24cm kodar för ett protein som ger kloramfenikolresistens. Därför om kolonier odlas på LB / ampicillin / kanamycin agarplattor den enda möjliga kombination som kommer att leda till resistenta kolonier är införlivandet av NPT1 / sacB kassett i plasmid B. - Rena båda preparaten (steg 3,4), ligera (steg 3,6), transform (steg 3,7) och inrätta fyra 2 ml flytande LB-kulturer med ampicillin (100 mikrogram / ml) och kanamycin (30 pg / ml) från separata kolonier för plasmidrening via minipreparat (steg 3,8).
- Kontrollera för insättning av NPT1 / sacB kassetten i plasmid B via BamHI-digerering (steg 3,5) och gelelektrofores (steg 3,3). Band av 4688 bp och 3894 bp indikerar korrekt insättning av the infoga i plasmiden (se detta som plasmid A).
- Alternativt trubbig ände den NPT1 / sacB kassetten och klon till en annan restriktionsendonukleasställe mellan de vänstra och högra fragment i plasmid B. NPT1 / sacB kassett måste klonas mellan de vänstra och högra fragment.
Obs: Om expression av en främmande kassett krävs då detta bör införas mellan vänster och höger fragment av plasmid B. Denna plasmid används sedan i omärkta knockout steg.
4. Framställning av Markerade Synechocystis och Synechococcus mutanter
- Inrätta en ny kultur genom att ympa en slinga full av celler i 30-50 ml BG11 medium. Odla kulturen i 2-3 dagar till OD 750 nm = 0,2 till 0,6.
Notera: Typiskt individuella kolonier är för små för att använda för inokulering och exponering av enskilda celler för att även låga nivåer av ljus kommer att resultera i photoinhibition och urvalför lätta resistenta mutanter. - Centrifug 1-2 ml av kulturen vid 2300 xg under 5 min och kasta bort supernatanten. Centrifugera inga blågrön kulturer vid> 2300 xg eftersom det kan skada cellerna. Tvätta pelleten en gång med BG11 medium.
Obs: Inte suspendera cellerna genom att vortexa eftersom det kan leda till förlust av pili som är nödvändiga för DNA-upptag. Resuspendera cellerna genom försiktig pipettering. - Lägg BG11 medium till en slutlig volym av 100 | il. Överför celler till en 14 ml rundbottnad rör.
- Lägg 1 pg av plasmiden A till cellerna och blanda genom försiktig knackning. Lägg <10 pl plasmid.
Notera: Företrädesvis bör plasmiden vara vid en koncentration av> 100 ng / ul men koncentrationer lägre än detta är tillräckliga för framgångsrik transformation. - Lägg rören ner horisontellt i inkubatorn. Inkubera kulturer i 6/4 h.
Obs: Celler kan kort blandas genom att knacka varje 1-2 h, men detta är inte väsentligt. Prover kan placeras i enskakinkubator även om detta inte avsevärt förbättra effektiviteten. - Sprid alikvoter av cellodlings / plasmid DNA-blandningen på BG11 agarplattor utan antibiotika. Typiskt 20 pl och 80 pl alikvoter är spridda på separata plattor.
- ~ 24 h senare, tillsätt 2,5-3 ml 0,6% agar-lösning i vatten innehållande kanamycin (per 20 ml: 0,12 g agar, 100 | il av 100 mg / ml kanamycin) till agarplattan. Kyla denna lösning till ~ 42 ° C, och tillsätt till kanten av agarplattan. Luta plattan så att lösningen bildar en jämn "toppagar 'skikt på ytan.
- Inkubera agarplattor för en ytterligare tid. Kolonier bör vara synlig efter cirka 7 dagar.
Obs: Agar plattor kan staplas 3 hög i en inkubator. Typiskt hundratals kolonier erhålls per transformation. - Streak individuella kolonier på BG11 + kanamycin (30 | ig / ml) agarplattor. Dela upp agarplatta i 6 sektorer och använda en trubbig ände tandpetare till strimma utkolonierna över varje enskild sektor. Erhålla enstaka kolonier är inte viktigt, bara tillväxten av transformanterna.
- Bekräfta markant knockout genom PCR med användning av Taq DNA-polymeras i enlighet med tillverkarens instruktioner. Tillsätt 2 pl MgCl2 (25 mM) per reaktion.
- Avlägsna en liten andel av cellerna och överför till ett rör innehållande 50 | j, l vatten och ~ 20 425-600 ^ m glaspärlor. Skaka i en vibrator för 5 min vid ~ 2000 rpm. Centrifugera vid 15.700 xg under 5 min och använder 5 | il av supernatant per 50 | al PCR-reaktion.
