

Summary
ここでは、単一分子感度と〜15ミリ秒の時間分解能で、偏TIRFMを用いたマイクロ流体チャネルにおけるリポソームおよびサポートされている二重層の間の単一、SNARE媒介融合事象を検出するためのプロトコルを提示します。脂質可溶性貨物放出を同時に検出することができます。リポソームサイズは、脂質拡散、および融合細孔特性が測定されます。
Abstract
膜融合のユビキタス過程で融合孔の開口部は2以前は別々の区画との間の第1の接続を確立します。エキソサイトーシスを介した神経伝達物質やホルモンのリリース時には、融合孔が一過開閉することができ、繰り返し、貨物の放出動態を制御します。細孔ダイナミクスも小胞のリサイクルのモードを決定します。過渡、「キス・アンド・ラン」の融合で不可逆的な再スケーリング結果、拡張が完全融合につながる一方。より良好な細孔力学を支配するものの要素を理解するために、我々は、単一分子感度と〜生化学的インビトロ系内で明確に定義された15ミリ秒の時間分解能で偏光全反射蛍光(TIRF)顕微鏡を用いて膜融合をモニターするためのアッセイを開発しました。 Fusionは、蛍光軟質ポリマークッション(T-SBL、トン・サポート二重層上に支持され、T-SNAREとベアリング平面二重層とのv-SNAREタンパク質(V-SUV車)を含む単層小胞を標識しました)、監視されます。アッセイは、SUV車の一定の密度を供給しながら、最小限のサンプル消費量を確保するためのマイクロ流体流路を使用しています。融合時のSBLへのSUVからの脂質のラベルの転送時に迅速なシグナル増強を利用して、脂質色素転写の動態を監視します。 TIRF顕微鏡法の感度は、脂質拡散とSUVのサイズはすべての融合イベントを推定することが可能な単一の蛍光脂質標識を追跡することができます。脂質色素放出時間は、恒久的に開気孔を通じて妨げられずに通過させるために予想よりもはるかに長くすることができます。脂質リリースの遅延がちらつき細孔によるものであると仮定したモデル、細孔「開放性」、時間の割合を使用して、細孔が融合中に開いたまま、推定することができます。可溶性マーカーは、脂質および可溶性貨物リリースの同時モニタリングのためのSUVの中にカプセル化することができます。このような測定は、いくつかの細孔が可溶性貨物の一部を失った後、再密閉することができる示します。
Introduction
膜融合は、細胞内の脂質とタンパク質の輸送、分泌、受精、発育、および宿主生物1-3へのウイルスの侵入をエンベロープするために必要な普遍的生物学的プロセスです。エキソサイトーシスを介したホルモンや神経伝達物質の放出を含むほとんどの細胞内の核融合反応では、2つの脂質二重層を融合させるエネルギーが内に固定、同族可溶性N-エチルマレイミド感受性因子付着タンパク質受容体 (SNARE)タンパク質間の4ヘリックスバンドルの形成によって提供されています小胞(V-SNARE)及び標的膜(T-SNARE)4、。シナプス小胞のエキソサイトーシスは、最も厳密に制御核融合反応であり、活動電位1,4,5の到着後ミリ秒以内に行われます。融合孔、2融合区画間の最初の接続は、オープンちらつきおよび再密封または5-7不可逆的に拡大する前に複数回を閉じすることができます。前者の結果過渡、「キス&ラン」の融合で、完全な融合に後者リードしながら。これら2つの融合のモードと細孔のちらつきを調整する機構との間のバランスを支配する要因はよく5,8を理解されていません。
SNAREタンパク質は、エキソサイトーシスのために必要とされます。シナプス小胞の融合は、神経毒9でのSNAREの切断の際に廃止されています。小さ な単層ベシクル(SUV車)を使用してバルク融合実験は、膜融合10を駆動するのに十分のSNAREのみが必要とされないことを示したが、。このバルクアッセイでは、V字のSNARE(V-SUV)で再構成のSUVは、蛍光リン脂質(Nでドープされた- (7-ニトロ2-1,3-benzoxadiazol -4-イル)-phosphoethanolamine(NBD-PE)及び( N - 。(リサミンローダミンBスルホニル)-phosphoethanolamine(LR-PE)およびt-SNAREと(T-SUV)を含む非標識小胞と混合当初のv-SUV車でNBD-PEの蛍光は蛍光共鳴エネルギー移動により消光される(FRET )LR-PEへ。研究室として、ELED V-SUVが非標識T-SUV車と融合、今組み合わせた膜中の蛍光体表面密度が減少し、NBD-PEの蛍光の結果の増加は、脂質が10を混合の程度を報告します。バルクアッセイがセットアップし、分析することが容易であるように、広くSNARE媒介融合10-14のメカニズムを研究するために使用されています。しかし、そのような低感度と悪い時間分解能など、いくつかの制限を有します。ドッキングおよび融合との識別だけでなく、困難なhemifusion中間体の検出を行うすべてのイベントで最も重要なのは、アンサンブル測定など、その平均値をもたらします。
私たちを含め、過去十年間、いくつかのグループ、の上、単一小胞レベル15-27で融合イベントを監視するための新しいアッセイを開発しました。ハらは、表面に係留さV-SUV車を使用し、無料のT-SUV車18,19との融合を監視しました。脂質混合は脂質結合蛍光団のemのペア間のFRETを用いてモニターしました全内部反射蛍光(TIRF)顕微鏡18を用いて、それぞれ、VおよびTのSUVにベッド。その後、Brungerの研究室では、脂質と内容が20,28の混合の同時検出のためのコンテンツマーカーと一緒に単一の脂質ラベル種を使用しました。脂質およびコンテンツマーカーの両方が高い、自己消光濃度で含まれていました。非標識のSUVとの融合は、20,28の脱消光蛍光をもたらしました。
その他がt-SNAREと15-17,21-27,29で再構成平面二重層にV-のSUVを融合させています。ターゲットの平面形状(T-SNARE含む)二重層より良い模倣フラット形質膜と小、大きく湾曲した小胞の生理的な融合プロセスを。 T-のSNAREで再構成細孔貫通膜を用いスタイネム基は、多孔質窒化ケイ素基板上に懸濁させ、共焦点レーザー走査顕微鏡23を使用して個々 の V-SUV車との融合を検出しました。その他FT-SNAREとで再構成平面二重層するために使用されるV-SUVが、ガラス基板15-17,21,22,24-27,29でサポートされています。サポートされている二重層(SBLs)を使用することの大きな利点は、マイクロフルイディクスを使用しても使用してシングルイベント分解能を提供するが、TIRF顕微鏡法は、優れた信号対雑音比と、無料V-SUV車からの干渉なしにドッキングおよび融合事象を検出するために使用することができるということです標準の遠視野落射蛍光顕微鏡24。
主な関心事は、どのように基板-二重層相互作用がサポートされている二重層の品質と融合プロセスに影響を与えるかどうかです。初期の研究は、直接ガラスや石英基板15-17でサポートされていました平面SBLsを利用しました。これらSBLsは、基板上に拡散し、T-SUV膜の融合、破裂、吸着することによって作製しました。これは、すぐにキートン-SNAREコンポーネントを省略し、SNAP25は、このようにして調製SBLsからのv-SUVのドッキングおよび融合反応速度indistinguisをもたらしたこと、しかし、実現しました完全なT-SNAREと17を使用して得られたものからhable。 SNAP25は絶対に生体内 30,31 に融合するために必要とされるので、これらの初期の試みの生理学的関連性は疑問に入れました。タムのグループは、より良好に制御サポート二重層形成21を使用することで、この課題を克服しました。これは、T-SUV車21と、その単分子膜の融合が続くSBLのタンパク質を含まない最初のリーフレット、ためにラングミュア-ブロジェット堆積を使用していました。これは、SNAP25依存融合をもたらしました。