Obs: inte suspendera lösningen. Celldebris behöver stanna på botten av röret.
- Avlägsna en liten andel av cellerna och överför till ett rör innehållande 50 | j, l vatten och ~ 20 425-600 ^ m glaspärlor. Skaka i en vibrator för 5 min vid ~ 2000 rpm. Centrifugera vid 15.700 xg under 5 min och använder 5 | il av supernatant per 50 | al PCR-reaktion.
- validera mutanter
- Design primers som spänner över knockout regionen med hjälp av primer design mjukvara (t.ex. Primer3). Design primers som börjar på ~ 200 bp vardera sidan av knockout regionen.
Obs: Primers för kontroll av cpcC1C2 mutanten beskrivs i tabell 2och benämns cpcC1C2for och cpcC1C2rev. - Amplifiera produkter med hjälp av ett program som består av ett initialt denatureringssteg av 95 ° C under 2 min, 35 omgångar 95 ° C under 1 min, 60 ° C under 1 min, 72 ° C under 1 min per kb av sekvens, följt av en slutligt förlängningssteg av 72 ° C under 5 min. Inkluderar en styr vildtyp. Detta ger vanligtvis konsekvent produkter.
- Kontrollera genotypen via gelelektrofores. Markerade knockout transformanter kommer att visa ett band av ~ 4 kb (0,2 kb från både vänster och höger fragment plus NPT1 / sacB kassett) och frånvaron av vildtyp band (Figur 2).
Obs: I vissa fall en ~ 4 kb band inte observeras i den markerade mutanten på grund av den stora storleken på denna PCR-produkt. Men om ett band motsvarande den förväntade storleken av vildtyp inte observeras sedan typiskt denna stam är en markant knockout.
- Design primers som spänner över knockout regionen med hjälp av primer design mjukvara (t.ex. Primer3). Design primers som börjar på ~ 200 bp vardera sidan av knockout regionen.
- Om en vild-typ-bandet är fortfarande närvarande sedan åter strimma påfrestningen på enfärsk BG11 + kanamycin (30 | ig / ml) agarplatta och upprepa PCR. Upprepa åter strimmor processen tills mutanten är segregerad så att ingen vildtyp band observeras i PCR-reaktionen.
Obs: Att öka mängden kanamycin till en koncentration av 50 pg / ml, därefter 100 mikrogram / ml är ibland nödvändigt för att avskilja en markant mutant helt. - Om stammen visar en markant mutant profil via PCR, sedan åter strimma på en färsk BG11 + kanamycin (30 | ig / ml) agarplatta. Använd denna stam för att generera den omärkta knockout.
Notera: Protokollet kan användas för att generera märkta mutanter med bara en antibiotikaresistens kassett dvs genom att ersätta NPT1 / sacB kassett med precis NPT1 kassetten från pUC18K 20 mellan de vänstra och högra fragment..
5. Generation omärkta Synechocystis mutanter
- Inrätta en ny kultur av den markerade knockout genom ympning en slinga full av calnar till 30-50 ml av BG11 medium. Odla kulturen i 2-3 dagar till OD 750 nm = 0,2 till 0,6.
- Centrifugera 10 ml av kulturen på 2300 xg under 5 min och kassera supernatanten. Tvätta en gång med BG11 medium.
Obs: Inte suspendera cellerna genom att vortexa eftersom det kan leda till förlust av pili som är nödvändiga för DNA-upptag. Resuspendera cellerna genom försiktig pipettering. - Lägg BG11 till en slutlig volym av 200 | il. Överför celler till en 14 ml rundbottnad rör.
- Lägg 1 ^ g av plasmid B-DNA till cellerna och blanda genom försiktig knackning.
- Inkubera proverna för 4-6 timmar. Lägg rören ner horisontellt.
Obs: Celler kan kort blandas genom att knacka varje 1-2 h, men detta är inte väsentligt. Prover kan placeras i ett skakande inkubator även om detta inte förbättra effektiviteten. - Lägga 1,8 ml av BG11-medium och inkubera proverna under totalt 4 dagar med skakning. Detta är tillräckligt med tid för att tillåta rekombination sker i flera kromosomala kopior.
- Platt alikvoter av transformationsblandningen på BG11 / 5% sackaros agarplattor. Plattan 50 pl, 10 pl och en il per agarplatta. Om en koloni gräsmatta visas på alla dessa agarplattor späda lösningen vidare och delprov på färska plattor. Kolonier bör vara synlig efter cirka 7 dagar.