直接ラングミュア-ブロジェット法を使用することなく、ガラス基板上に支持二重層に関連する潜在的なアーチファクトを避けるために、Karatekin らは 、二重層と基板24との間に柔らかい、水和したポリ(エチレングリコール)(PEG)クッションを導入しました。この変形例はまた、SNAP25依存融合24を生じました。軟質ポリマー層上にクッション二層は、より良好な貫通を維持することが知られていましたタンパク質の移動度と機能32、およびウイルス33との融合の研究で使用されていました。また、PEG化された二重層は、自己治癒すると34,35非常に堅牢であるにいくつかの能力を保持するように見えます。まず、市販の、脂質結合PEG鎖の画分は、T-SUV膜に含まれています。これらのT-SUVのバーストと、ガラス基板上に平面二重層を形成する場合、PEGブラシは、平面二重層の両方の弁尖を覆います。平面二重層の形成は親水性のガラス表面上のT-SUV車の周囲のPEG鎖の付着によって駆動されるので、リポソームの破裂及び平面二重層の形成は、使用される脂質組成物に対して比較的鈍感です。コレステロールの大量に含まれている場合しかし、SUV車の凝集性を増加させる、SUVが自然に破裂しないことができます。この場合、浸透圧ショック、又は二価のイオンは、平面二重層の形成25を助けるために使用することができます。
このAPに、上記のようにPEGブラシが平面の両側をカバーローチ、二重層を支持しました。マイクロ流体流路に面するブラシはまた、通常、PEGの層で覆われている着信V-SUV車の非特異的付着を防止するのに役立ちます。 V-とt-SNARE複合体の形成は、膜近位領域36に向かって段階的に膜遠位N末端 および進行から始まります。 V-SUV車がt-SBL、V-およびアッセイの条件下でケースのように思われるPEGブラシ、上に突出するT-SNARE N末端必要とやり取りするために。ブラシの高さは、PEG化脂質の濃度及びPEG鎖長37,38を変化させることによってのSNARE以外のタンパク質を研究するために適合させることができます。融合二重層の近位表面を覆うPEGブラシのもう1つの利点は、平方ミクロン39あたり30,000〜40,000内在性膜タンパク質で充填されている生体膜の混雑した環境を模倣することです。ただ、このアッセイにおけるPEG鎖のように、repu生体膜をカバーlsiveタンパク質層が発生する核融合のための2つのリン脂質二重層の間の接触を可能にするために脇にプッシュする必要があります。
彼らはユニークな利点を提供するように、マイクロ流体流路は、このアッセイで使用されています。まず、マイクロ流体の流れがt-SBLを形成するために拡散し、融合するT-SUV車のより均一な成膜を可能にします。第二に、小さなチャネル容量(<1μl)をサンプルの消費を最小限に抑えます。第三に、必要な少量は、全実験は一定流量下で行うことを可能にします。流れは、弱いSBL 16からおそらく非特異的に、付着したV-のSUVを削除します。また、動力学的分析17を簡素化し 、T-SBLの上のv-SUV車の一定の密度を維持します。最後に、ドッキングされた小胞は、簡単に流れ25によって運ばれる無料のものと区別されています。第四に、いくつかのマイクロ流体チャネルは、各々が異なる状態をプロービング、同じカバーガラス上で使用することができます。これは、条件ドゥリの比較を可能にします同じ実験ランをngの。同様のアプローチは、インフルエンザウイルスとクッションSBLs 33との間の融合を研究するためにバンOijenグループが使用されてきました。
TIRF顕微鏡では、(〜100 nmの減衰定数を持つ)エバネセント場の指数関数的減衰は、ガラスバッファインタフェースの非常に近接している分子に蛍光励起を閉じ込めます。これは、遠く離れている蛍光分子の寄与を最小にする信号対雑音比を増加させ、10〜40ミリ秒のフレームの露光時間を有する単一分子感度を可能にします。エバネッセント場はまた、融合時の信号の増加につながる:SBLへのSUVからの標識脂質転送として、彼らはより強力な励起場で、平均して、自分自身を見つけます。蛍光の増加は、より大きなリポソームのために強いです。
偏光がエバネッセント場を生成するために使用される場合、付加的な効果は、T際の蛍光の変化に寄与するSBLへのSUVからのラベルのransfer。いくつかの脂質染料は、それらが埋め込まれた二層に対して好適な平均の角度で配向遷移双極子を持っています。偏光ビームが異なる二つの膜に色素を励起するので、これは、彼らがSBL 対 SUVにあるときに蛍光団によって放出される蛍光量の差を作成します。後者のために、双極子の向きが平坦SBLジオメトリによって制限される一方、前者の場合、励起光は、球状のSUVの周りに配向遷移双極子と相互作用します。染料は、膜29,40の脂質色素遷移双極子配向並列用SUVよりもSBLにあるとき、例えば、s偏光(入射面に垂直な偏光)の入射光を用いる場合には、励起がより効率的です(そのようなのDiIのことやDID 41-43など)。このような蛍光体でドープされたSUVは、ときにドックSBL( 図7、Representatに暗く表示されますアイブ結果)。融合孔が開き、SUVとSBL膜を接続するように、蛍光プローブは、SBL内に拡散し、s偏光エバネッセント場25,27,29によって励起される可能性が高くなります。その結果として、融合部位の周囲に統合蛍光シグナルは、SBL 27( 図3及び図7)にSUVの色素転写中に急激に増加します。 SBLに移す際に、それらが希釈されるように融合を伴う変更を知らせるために貢献する追加の要因は、蛍光標識の脱消光です。なぜなら小さな割合のみ(脱消光の寄与は、ここに記載されたアッセイにおけるエバネッセント場の減衰や偏光効果に比べて、通常軽微であります )脂質のは標識されています。
)脂質のは標識されています。
融合時のシグナル増加は、時間を比較することにより、融合細孔特性を推定するために利用することができ、1 "SRC =" /ファイル/ ftp_upload / 54349 / 54349eq2.jpg "/>、脂質は実際の解放時間に脂質が自由に透過性である細孔を通って脱出するために必要な、  。二つの時間スケールが同等である場合、細孔が脂質流れに対してほとんど抵抗を示すと結論されます。実際の放出時間が長い拡散制限のリリースのための時間よりも大幅にある場合は、これは脂質の放出を遅らせる、このような細孔ちらつきとして、プロセスを示すであろう。拡散制限解除時間、
。二つの時間スケールが同等である場合、細孔が脂質流れに対してほとんど抵抗を示すと結論されます。実際の放出時間が長い拡散制限のリリースのための時間よりも大幅にある場合は、これは脂質の放出を遅らせる、このような細孔ちらつきとして、プロセスを示すであろう。拡散制限解除時間、  、定着リポソームおよび脂質拡散の大きさに依存します。その推定は、これら2つのパラメータを定量化することが必要です。アッセイの単一分子感度は、脂質拡散率は、すべての融合イベント26用SBLへのそれらのリリース後に、いくつかの単一の脂質の蛍光団を追跡することにより測定することができます。すべての融合小胞の大きさ(I)単脂質色素の強度、SUVの(ii)のすべてのフルオロフォアが融合時SBLに転送された後、ドッキング部位の周囲の合計の蛍光の変化、(III)は、公知の標識密度を組み合わせることにより、27を推定することができます脂質、および(iv)脂質当たりの面積。均一なSUVのサイズ44を想定して先に述べたように、多くの融合イベントでは、実際の脂質の放出時間は、拡散制御放出27で予想よりはるかに遅いことが判明しました。脂質リリースの遅延がちらつき細孔によるものであると仮定すると、定量的なモデルは「ポア開放性」の推定を可能にする、時間の割合は、細孔が融合27の間、開いたままになります。
、定着リポソームおよび脂質拡散の大きさに依存します。その推定は、これら2つのパラメータを定量化することが必要です。アッセイの単一分子感度は、脂質拡散率は、すべての融合イベント26用SBLへのそれらのリリース後に、いくつかの単一の脂質の蛍光団を追跡することにより測定することができます。すべての融合小胞の大きさ(I)単脂質色素の強度、SUVの(ii)のすべてのフルオロフォアが融合時SBLに転送された後、ドッキング部位の周囲の合計の蛍光の変化、(III)は、公知の標識密度を組み合わせることにより、27を推定することができます脂質、および(iv)脂質当たりの面積。均一なSUVのサイズ44を想定して先に述べたように、多くの融合イベントでは、実際の脂質の放出時間は、拡散制御放出27で予想よりはるかに遅いことが判明しました。脂質リリースの遅延がちらつき細孔によるものであると仮定すると、定量的なモデルは「ポア開放性」の推定を可能にする、時間の割合は、細孔が融合27の間、開いたままになります。
たびに実用的な、脂質および可溶性内容のラベルの両方を使用して、融合メカニズムをテストすることが重要です。例えば、脂質リリースは、このようなトンを囲むSNAREタンパク質による脂質拡散の制限として細孔ちらつき以外のプロセスによって遅らせることができました彼は細孔。この場合であれば、次に、脂質標識の放出に先行することになる内容物の放出孔可溶性プローブの通過を可能にするのに十分な大きさで提供。アプローチで、より根本的な欠陥は、SBLに対する標識脂質の転送が大きく、その融合前の形状を保持している小胞にSBLを接続する狭い融合孔を介して起こることを前提である可能性があります。以前脂質放出データだけでは29に基づいて提案されているようSBLへの脂質の転送はまた、SBL膜に付随するとの融合細孔の急速な拡張、SUVの非常に急速な崩壊から生じる可能性があります。両方の脂質を監視し、内容物を同時に放出し、それは多くの細孔がすべての脂質のラベルを解放した後再密封することを発見しましたが、その可溶性貨物27の一部を保持しました。これは、少なくともいくつかのリポソームが融合後SBLに崩壊し、SBLへの脂質色素転写が融合孔を介して発生することはありませんことを示しています。また、LIPIDと内容のリリースでは、脂質の放出の遅延は、細孔45を囲むSNAREタンパク質による脂質拡散の障害のために起因したこと、それはそうなって、同時に27が発生しました。
可溶性コンテンツのリリースを監視しませんでしたSUV-SBL融合プロトコルは、以前Karatekinとロスマン25によって出版されました。ここでは、より最近の開発は、含まれる脂質のすなわち同時モニタリング及び内容は解放し、SUV、脂質、および融合細孔特性27の推定されています。プロトコルは、カバーガラス25との溝を含むポリ(ジメチルシロキサン)(PDMS)エラストマーブロックを接合することによって作られたマイクロ流体細胞を調製するための命令で始まります。次に、脂質及びコンテンツマーカーの両方を有するV型のSUVの調製を説明します。セクション4および5は、電圧vSの導入を、マイクロ流体セルを組み立て、その場で SBLsを形成し、欠陥および流動性をチェックするための命令を提供しますフローセルと融合事象の検出にUVS。第6節は、データ分析のための手順を説明します。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
マイクロ流体チャネルを形成するために、PDMSブロックの調製

フローセルテンプレートとPDMSブロックの準備の 図1. 微細加工。(A)24×60ミリメートルのガラスカバースリップ(下)に適合した4チャンネルフローセルの設計。六同じ設計は10cmのシリコンウエハー(上面)に収まるように配置されています。 (B)穴パンチャーで約5〜8ミリメートルの厚さのPDMSのブロックをカット。 (C)ピンセットを用いて、パンチ穴にチューブの挿入が。 この図の拡大版をご覧になるにはこちらをクリックしてください。
- このような図1Aの一つとして、フローセルテンプレートを入手します。典型的なチャネルは、70から100ミクロン、高、0.3〜1ミリメートル幅であり、1〜2センチ。テンプレートたちを作製クリーンルームへのアクセスが利用できない場合クリーンルーム施設25または順序で標準的なフォトリソグラフィ技術をる。また、マシンの適切な材料から大きな寸法を持つテンプレート。
注:クリーンルームのスタッフがデザインし、発注マスクの、ウェハ洗浄、およびフォトリソグラフィに(クリーンルーム施設へのアクセスが常に適切な訓練を持つユーザーに制限されている)未経験のユーザーを訓練し、導くことができます。テンプレートが得られると、それは繰り返してクリーンルーム外で使用することができ、それは閉じた皿に保持され、注意がダストを排除するために取られました。 - 使い捨てのプラスチックカップに〜100ミリリットルのPDMS(シリコーンエラストマーベース)と〜架橋剤の10ミリリットル(硬化剤)の混合物を準備します。より簡単に粘性PDMSを処理するために、使い捨てピペットの先端を折り。ピペット操作が正確でないなどのプラスチックカップにPDMSを計量。
- よく、混合液を撹拌しました。 (約20真空デシケーター中で脱気することにより、多くの気泡を除去分)。カップが真空下に置かれるように、気泡が最初に大幅に混合物の体積を増加させる、サイズが大きくなります。適用される真空を制御し、カップが流出を回避するのに十分な深さであることを確認してください。
- ガラスシャーレ(150ミリメートル×20ミリメートル)に脱気したPDMS混合物の大幅な低下を注ぎ、テンプレートを上に向けて、PDMS上にウエハを押してください。これは、ウェハの下に閉じ込められる気泡を回避することができます。閉じ込められた気泡が拡大すると、皿をオーブンに置かれたときに、ウエハを傾けることができます。それはPMDSの約5〜8ミリメートルで覆われているまで今ウエハの上に、テンプレート上に脱気したPDMSを注ぎます。気泡が発生した場合は静かにピペットチップでそれを削除します。
- 3時間、60℃のオーブンでPMDS焼きます。料理はレベルであることを確認してください。
- 成形されたチャネル構造を含むPDMSブロックを切り出すために、新しいメスの刃を使用してください。切り出されたブロックは、カバーガラスの上にフィットする必要がある(ステップ4.1.9を参照してください)。
- 皿から切り出したブロックアウトピールとPLきれいなアルミ箔の部分にそれをエース。
注:架橋PMDSブロックは、数ヶ月間維持することができます。単一のウエハ上に、流路( 図1A)の6組が存在するので、6 PDMSブロックは、単一のPDMS成形で製造することができます。 、PDMSのきれいな緩い部分をPDMSブロックのすべての6を切り出した後、それ以外は注ぎ、最初のバッチのために使用される唯一の約半分PDMSを必要とする次のバッチを、架橋の前に残りのPDMSを削除しないでください。良いテンプレートは数年続くことができます。 - 1直線運動( 図1B)にPDMSブロックを通してドリル穴パンチャーを使用してください。チャンネル溝の側面に起動します。 PDMSの打ち抜か部分を削除してください。 4チャネル設計のすべての8つの穴のためにこれを繰り返します。