- Patch 30-50 individuella kolonier på BG11 + kanamycin (30 | ig / ml) agarplattor första och BG11 / 5% sackaros agarplattor andra, med hjälp av en trubbig ände tandpetare. Några bakterier som växer på BG11 / 5% sackaros plattor men inte BG11 + kanamycin plattor är potentiella omärkta knockouts. Bakterier som växer på båda plattorna är sannolikt att vara sackaros resistenta på grund av en mutation i sacB-genen.
- Kontrollera omärkta knockouts med användning av samma primrar och metod som användes för att kontrollera de markerade knockouts. T.ex. cpcC1C2for och cpcC1C2rev (tabell 2) för att verifiera cpcC1C2 omärkta knockout. En omärkt knockout kommer att visa ett band på en agarosgel som motsvarar to vildtyp storlek minus den borttagna regionen (Figur 2).
- Om stammen visar en omärkt mutant profil via PCR (steg 4.11.2) och gelelektrofores (figur 2), sedan åter strimma på en ny BG11 agarplatta utan antibiotika.
6. Långsiktig Lagring av stammar
- Inrätta en ny kultur av stammen genom ympning en slinga full av celler i 30-50 ml BG11 medium. Odla kulturen i 3-4 dagar till OD 750 nm = 0,4-0,7.
- Tvätta cellerna en gång med BG11 och återsuspendera i ~ 2 ml BG11.
- Lägg 0,8 ml koncentrerad celler till ett rör. Tillsätt sedan 0,2 ml av 80% filtersteriliserad glycerol.
- Valfritt: Lägg 0,93 ml koncentrerade celler till ett annat rör. Lägg 0,07 ml DMSO till detta rör.
VARNING: DMSO är giftig och bör hanteras med lämpligt skydd. - Lagra både rören vid -80 ° C. Att återuppliva stammar bort röret och skrapa bort några celler med en trubbig tandplocka på en agarplatta utan antibiotika. Strimma ut som vanligt med hjälp av en steril ögla.

Figur 1: Plasmid konstruktion för generering av märkta och omärkta knockouts, t.ex. cpcC1 och cpcC2 i Synechocystis (A) region Synechocystis-genomet där (B) cpcC1 och cpcC2 och angränsande gener är belägna.. Markerad i svart är den region av genomet som ska tas bort i mutanten. (C) områden av genomet som amplifieras genom PCR. Den 5 'flankerande regionen (indikeras i blått) och 3' flankerande regionen (markerade med rött) amplifieras med restriktionsendonukleasställen för kloning in i pUC19. Den 5 '(eller 3') flankerande regionen skärs ut ur pUC19 och sattes in i pUC19 + 3 '(eller 59;) flankerande regionen plasmid för att generera plasmid B. (D) Den NPT1 / sacB kassett från pUM24 skärs via BamHI digestion och in mellan 5 'och 3' flankerande regioner för att generera plasmid A. Klicka här för att se en större version av denna siffra.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Plasmid design är avgörande för framgångsrik alstring av både märkta och omärkta mutanter. Figur 1 visar ett exempel på plasmid A och B används för att generera en deletionsmutant i Synechocystis generna cpcC1 och cpcC2 13. I varje enskilt fall de 5 'och 3' flankerande regioner är ungefär 900-1,000 bp. Minskad flankerande regionerna kan användas även det minsta vi har framgångsrikt provat har varit cirka 500 bp. Plasmid B kan även innehålla ett genkassetten mellan de 5 'och 3' ~ 1 kb flankerande regioner eller en modifierad version av den nativa gensekvensen.