- アルミニウム箔の新しい、しわのない片上にダウンPDMSブロックチャネル側に配置します。理想的にデシケーター中で、ドライボックス中で数ヶ月までのためのブロックを格納します。
- ピンセット( 図1C)のペアを使用して、パンチ穴に約1/3チューブ(内径0.25mm、0.76ミリメートルのOD)を押してください。簡単に挿入するための傾斜チューブをカットします。それぞれ、SUVリザーバーシリンジポンプに到達するのに十分な長さのチューブを残します。彼らはあまりにも長い場合、顕微鏡上に組み立てチップを配置した後、再びチューブをカット。
- ポンプのシリンジに接続するには、より大きなシリコンチューブ(0.51ミリメートルのID、2.1ミリメートルのOD)の短い部分をカットし、一方の側に薄いチューブを挿入します。 PDMSのこのすぐに使えるブロックは数ヶ月間維持することができます。
2.カバースリップクリーニング
- 硫酸(H 2 SO 4)と過酸化水素の強力な酸化剤混合物を使用してKaratekinとロスマン25に記載のプロトコールに従って清浄なカバーガラス(H 2 O 2)。クリーンルームでは、この手順を実行し、適切な安全注意事項を守ってください。また、クリーンルーム清浄Cを使用(材料リストを参照してください)市販されているoverslip。
脂質とコンテンツラベルの両方を含むV-のSUVの調製

SUVの調製図 2の 回路図。脂質はガラス管(1)中で混合し、溶媒を水浴中でチューブを回転させて脂質フィルムを形成するために蒸発させる(2)。溶媒の残りのトレースは、高真空(3)の下で除去されます。脂質膜をボルテックス(4)しながら、界面活性剤およびタンパク質を含む再構成緩衝液で水和されます。コンテンツ色素がカプセル化される場合、それは、この段階で、ならびに希釈工程(5)に含まれています。臨界ミセル濃度以下の界面活性剤濃度の希釈液は、リポソーム形成につながります。界面活性剤は、(6)一晩離れて透析します。 NBD-PE(緑色)を含むSBL形成のためのTのSUVは、小胞は、密度勾配に浮遊しているとで収集します2層(7A)との間のインタフェース。遊離色素からカプセル化されたコンテンツマーカーとV-のSUVを分離するために、サンプルはサイズ排除カラム上で実行し、0.5ミリリットルの分画(図7b)に回収される。 この図の拡大版をご覧になるにはこちらをクリックしてください。
- 25mMのHEPES-KOH、140 mMのKClを、100μMのEGTAおよび1mM DTT、pHが7.4を含む、再構成バッファの4 Lを準備します。透析および他のステップのために100ミリリットルのためのバッファのほとんどを使用してください。
他の緩衝液を用いることができるが、実験全体を通して一貫した緩衝浸透圧を維持し、タンパク質が機能的である、条件を選択することが重要です。注意してください 。唯一の脂質ラベルを含むV-SUVとT-のSUVを調製するために、Karatekinとロスマン25に従ってください。 - 脂質ストックをクロロホルムまたはクロロホルム中で-20℃で保存される:メタノール(2:1、v / v)を、バイアルを開ける前に室温に到達させ結露を避けます。
- クロロホルムとメタノールの混合物中に1マイクロモルの最終量と所望の割合で、界面活性剤の微量を除去し、脂質を混合し、クロロホルムでガラス管をすすぎ(2:1、v / v)でした。唯一の有機溶剤に溶解/脂質を処理するために、ガラス注射器/チューブを使用します。
注:ここで使用される脂質は、POPCされている:57.4のモル比でDID:SAPE:DOPS:コレステロール:PEG2000-DOPE:12:10:15を4.6:1のv-SNAREのための小胞およびPOPC含む:SAPE:DOPS:コレステロールを:脳のPI(4,5)P 2:PEG2000-DOPE:NBD-PE(54.9:15:12:10:3:4.6:0.5)T-SNAREのSUVのため。詳細については、資料を参照してください。他の脂質組成物を使用することができます。 - 穏やかな窒素流の下で、またはロータリーエバポレーターで溶媒を蒸発させ。均質な脂質膜を得るために、溶媒を蒸発させながら、温度の大きな変化を回避するために、脂質の最高溶融温度以上に加熱水浴(37°C)チューブの先端を浸します。周りで真空を開始脂質フィルムが形成されるまで300ミリバール、次にロータリーエバポレーターで2〜分間可能な最高の真空を継続します。
- ガラス管を外し、光の露出を避け、少なくとも2時間、高真空下でデシケーター中にチューブを配置することによって、溶媒の残りの痕跡を除去するためにアルミホイルで包ん。このステップは、夜間に実行することができます。
- 乾燥した脂質フィルムを再水和するために、再構成緩衝液中の界面活性剤の混合物(n-オクチルβ-D-グルコピラノシド、OG)、タンパク質(V-SNAREと)、およびSRB(500μlの最終容量)を準備します。臨界ミセル濃度(CMC、OGのために20〜25 mM)の上に1〜2回、最終的な界面活性剤濃度を維持し、脂質比に洗剤が> 10であるように、最終的な音量を調整します。穏やかに溶液を振盪することにより10〜50 mMのSRBの最終濃度を得るために、タンパク質 - 界面活性剤溶液にスルホローダミンB(SRB)粉末を溶解します。 SRBの高濃度を追加するときの共同を低減することにより、再構成バッファの浸透圧を調整しますそれに応じて塩化カリウムのncentration。
注:(。平方あたりLP〜20,000〜70トン-SNAREとミクロン25)T-SNAREとが高いため脂質対タンパク質比(LP)。 SBLの方が有意に高かっトン-SNARE密度が大幅に融合率17,24を低減さと不活性なタンパク質凝集体につながる可能性があります。 V-のSNAREのためのLPは(〜、μmの2当たり7000のv-SNAREとLP〜200)シナプス小胞46,47で検出されたのv-SNAREの密度に近く、はるかに低いです。組換え発現とV-とt-SNAREとの精製後のタンパク質の機能を確実にするためには、単一の小胞融合アッセイの全ての工程を経る前に、簡単なバルク融合アッセイ48を実行するために有用です。 - ステップ3.6から蛋白質 - 洗剤 - SRB溶液を添加しながら、ゆっくりと乾燥した脂質フィルムを含む予熱したガラス管を振ります。泡を作成しないようにしてください。 37℃で15分間振とうし続けています。
- 再構成を追加することにより、5倍の洗剤を希釈急速に濃度勾配を避けるために、ボルテックスしながらバッファーを含むSRB(2ミリリットル、2.5ミリリットル、最終容量を追加します)。 37℃で1時間に15分間振り続けます。
注:長いインキュベーション増加タンパク質の再構成効率を。迅速な希釈は、CMC以下の界面活性剤濃度を低下させ、小さなリポソームの形成をもたらします。 - 2万MWCO透析チューブやカセットを使用して、ポリスチレン吸着剤の4グラムと4℃で一晩周囲温度で1〜2時間の再構成バッファの〜1 Lに対して最初のベシクル懸濁液を透析した後、再構成バッファの3 Lに対して。交差汚染を防止するために、SRBとない小胞の透析に異なるビーカーを使用してください。
- 再構成緩衝液を用いてゲルろ過カラムを平衡化します。カプセル化されたSRBでV-SUV車からフリーのSRBを分離するためにカラムに通してベシクル懸濁液を実行します。試料全体が持っていたら、溶離液としてSRBせずに再構成バッファを使用します列を入力しました。 0.5ミリリットル画分中のv-のSUVを収集します。
- 小胞16のアリコートに界面活性剤の添加の前と後にSRBの蛍光を測定することにより、自己急冷濃度で成功したSRBカプセル化を確認します。蛍光分光計を用いて、550 nmでサンプルを励起し、570 nmおよび630 nmのSRB放射をスキャンします。膜が可溶化され、解放さSRBが希釈されるように界面活性剤添加の際にカプセル化された量に応じて、SRB蛍光の4-8倍の増加を期待しています。
- それぞれ、蛍光分光法24とSDS-PAGEゲル電気泳動を用いて、脂質とタンパク質回収を特徴付けます。特に高LPトン-SUVサンプルのため、敏感な染色方法を使用します(材料リストを参照してください)。典型的には、脂質とタンパク質の両方の入力の約50%は、公称値に近いLPに得られた調製物中に失われています。
- 24または電子microscoを動的光散乱を使用して、SUVのサイズを特徴付けますPY 48。 〜3-4日まで4℃でのv-のSUVを含むストアSRB。凍結融解は、SRBをカプセル化膜とのリリースを破壊するとして、凍結しないでください。
4. SUV-SBL融合アッセイは、脂質リリースのみを監視します
- マイクロ流体流路内に係留サポート二重層の形成
- 溶存ガスを除去するために、実験前に少なくとも20分間、高真空下でのPDMSブロック(ステップ1.11)を配置します。これは、非常に融合実験中のマイクロ流体チャネル内の気泡の危険性を減少させます。
- 顕微鏡の設定をオンにして、所望の温度にステージと試料ホルダー( 図3と図 4)を加熱。
- 0.45ミクロン以下の細孔径を有するフィルターを通して小胞を希釈するために使用される再構成バッファをフィルタリングします。
- 希釈〜NBD-PE30μlのがt- SUV車または無タンパク質(PF-SUV車)を標識対照リポソーム(ストックバッファの〜60μlの持つソリューション0.5-1 mMの脂質)。最終濃度は、ここでは重要ではありません。
- ドガ3ミリリットル注射器を使用して、この混合物。垂直方向に注射器を保持しながら、サンプル上の空気のほとんどを押してください。パラフィンフィルムを使用して(無針)の先端を密封し、プランジャーを下に引っ張ることにより、真空を作り出します。液の脱気を促進するためにシリンジバレルをタップします。真空が適用されたときにそれ以上の気泡が発生しなくなるまで、このプロセスを数回繰り返します。
- マイクロチューブのキャップに穴をパンチするためにPDMSブロックに取り付けられたチューブよりもわずかに大きい直径の皮下注射針を使用してください。それは、後にマイクロ流体チャネルの入口に接続された配管を詰まらせる可能性があるようなプラスチックの打ち抜い片がチューブの内側ではないことを確認してください。
- キャップに穴を有するマイクロ遠心チューブに脱気SUV液を記入し、設定温度に平衡化する顕微鏡ステージ上にホルダーにそれを置きます。
- PLプラズマクリーナーで以前に清掃カバースリップ(セクション2)をエースし、約5分間、空気プラズマを実行します。クッションとして、いくつかの糸くずの出ない組織の上にプラズマ処理されたカバースリップを(処理された側を上に)置きます。
- 脱気したPDMSブロックからアルミホイルを外し、カバーガラスの上にブロックを配置します。再利用する場合PDMSブロックは、上に置き、それをきれいにするために、チャネル側に粘着テープ片を取り外します。 PDMSダウンを押して、それを固執するためにピンセットを使用してカバースリップ上にブロックが、ガラスを壊すかもしれないとして、あまりにもハード押さないでください。
- 顕微鏡ステージ上に組み立てフローセルを配置し、それぞれ、SUVリザーバーシリンジポンプにチューブを接続します。ステージ( 図3B)にカバースリップをテープで固定します。
- 解決策は、(75ミクロン×300ミクロンのチャネル断面のための〜2.25ミリメートル/秒)完全にチャンネルをいっぱいになるまで3μL/ minでのSUVを吸引開始します。すべてのチャネルのためのソリューションは、目を上に移動を開始すると出口側で電子管は、0.5μL/ minに流量を減少させ、30-45分間インキュベートします。
注:プラズマ処理の効果は一時的であるように、プラズマは、スライドガラスを処理し、チャンネルにSUV車を流れる間の時間は10-20分を超えてはなりません。 - 任意のリークのためのチャンネルを確認してください。 T-またはPF-のSUVを含むNBD-PEからNBD-PEの蛍光を観察するために10-20X空気の目標を使用します。エキサイトNBDは、488nmのレーザーを用いてフルオロフォア。漏れは、明視野照明を使用して検出することが困難です。
- ホルダーに脱気再構成緩衝液を用いてチューブを入れて、温度がSBLに欠陥が発生する可能性があり、温度のように急速な変化を平衡化しましょう。流れを停止し、流れが完全に停止していると全く気泡が脱気したバッファーへの入口管を切り替える前に、チューブ内に吸引されませんを確認するために、〜1分を待ちます。
- 未結合のSUVを洗い流すために脱気したバッファーを持つすべてのチャンネルを洗い流します。
- higheに切り替えR倍率TIRF対物レンズ(60X、油、NA 1.45から1.49)とは、二層が均質に見えるし、そのような暗いパッチやSBLからはみ出し脂質細管など、明らかに大規模な欠陥のないことを確認してください。
- 二重層の流動性を確認してください
- 専用のFRAPユニットが利用できない場合、または次のようにFRAPシーケンスは、その後、定性的試験膜の流動性をプログラムすることができない場合。
- 小さ いサイズ(〜40μmの直径)に視野絞りを閉じて、露出した中でNBD-PE蛍光を漂白する(20-80μW、または15から60ナノワット/μmの2)ソフトウェアを使用して488 nmの励起光強度を調整しますエリア著しく、完全ではありません。無傷のNBD-PE分子は露出面積を入力して、漂白前に一定の距離を拡散するように、流体二重層の場合、定常状態で、露光領域の中央に蛍光強度は、エッジにおけるよりも低くなければなりません。対照的に、表面付着S場合UVは破裂することができなかった、またはサポートされている二重層は、流体ではない他のいくつかの理由で、露光領域内のすべてのフルオロフォアが漂白する必要があります。
注:これ以降の工程におけるレーザー強度値は、粗出発点として与えられる条件の所与のセットについて最適化されるべきです。 - 照明を停止し、定常状態の測定の結果を検証するために、数分後に再度起動
- 小さ いサイズ(〜40μmの直径)に視野絞りを閉じて、露出した中でNBD-PE蛍光を漂白する(20-80μW、または15から60ナノワット/μmの2)ソフトウェアを使用して488 nmの励起光強度を調整しますエリア著しく、完全ではありません。無傷のNBD-PE分子は露出面積を入力して、漂白前に一定の距離を拡散するように、流体二重層の場合、定常状態で、露光領域の中央に蛍光強度は、エッジにおけるよりも低くなければなりません。対照的に、表面付着S場合UVは破裂することができなかった、またはサポートされている二重層は、流体ではない他のいくつかの理由で、露光領域内のすべてのフルオロフォアが漂白する必要があります。