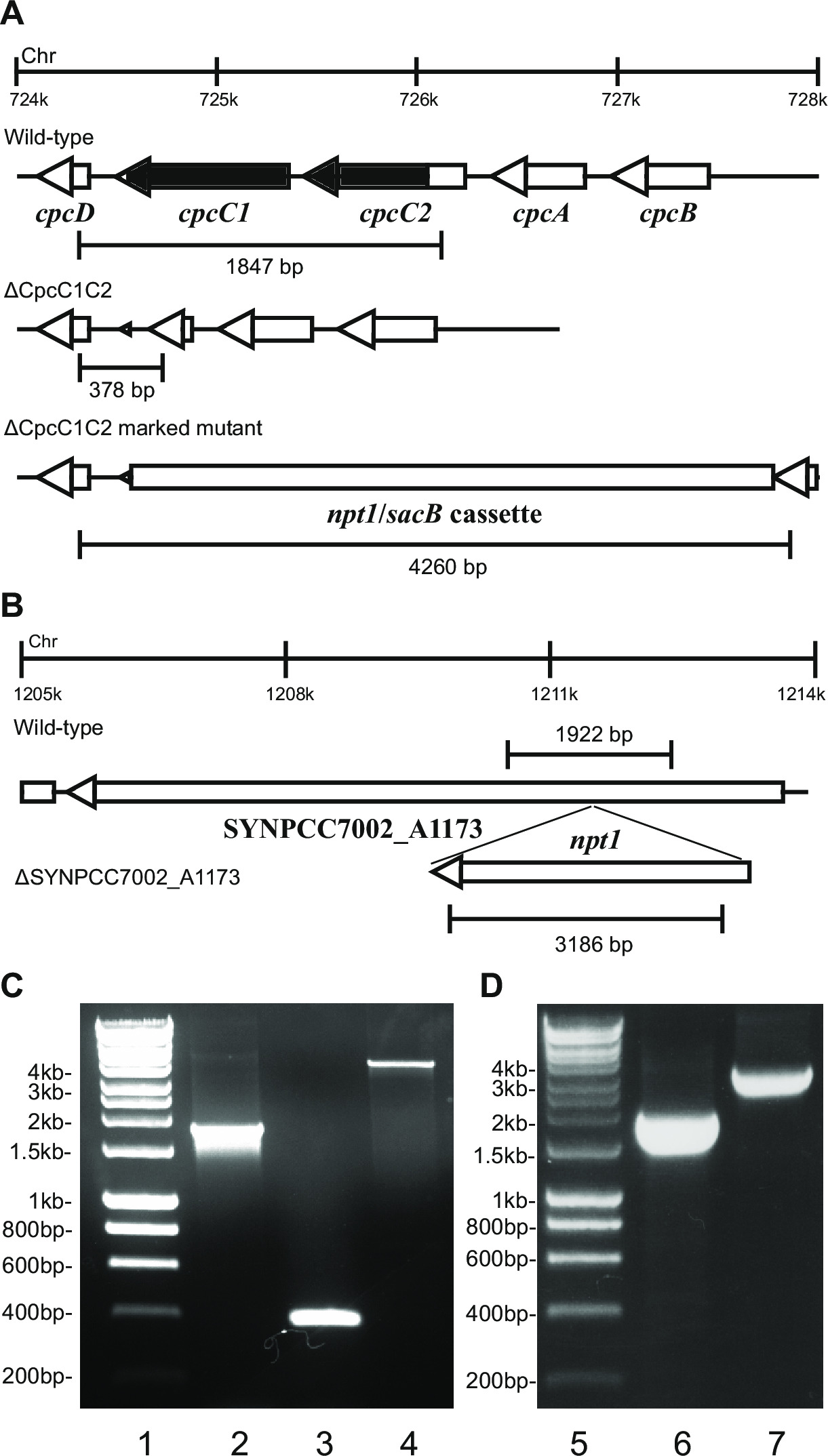
Figur 2: Kontroll av märkta och omärkta mutanter, t.ex. cpcC1 / cpcC2
Efter transformation av plasmid A in i cellerna, kommer typiskt flera hundra kolonier visas på en platta efter cirka 7-10 dagar. Kolonier är <1 mm i diameter och kommer inte att öka i storlek för de närmaste veckorna. Därför är det viktigt att använda en trubbig ände tandpetare för att avlägsna kolonin och strimma det på en ny BG11 + kanamycin agarplatta. Ungefär hälften av åter strimmiga kolonier kommer att växa efter 4-6 dagar. Om gener är icke-essentiell och mutanter visar tillväxt liknar den vilda stammen under kontinuerlig ljus av 20-40 fimol fotoner m -2 sek -1 (t.ex. terminaloxidas mutanter i Lea-Smith et al., 2013 14) (Figur 3), sedan alla kromosomer bör innehålla en kopia av t han NPT1 / sacB kassett sekvens, som bestäms via PCR. Om gener är icke-essentiell och mutanter visar en långsam tillväxt fenotyp under kontinuerlig ljus av 20-40 fimol fotoner m -2 s -1 (t.ex. phycobilisome mutanter i Lea-Smith et al., 2014 13) (Figur 3), sedan flera omgångar åter strimmor på BG11 agarplattor med gradvis ökade mängder av kanamycin är nödvändiga för att erhålla ett segregerat markerade mutant. När en segregerad mutant erhålles detta bör åter ströks ut på en ny BG11 plus kanamycin agarplatta för att säkerställa att segregationen är klar. Om upprepade rundor av strimmor inte resulterar i ett segregerat markerade mutant då genen är sannolikt nödvändigt för överlevnad. Figur 4 ger en översikt av de experimentella stegen i omärkt mutant generation.