- 可能であれば、より定量的な測定のためのFRAPシーケンスをプログラムします。補足ファイルや詳細については、対応する凡例を参照してください。
注意:時々のSUVは、カバーガラスに付着するが、破裂や液二重層を形成するために失敗します。この問題が発生した場合は、サポートされている二重層形成を助けるために10 mMのマグネシウム2+含む脱気再構成緩衝液でチャンネルをすすぎます。 4.2のように二層の流動性を評価するために、蛍光漂白を使用してください。流体二重層が形成されると、フリーRECO の Mg 2+で洗い流しnstitutionバッファ。
- 専用のFRAPユニットが利用できない場合、または次のようにFRAPシーケンスは、その後、定性的試験膜の流動性をプログラムすることができない場合。
- マイクロ流体流路にV-SUV車を導入
- ドガの再構成バッファとv-SUVストック濃度に応じて、10 5〜約10 3倍のv-SUV原液を希釈するために使用します。視野の60秒以内に約10-100融合現象が生じる希釈を目指します。
注:あまりにも多くの融合は(各イベント堆積物LR-PE以降またはSBLへの脂質のラベルをした)バックグラウンド蛍光を増加させ、融合事象の検出および分析を困難にします。これとは対照的に、貧しい統計が低すぎた結果であるか、より多くの映画の取得を必要と融合率。 0.1 mMの脂質のV-SUVの濃度については、995μlの再構成緩衝液に5μlのSUVの株式を希釈することにより開始し、950から995μlの再構成バッファにこれを5-50μLを希釈します。 - 温度は流れとinserを停止する前に平衡化してみましょうティン希釈されたV-SUV溶液中に注入管。
- 次のようにTIRF角と偏光を調整します。
- 励起ビームを回転させることにより、所望の偏光を設定した後、ソフトウェアを介してステッピングモータを介して、ビームの位置を指示するミラーの傾きを調整します。ゆっくりとオフセンター1側へのバック焦点面での目的の中心からレーザビームの位置を移動させます。位置は中心から外れ、さらに移動すると、手前側の目的からの光を観察するには、客観的な軸に対して増加する角度で出てきます。
- TIRが最初に達成されたときに出たビームは最初、 すなわち 、客観的に消滅したときにモータの位置に注意してください。
- ゆっくりと表面からの蛍光をモニターしながらオフセンターさらにビーム位置を動かし続けます。ビームが中心から外れすぎ移動したときに表面蛍光が消失したときモータの位置に注意してください。
- ビームpositioを選択します。上記決定された2つの限界の間のn。融合の際に最良の信号対雑音比と複数の信号増強のために、まだ視野の均一な照明を提供する浅い浸透深さ(近い目的のエッジにビーム位置)を選択します。
注:これは、設定が最適化された後、すべての実験のために同じTIRビーム位置(同じ侵入深さ)を維持するのが最善です。ビーム位置の変化が生じない偏光を回転することを確認してください。
- 75ミクロン×300ミクロンの断面1.5 mm /秒〜の平均線流速に対応する、2μL/分の流速でチャネルにV-SUVの流れ。混合脂質監視するために励起/発光の設定に切り替えます(LRをかだけでした)。
- ドガの再構成バッファとv-SUVストック濃度に応じて、10 5〜約10 3倍のv-SUV原液を希釈するために使用します。視野の60秒以内に約10-100融合現象が生じる希釈を目指します。
- シングルV-SUVとSBLの間に観察融合
.JPG "/>
図3. 実験pTIRFセットアップ。ガラス基板上にV-SUVおよびt-SBLの(A)概略図。脂質のドッキングを示す単一SUV-SBL融合事象の偽色TIRFM画像(1)とSBLに脂質色素の放出(2)漂白及び蛍光強度の減少が続く(3)。総蛍光強度(5.3ミクロンのx 5.3μmのボックス内の画素値の和)信号が示されています。 (B)PDMSブロックに結合したカバーガラスは、加熱されたステージ上にテープで固定されています。マイクロ流体チャネルのための入口管は、シリンジポンプ(左)により吸引(右)金属試料ホルダーにチューブからサンプルを引き出します。ポンプの下に両面発光ユニットである。 この図の拡大版をご覧になるにはこちらをクリックしてください。
OAD / 54349 / 54349fig4.jpg "/>
実験の 図4 の回路図。エバネッセント波は、マイクロ流体チャネル内のガラス・バッファインタフェースで作成されます。 SBLは、ガラス上に形成され、V-SUVは(左上)をシリンジ内にチャネルを介して金属試料ホルダー(右上)から吸引されます。 M、ミラー; DM、ダイクロイックミラーと、 L、レンズ; F、フィルター; P、偏光板。 この図の拡大版をご覧になるにはこちらをクリックしてください。
- 継続的に励起し、V-のSUVに組み込まれた蛍光団に応じてV-SUVの(LR-PE用)561ナノメートルを使用して蛍光または(DID用)638 nmのレーザーを監視します。 V-のSUVは、流路に達し、上にドッキングすると、SBLと融合したように、バックグラウンド蛍光シグナルが蓄積し始めます。
- 継続的背景を漂白するソフトウェアを介して励起レーザ強度を調整します定常状態で新たなドッキングおよび融合事象が容易に観察できるように、蛍光。
注:漂白が遅すぎる場合は、バックグラウンド蛍光があまりにも高くなります。漂白が速すぎる場合は、ドッキングのSUVからとSBLに放出蛍光団からの信号は、ドッキングのSUVの融合を検出することができ、またはフルオロフォアを追跡することができ、その間ウィンドウを短く、急速にフェードします。 561 nmで励起されたLR-PEについては、190μmの直径の円(100から250ナノワット/μmの2)を照明2.5から7.5 mWのパワーが開始するための妥当な値です。 DIDの638 nm励起、190μmの直径の円(30-60ノースウエスト/μmの2)の上に0.8から1.6 mWのパワーの初期試験のために使用することができます。 - 与えられたチャネルの異なる位置にいくつかの映画を取得します。 SBLは、これらの位置での欠陥を持っていないことを確認するために、NBD-PE蛍光を確認してください。高いフレームレートで最大速度(〜50フレーム/秒)、または関心のトリミングされた領域にフルフレーム映画を取得(最大に〜100 Hz)で60秒間。
- 別のマイクロ流体チャネルに移動し、他の条件のために録音を繰り返します。このようなタンパク質を含まないSBLやSUVの陰性対照として含まれ、阻害剤としてのv-SNARE VAMP2(CDV)の可溶性細胞質ドメインを追加したり、1つ以上において破傷風神経毒(37℃で30分)で、V-SUVを扱います同じカバースリップ上のチャネルの。
- クリーンアップとPDMSのリサイクル
- PDMSブロックを再利用するためには、5μL/分の流速での70%エタノール〜200μlでマイクロ流体チャネルをすすぎます。最後に、チューブおよびチャネルを介して空気を吸引。
- 少し絞ることによってカバースリップから静かにPDMSブロックを外します。アルミホイルのきれいな作品の上に置きます。真空デシケーターに保管してください。これは、複数回再利用することができます。
- より徹底した清掃のため、70%エタノールの前に洗剤または水酸化ナトリウム溶液でチャンネルをすすぎます。また、あちこちにすべてのチューブを削除します PDMSをMと乾燥して、新しいチューブを挿入する前に、イソプロパノール中で30分間ブロックを超音波処理します。
5. SUV-SBL融合アッセイは、同時に脂質とコンテンツのリリースを監視します
- 同時脂質および可溶性内容のセクションで説明したように、使用の両方の脂質(DID)で標識したリポソームおよび可溶性内容(SRB)ラベルを除いて、セクション4と同じ手順に従って、解放のデュアルカラーの監視のために3 SRBが封入されています10mMで、最初は非常に自己クエンチしています。
- 図4に概略的に示す構成を使用して、SRBを励起し、それぞれ、561 nmおよび638 nmのレーザーを用いて同時に蛍光をしました。ダイクロイックミラー(640 nm)はSRBを検出するために、短い(50分の595 nm)を長く(75分の700 nm)の波長フィルタを介して実行し、それぞれの排出量を、DID 2つのビームに発光を分割します。 2つの発光ビームは、EM-CCDチップ上に並んで突出しています。
- FRAPデータの分析
- 脂質拡散係数、Dを推定するために補足情報で提供MATLABプログラムを使用してください。プログラムは、OME-TIFFファイル49のリストを読み出し漂白領域を検出し、時間の関数としての漂白領域の平均画素値をプロットし、回復時間を抽出するSoumpasis 50によってモデルに生じる回復曲線にフィット
 ここで、wは漂白円の半径です。
ここで、wは漂白円の半径です。
注意:MATLABプログラムは、ニコンND2ファイルを使用して、FRAP映画の分析のために、以前の25提供されました。ほとんどのファイルフォーマットは容易にOME-TIFFに変換することができるので、現在のプログラムは、OME-TIFFファイル49を読み込みます。漂白は、bleac瞬間であるときFRAPデータの定量分析は、最も簡単で正確ですHED領域は円形であり、読み出し時の漂白は無視できます。これらの基準は、ここで説明する単純なFRAP測定において厳密に満足していないが、拡散係数の妥当な推定値を得ることができます。より正確な推定のために、単一の脂質染料(6.3節)の追跡を使用しています。
- 脂質拡散係数、Dを推定するために補足情報で提供MATLABプログラムを使用してください。プログラムは、OME-TIFFファイル49のリストを読み出し漂白領域を検出し、時間の関数としての漂白領域の平均画素値をプロットし、回復時間を抽出するSoumpasis 50によってモデルに生じる回復曲線にフィット
- ドッキングレート、融合率とドッキングツー融合遅延時間の分析
- 使用ImageJが明らかに小胞のドッキングおよび融合イベントを識別するために分析し、明るさとコントラストを調整するムービーを開きます。 SpeckleTrackerJ 26プラグインを起動します。使用方法についてはSpeckleTrackerJためSmithら 26およびオンラインマニュアルを参照してください。
- SpeckleTrackerJ内のすべての新たにドッキングさのSUVを識別します。 SBL跳ね返るものからしっかりとドッキングのSUVを区別するために、数フレームの最小ドッキング期間を課します。トラックを保存し、すべての映画のために繰り返します。
注意:ドキュメントについて王率は、重要なことはすべて、SUVがドッキングするとき、SUVのは、これらのトラックに記録するニーズをドッキングされているので、最初のフレームのみを識別することです。トラックの残りの部分は、分析において考慮されません。しかし、自動追尾、それは漂白またはヒューズまで各SUVは既に追跡されているマークのSUVをするのに役立ちます。 - すべての融合小胞を特定します。これらの場合、トラックは最初のフレームの融合が追跡スポットの蛍光の急激な増加によって明らかである第1のフレームまでドッキングしたSUVからのすべてのフレームを含むべきです。これらのトラックの持続時間は、ドッキング・ツー・融合遅延を計算するために使用されます。すべてのトラックを保存します。すべての映画のために繰り返します。
- ドッキングまたは融合データのバッチ分析のために、関連する映画から軌道ファイルのリストをコンパイルし、Karatekinとロスマン25が設けられMATLABプログラムを実行します。プログラムと一緒に提供されている指示に従ってください。
注:プログラムは、兼をプロット軌道ファイルから抽出された情報に基づいて、時間の関数としてulativeドッキングおよび融合イベント。ドッキングおよび融合率は、これらのプロットの傾きから推定されます。融合データについては、ドッキング・ツー・融合の遅延も計算されており、その分布は、生存プロットとしてプロット、 すなわち 、確率は融合はまだドッキングした後、所定の遅延によって発生していません。
- 脂質拡散
- 彼らが認識できるになると、彼らは十分に離れて融合部位( 図6)から拡散した場合に、各融合イベントでは、単一の蛍光脂質を追跡します。トラッキングのためSpeckleTrackerJを使用し、MATLABを使用して、さらなる分析のためのトラックを保存します。定着小胞の大きさに応じて、典型的には3-30単一のフルオロフォアを追跡することができます。長いトラックの計算のために望ましいものであるので
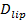 、長いような単一分子を追跡してみてください可能な限り、必要に応じて軌道の手動補正を使用して。
、長いような単一分子を追跡してみてください可能な限り、必要に応じて軌道の手動補正を使用して。 - 平均は(〜1.5秒程度)> 40〜50フレーム続く単一の脂質マーカーの軌跡のために変位(MSD)を乗を計算します。脂質拡散係数を計算するためにMSDを使用して、 。 Smithら 26とストラットンら 27の詳細を参照してください。
- 彼らが認識できるになると、彼らは十分に離れて融合部位( 図6)から拡散した場合に、各融合イベントでは、単一の蛍光脂質を追跡します。トラッキングのためSpeckleTrackerJを使用し、MATLABを使用して、さらなる分析のためのトラックを保存します。定着小胞の大きさに応じて、典型的には3-30単一のフルオロフォアを追跡することができます。長いトラックの計算のために望ましいものであるので
- 単一脂質色素強度、SUV-SBL強度減少係数、および小胞サイズ
- 単一ステップでのマーカー漂白剤の前と後〜15フレームのマーカーの周りの3×3画素(0.8ミクロン×0.8μm)の領域内の脂質マーカーの画素値の合計を測定します。バックグラウンド強度が追跡脂質マーカーの強度を得るために、漂白前に平均プリ漂白強度からポスト漂白フレームにわたって平均引きます。与えられたためにできるだけ多くのマーカーの測定を繰り返し、映画。
- 単一の脂質ラベル強度の分布をプロットし、
 、および平均値を推定するガウシアンにフィット。
、および平均値を推定するガウシアンにフィット。 - そのイベントにリリースされた単一の脂質マーカーの軌跡が一段階漂白で終わったとき融合が発生したときの間の遅延を計算します。 SBLにおける蛍光団のための漂白時間を入手し、
 、生き残り遅延の関数と指数関数にフィッティングをプロットすることもできます。
、生き残り遅延の関数と指数関数にフィッティングをプロットすることもできます。
注:偏励起磁場カップルのでより弱くSUV上の蛍光団に、SUVの漂白時間は、通常27はるかに遅いです。 - 見積もり
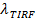 、ときにストラットン27以下、SBLにあるときにSUV相対脂質色素に対する強度減少係数(tp_upload / 54349 / 54349eq8.jpg "/> SUVにおける単一の色素の強度)です。