/54001/54001fig3highres.jpg "Width =" 700 "/>
Figur 3:. Tillväxt av Synechocystis mutanter Exempel på mutanter som visar (A) liknande tillväxt vildtyp och (B) en långsammare tillväxt än vildtypen. Den ΔCOX mutanten saknar cytokromoxidas grund av radering av CtaC1D1E1 gener. Den ΔCyd mutanten saknar kinol oxidas på grund av radering av CydAB gener. Oliv mutanten saknar ett parti av den phycobilisome på grund av deletion av de CpcABC1C2D gener. Prover i (B) bubblades med luft för att underlätta tillväxten. Reproduceras från uppgifter som publicerats i Lea-Smith et al, 2013 14 och 2014 13 (www.plantphysiol.org, Copyright American Society of Plant Biologer).. Klicka här för att se en större version av denna siffra.
t ex Figur 2. Om en gen kassett införes i kromosomen sedan typiskt en högre andel av kanamycinresistenta och sackaros-resistenta kolonier observeras. Dessa mutanter kan växa på sackaros på grund av en mutation i sacB-genen. Om inga kanamycin känsliga och sackaros-resistenta kolonier genereras därefter genkassetten är skadlig för cellen.

Figur 4: Framställning av märkta och omärkta mutanter i Synechocystis Schematisk detaljer (A) rekombination och (B) experimentella stegen i mutant generation.. Plasmid A först blandas med celler. Efter inkubation på agarplattor innehållande kanamycin, kolonier i vilka en rekombinationshändelse inträffar mellan 5'end 3 'flankerande regioner (indikeras i blått och rött, respektive) och den homologa sekvensen i kromosomen, är isolerade. Dessutom är den NPT1 / sacB kassett mellan de 5 'och 3' flankerande regioner införas i kromosomen. Efter segregering en markant mutant genereras. Märkta mutanta celler blandas sedan med plasmid B, som kan innehålla antingen (C) 1: de 5 'och 3' flankerande regioner; 2: 5 'och 3' flankerande regioner med en expressionskassett innehållande gener av intresse införas mellan dessa sekvenser; 3: 5 'och 3' flankerande regioner med vildtypssekvensen med de önskade nukleotidförändringar införda mellan dessa sekvenser. En andra homolog rekombinationshändelse sker mellan 5 'och 3' flankerande regioner och de homologa regionerna i kromosomen, vilket resulterar i avlägsnande av NPT1 / sacB kassetten och antingen den omärkta knockout eller en mutant med en inkopplings eller förändrad vild-typE-regionen infördes i kromosomen. Klicka här för att se en större version av denna siffra.
| Stamlösning recept | |
| Kemisk | Mängd (g) |
| 100x BG11 (per liter) | |
| NaNO 3 | 149,6 |
| MgSO 4 .7H 2 O | 7,49 |
| CaCl2 .2H 2 O | 3,6 |
| Citronsyra | 0,6 |
| Lägga 1,12 ml 0,25 M Na2EDTA, pH 8,0 | |
| 0,25 M Na2EDTA, pH 8,0 (per 100 ml) | |
| na 2 | 9,3 |
| Spårelement (per 100 ml) | |
| H 3 BO 3 | 0,286 |
| MnCl2 .4H 2 O | 0,181 |
| ZnSO 4 .7H 2 O | 0,022 |
| Na 2 MoO 4 .2H 2 O | 0,039 |
| CUSO4 5H 2 O | 0,008 |
| Co (NO3) 2 .6H 2 O | 0,005 |
| Järn lager (per 100 ml) | |
| Jämammoniumcitrat | 1,11 |
| Fosfatstam (per 100 ml) | |
| K 2 HPO 4 | 3,05 |
| Na 2 CO 3 lager (per 100 ml) | |
| Na 2 CO 3 | 2 |
| TES-buffert, pH 8,2 (per 100 ml) | |
| TES | 22,9 |
| NaHCOs 3 lager (per 100 ml) | |
| NaHCOs 3 | 8,4 |
| HEPES, pH 8,2 (per 500 ml) | |
| HEPES | 119,15 |
| Vitamin B12 (Per 50 ml) | |
| cyanokobalamin | 0,02 |
| Luria Bertani medium (Per 500 ml) | |
| Luria Bertani-buljong | 12,5 |
| 1 M MgCl2 (Per 100 ml) | |
| MgCl2 .6H 2 O | 20,33 |
| MnCl2 .4H 2 O | 0,395 |
| CaCl2 .2H 2 O | 1,47 |
| 2- (N morfolino) etansulfonsyra hydrat, 4-morfolinetansulfonsyra syra (MES) | 0,4265 |
| Lösning A + glycerol | |
| 10 ml lösning A | |
| 1,5 ml glycerol |
Tabell 1: Lösningar som används i denna studie.
| Primer | Sekvens |
| cpcC1C2leftfor | GTAC TCTAGA GCGGCTAAATGCTACGAC |
| CPCC1C2leftrev | GATC GGATCC GCGGTAATTGTTCCCTTTGA |
| cpcC1C2rightfor | GATC GAGCTC TGCACTGGTCAGTCGTTC |
| cpcC1C2rightrev | GACT GAATTC ATCGTTGCTTGAACGGTCTC |
| M13 framåt | TGTAAAACGACGGCCAGT |
| M13 omvänd | CAGGAAACAGCTATGAC |
| cpcC1C2for | GTTTTCATTGGCATCGGTCT |
| cpcC1C2rev | ATGTCCCAGGAACGACTGAC |
| A1173for | AGCAAACCGTTTTTGTGACC |
| A1173rev | TGCAAGGTGGCGAACTGTAT |
Tabell 2:. Primrar som användes i denna studie restriktionsendonukleasställen är understrukna.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
De mest kritiska stegen i generering av omärkta mutanter är: 1) noggrann plasmid design för att säkerställa att endast målregionen förändras; 2) se till att proverna förblir axenisk, särskilt när de odlas på sackaros; 3) plätering transformerade celler för märkt mutant generationen initialt på BG11 agarplattor som saknar antibiotika, följt av tillsats av agar plus antibiotika 24 h senare; 4) odling märkta mutanter för 4 hela dagar före plätering på BG11 plus sackaros agarplattor: 5) se till att märkta mutanter är helt åtskilda och 6) noggrant bekräftar genotyp mutantstammar. För detta sista steg, ytterligare primers utformade för att amplifiera en del av den deleterade regionen, kan användas för att säkerställa att den har tagits bort. Southern blotting, medan mödosam, kan också användas. Men är vår erfarenhet att det förfarande som beskrivs i detta dokument är tillräcklig för korrekt kontroll av mutanter. Detta förfarande har också använts för att generera märkta mutanter i Synechococcus elongatus PCC7942. Emellertid har upprepade omvandling av denna cyanobakterien visat utmanande.
Om märkta mutanter inte kan separeras sedan olika miljöförhållanden hög CO2, svagt ljus (<20 fimol fotoner m -2 s -1) eller ytterligare näringsämnen (dvs glukos) kan testas. Till exempel, är avgörande för att skapa fotosystem II mutanter 21 tillsats av glukos. Om markerade mutanter aldrig segregera helt då genen är förmodligen nödvändig för viabilitet. Men det finns exempel från litteraturen där vissa forskargrupper har kunnat knockout en gen (till exempel Vipp i Synechocystis) 22, endast för andra grupper för att sedan visa att genen är inte nödvändigt 23. Detta kan bero på skillnader i vildtyp stammar eller felaktig plasmid konstruktion, vilket resulterar i polära effekter på angränsande, essentiella gener. Om en mutant inte heltsegregera vi rekommenderar att plasmiden innehållande NPT1 kassetten från pUC18K 20 mellan den vänstra och högra fragment användas för transformation. Det är lättare att verifiera närvaron av band motsvarande vildtypen och mutant genom PCR, eftersom detta fragment är ca 1,2 kb, jämfört med de 3,8 kb NPT1 / sacB kassett. Detta resultat är en viktig del av bevis som visar att genen är väsentlig.
Generation omärkta mutanter med insatta uttryckskassetter är i allmänhet mer utmanande än utvecklingen av knockout stammar. Vi uttrycker i allmänhet gener under kontroll av den starka cpcBAC1C2D promotorn 13. I vissa fall kan detta minska riskerna för framgångsrik insättning av genkassetten, om överuttryck av ett protein är skadlig för cellen. Svagare promotorer bör sedan testas. I allmänhet har vi observerat att ju större genkassetten är, är det ännu svårare att iSERT det i genomet. Vi har inte kunnat att infoga genkassetterna större än 5 kb. Försiktighet måste även tas vid val av platser för att infoga expressionskassetter i genomet. Neutrala platser som inte påverkar cellviabiliteten eller tillväxten ska användas. Exempel i Synechocystis inkluderar phaAB och Phace, som kodar för proteiner som kodar för polyhydroxibutyrat biosyntesvägen 24,25. Mer nyligen en omfattande lista av neutrala platser i Synechocystis har identifierats 26.
Generation av omärkta mutanter i cyanobakterier är en långsam process, tar ca 5-7 veckor om alla steg genomför ordentligt. Detta är långsammare än standardmetoden för att generera markerade knockouts som används av de flesta forskargrupper undersöker cyanobakterier. Men flexibiliteten att kunna delvis införa ytterligare mutationer i omärkta mutanter kompenserar för detta, eftersom ytterligare plasmider fortsaining en rad kassetter som ger resistens mot olika antibiotika, inte behöver konstrueras. För forskningsändamål är ibland nödvändigt möjligheten att mutera flera gener för att fullt ut karakterisera den funktionella rollen av proteiner. Till exempel har vi identifierat en skadlig fenotyp endast vid radering av de två terminala oxidaselektron sänkor lokaliserade till tylakoid membranet, eftersom förlust av endast en av dessa komplex skulle kunna kompenseras genom aktivitet i den andra 14. Utveckling av en stam för industriella tillämpningar kommer också att kräva flera ändringar till en stam, inte bara för införande av främmande gener utan också för att öka fotosyntetiska effektivitet, ljus skörd optimering och radering av konkurrerande vägar för det önskade substratet.
Den viktigaste faktorn som begränsar hastigheten på omärkta mutant generation är den långsamma division tiden modell blågrön arter, mellan 8-20 timmar beroende på ljusförhållanden. Fnder högre ljusintensiteter och CO 2 koncentrationer är tillväxten snabbare. Det finns dock en risk för att mutanta stammar som inte kan tolerera antingen hög ljus eller CO 2 kommer att väljas mot, eller att muterade stammar kommer att genomgå oönskade förändringar före fenotypisk karakterisering. Därför är detta inte rekommenderas. Emellertid skulle det vara mycket fördelaktigt om en snabbare protokoll för att generera omärkta mutanter utvecklades. Sammantaget skulle detta underlätta utvecklingen av stammar för både grundforskning och tillämpade applikationer. Sådana stammar kan användas för biobränslen, biomassa eller kemisk produktion eller att förstå många aspekter av cyanobakterier biokemi, genetik och fysiologi.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Vi är tacksamma för Environmental Services Association Education Trust, Syntetisk biologi i Cambridge SynBio fond och ministeriet för social rättvisa och Empowerment, Indiens regering, för ekonomiskt stöd.
Materials
| Name | Company | Catalog Number | Comments |
| NaNO3 | Sigma | S5506 | |
| MgSO4.7H2O | Sigma | 230391 | |
| CaCl2 | Sigma | C1016 | |
| citric acid | Sigma | C0759 | |
| Na2EDTA | Fisher | EDT002 | |
| H3BO3 | Sigma | 339067 | |
| MnCl2.4H2O | Sigma | M3634 | |
| ZnSO4.7H2O | Sigma | Z4750 | |
| Na2MoO4.2H2O | Sigma | 331058 | |
| CuSO4.5H2O | Sigma | 209198 | |
| Co(NO3)2.6H2O | Sigma | 239267 | |
| Ferric ammonium citrate | Sigma | F5879 | |
| K2HPO4 | Sigma | P3786 | |
| Na2CO3 | Fisher | SODC001 | |
| TES | Sigma | T1375 | |
| NaHCO3 | Fisher | SODH001 | |
| HEPES | Sigma | H3375 | |
| cyanocobalamin | Sigma | 47869 | |
| Na2S2O3 | Sigma | 72049 | |
| Bacto agar | BD | 214010 | |
| Sucrose | Fisher | SUC001 | |
| Petri dish 90 mm triple vented | Greiner | 633185 | |
| 0.2 µm filters | Sartorius | 16534 | |
| 100 ml conical flasks | Pyrex | CON004 | |
| Parafilm M 100 mm x 38 m | Bemis | FIL003 | |
| Phusion high fidelity DNA polymerase | Phusion | F-530 | |
| Agarose | Melford | MB1200 | |
| DNA purification kit | MoBio | 12100-300 | |
| Restriction endonucleases | NEB | ||
| T4 ligase | Thermo Scientific | EL0011 | |
| Luria Bertani broth | Invitrogen | 12795-027 | |
| MES | Sigma | M8250 | |
| Kanamycin sulfate | Sigma | 60615 | |
| Ampicillin | Sigma | A9518 | |
| GeneJET plasmid miniprep kit | Thermo Scientific | K0503 | |
| 14 ml round-bottom tube | BD falcon | 352059 | |
| GoTaq G2 Flexi DNA polymerase | Promega | M7805 | |
| 425-600 µm glass beads | Sigma | G8772 | |
| Glycerol | Sigma | G5516 | |
| DMSO | Sigma | D8418 | |
| Fluorescent bulbs | Gro-Lux | 69 | |
| HT multitron photobioreactor | Infors |
References
- Zwirglmaier, K., et al. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol. 10, 147-161 (2008).
- Galloway, J. N., et al. Nitrogen cycles: past, present, and future. Biogeochemistry. 70, 153-226 (2004).
- Lea-Smith, D. J., et al. Contribution of cyanobacterial alkane production to the ocean hydrocarbon cycle. Proc Natl Acad Sci U S A. , (2015).
- Howe, C. J., Barbrook, A. C., Nisbet, R. E. R., Lockhart, P. J., Larkum, A. W. D. The origin of plastids. Philos Trans R Soc Lond B Biol Sci. 363, 2675-2685 (2008).
- Lea-Smith, D. J., Bombelli, P., Vasudevan, R., Howe, C. J. Photosynthetic, respiratory and extracellular electron transport pathways in cyanobacteria. Biochim Biophys Acta. , (2015).
- McCormick, A. J., et al. Hydrogen production through oxygenic photosynthesis using the cyanobacterium Synechocystis sp PCC 6803 in a bio-photoelectrolysis cell (BPE) system. Energy Environ. Sci. 6, 2682-2690 (2013).
- Bradley, R. W., Bombelli, P., Lea-Smith, D. J., Howe, C. J. Terminal oxidase mutants of the cyanobacterium Synechocystis sp. PCC 6803 show increased electrogenic activity in biological photo-voltaic systems. Phys Chem Chem Phys. 15, 13611-13618 (2013).
- Ducat, D. C., Way, J. C., Silver, P. A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 29, 95-103 (2011).
- Dismukes, G. C., Carrieri, D., Bennette, N., Ananyev, G. M., Posewitz, M. C. Aquatic phototrophs: efficient alternatives to land-based crops for biofuels. Curr Opin Biotechnol. 19, 235-240 (2008).
- Tan, L. T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 68, 954-979 (2007).
- Volk, R. B., Furkert, F. H. Antialgal, antibacterial and antifungal activity of two metabolites produced and excreted by cyanobacteria during growth. Microbiol Res. 161, 180-186 (2006).
- Scott, S. A., et al. Biodiesel from algae: challenges and prospects. Curr Opin Biotechnol. 21, 277-286 (2010).
- Lea-Smith, D. J., et al. Phycobilisome-deficient strains of Synechocystis sp. PCC 6803 have reduced size and require carbon-limiting conditions to exhibit enhanced productivity. Plant Physiol. 165, 705-714 (2014).
- Lea-Smith, D. J., et al. Thylakoid terminal oxidases are essential for the cyanobacterium Synechocystis sp. PCC 6803 to survive rapidly changing light intensities. Plant Physiol. 162, 484-495 (2013).
- Liu, X., Sheng, J., Curtiss, R. 3rd Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci U S A. 108, 6899-6904 (2011).
- Xu, H., Vavilin, D., Funk, C., Vermaas, W. Multiple deletions of small cab-like proteins in the cyanobacterium Synechocystis sp PCC 6803 - Consequences for pigment biosynthesis and accumulation. J Biol Chem. 279, 27971-27979 (2004).
- Castenholz, R. W. Culturing methods for Cyanobacteria. Method Enzymol. 167, 68-93 (1988).
- Mitschke, J., et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp PCC6803. Proc Natl Acad Sci U S A. 108, 2124-2129 (2011).
- Ried, J. L., Collmer, A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 57, 239-246 (1987).
- Vieira, J., Messing, J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 19, 259-268 (1982).
- Vermaas, W. F. J., Williams, J. G. K., Rutherford, A. W., Mathis, P., Arntzen, C. J. Genetically Engineered Mutant of the Cyanobacterium Synechocystis 6803 Lacks the Photosystem-Ii Chlorophyll-Binding Protein Cp-47. Proc Natl Acad Sci U S A. 83, 9474-9477 (1986).
- Westphal, S., Heins, L., Soll, J., Vothknecht, U. C. Vipp1 deletion mutant of Synechocystis: A connection between bacterial phage shock and thylakoid biogenesis? Proc Natl Acad Sci U S A. 98, 4243-4248 (2001).
- Zhang, S. Y., Shen, G. Z., Li, Z. K., Golbeck, J. H., Bryant, D. A. Vipp1 Is Essential for the Biogenesis of Photosystem I but Not Thylakoid Membranes in Synechococcus sp PCC 7002. J Biol Chem. 289, 15904-15914 (2014).
- Taroncher-Oldenberg, G., Nishina, K., Stephanopoulos, G. Identification and analysis of the polyhydroxyalkanoate-specific beta-ketothiolase and acetoacetyl coenzyme A reductase genes in the cyanobacterium Synechocystis sp strain PCC6803. Appl Environ Microbiol. 66, 4440-4448 (2000).
- Hein, S., Tran, H., Steinbuchel, A. Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch Microbiol. 170, 162-170 (1998).
- Ng, A. H., Berla, B. M., Pakrasi, H. B. Fine tuning of photoautotrophic protein production by combining promoters and neutral sites in Synechocystis 6803, a cyanobacterium. Appl Environ Microbiol. , (2015).