、ときにストラットン27以下、SBLにあるときにSUV相対脂質色素に対する強度減少係数(tp_upload / 54349 / 54349eq8.jpg "/> SUVにおける単一の色素の強度)です。
注意:漂白は無視できる程度であれば、ドッキングされた強度に等しくなります すべてのフルオロフォアが(破線は標識融合の際にSBLに堆積された後に到達した合計強度で割った( 図3A、右パネルのポイント(1))、
すべてのフルオロフォアが(破線は標識融合の際にSBLに堆積された後に到達した合計強度で割った( 図3A、右パネルのポイント(1))、  図3で)。しかし、SBLの急速な漂白のために、 一般的に到達したとの正確な推定されていませんその込み式27に放出動態を当てはめる必要があります前記UDEの両方 (6.4.3を参照) 。セパレート式はストラットンらに提供されている細孔限定されるものではなく、拡散限定発売動態の例のための27。ベストフィットの場合は、イベント(細孔限ら動態の場合の6.5.1を参照)にイベントごとに異なります。
図3で)。しかし、SBLの急速な漂白のために、 一般的に到達したとの正確な推定されていませんその込み式27に放出動態を当てはめる必要があります前記UDEの両方 (6.4.3を参照) 。セパレート式はストラットンらに提供されている細孔限定されるものではなく、拡散限定発売動態の例のための27。ベストフィットの場合は、イベント(細孔限ら動態の場合の6.5.1を参照)にイベントごとに異なります。 - 27から個々のイベントのための小胞面積を計算します
 ここで、 ドッキングされたSUVの強度があり、 SBLにおける単一の脂質色素の平均強度は、ありますこれはSUVでは、SBLにあるときを基準としたときに脂質色素に対する強度減少係数であり、かつエーション1 "SRC =" /ファイル/ ftp_upload / 54349 / 54349eq12.jpg "/>脂質色素の既知の面密度です。
ここで、 ドッキングされたSUVの強度があり、 SBLにおける単一の脂質色素の平均強度は、ありますこれはSUVでは、SBLにあるときを基準としたときに脂質色素に対する強度減少係数であり、かつエーション1 "SRC =" /ファイル/ ftp_upload / 54349 / 54349eq12.jpg "/>脂質色素の既知の面密度です。
- フュージョン細孔特性
- リリースは細孔限られていると仮定すると、27に脂質ラベル放出動態に合わせ
 ここで、
ここで、  ちょうど融合前にドッキングSUVの強度であり、他のパラメータは先に定義した通りです。の値を使用固定パラメータとして、6.4.3で得られた、とのための最適推定値を抽出しますそして
ちょうど融合前にドッキングSUVの強度であり、他のパラメータは先に定義した通りです。の値を使用固定パラメータとして、6.4.3で得られた、とのための最適推定値を抽出しますそして 。
。 - P 0は 、脂質リリースの仮定の位相差が27をちらつき細孔することによるものであり、孔が開いている時間の割合を計算します。60;
 =
=  VESは、小胞面積(セクション6.4.5)であり、bは、孔の高さは、(典型的には〜15nmであることが撮影したもの)であり、R P≈3 nmのは、拡散脂質ラベルで見られるように有効細孔半径であり、半分が含まれて二層の厚さ(〜2 nm)を、 (6.3で計算)脂質拡散率であり、 脂質は(6.5.1から)SBLにSUVから解放するための時間があります。
VESは、小胞面積(セクション6.4.5)であり、bは、孔の高さは、(典型的には〜15nmであることが撮影したもの)であり、R P≈3 nmのは、拡散脂質ラベルで見られるように有効細孔半径であり、半分が含まれて二層の厚さ(〜2 nm)を、 (6.3で計算)脂質拡散率であり、 脂質は(6.5.1から)SBLにSUVから解放するための時間があります。 - 公称P 0> 1はストラットンらの4を式に強度の時間経過に合わせ、P 0 = 1、全開気孔を示している。27、永久にオープンポアの予測動態を確認します。このフィットはfittinより良いはずですグラム恒久的に開気孔のための6.5.1で表現。
- リリースは細孔限られていると仮定すると、27に脂質ラベル放出動態に合わせ
 ここで、wは漂白円の半径です。
ここで、wは漂白円の半径です。  、生き残り遅延の関数と指数関数にフィッティングをプロットすることもできます。
、生き残り遅延の関数と指数関数にフィッティングをプロットすることもできます。  すべてのフルオロフォアが(破線は標識融合の際にSBLに堆積された後に到達した合計強度で割った( 図3A、右パネルのポイント(1))、
すべてのフルオロフォアが(破線は標識融合の際にSBLに堆積された後に到達した合計強度で割った( 図3A、右パネルのポイント(1))、  図3で)。しかし、SBLの急速な漂白のために、
図3で)。しかし、SBLの急速な漂白のために、  ここで、
ここで、  ここで、
ここで、  ちょうど融合前にドッキングSUVの強度であり、他のパラメータは先に定義した通りです。の値を使用
ちょうど融合前にドッキングSUVの強度であり、他のパラメータは先に定義した通りです。の値を使用 =
=  VESは、小胞面積(セクション6.4.5)であり、bは、孔の高さは、(典型的には〜15nmであることが撮影したもの)であり、R P≈3 nmのは、拡散脂質ラベルで見られるように有効細孔半径であり、半分が含まれて二層の厚さ(〜2 nm)を、
VESは、小胞面積(セクション6.4.5)であり、bは、孔の高さは、(典型的には〜15nmであることが撮影したもの)であり、R P≈3 nmのは、拡散脂質ラベルで見られるように有効細孔半径であり、半分が含まれて二層の厚さ(〜2 nm)を、 Subscription Required. Please recommend JoVE to your librarian.
Representative Results
SBLの品質
融合実験の前にSBLの品質および流動性を検証することが重要です。マイクロ流体チャネルの下、ガラス側での蛍光は、任意の明白な欠陥のない、均一でなければなりません。気泡がチャネルも合格した場合、それは通常、SBLに見える傷跡を残します。このような大規模な傷跡/欠陥がある場合は、そのチャネルを使用しないでください。時々、SUVが基板上に付着するが、連続SBLを破裂し、形成するために失敗することがあります。その場合は、蛍光はまだ非常に均一に見えるかもしれませんが、小さな領域を漂白した後に回復することはありません。導入を10mMのMg 2+は、多くの場合、この問題を解決するのに役立ちます。他の薬剤は、他の二価の陽イオン51、高分子溶液52、または浸透圧ショック25として、SUVの破裂を助けるために導入することができます。
脂質によって占有される平均面積は、温度に依存するため、温度変化はSBL領域の変化をもたらします。 5〜10℃以上53それぞれの表面から押し出され、チューブ、または表面上に現れる暗い、SBLフリースポットに入る過剰な領域をもたらすことができることにより、温度を上昇させるか、減少させます。最小の温度変動を維持することが重要です。
図5に示されています。

SBLの流動性を調べるために 、図5 FRAP。前に(最初のフレーム)と、強い光パルスで漂白した後、閉じた視野絞り(直径49ミクロン)で区切られた照明領域を示す一連の画像。面積は、脂質標識NBDの拡散による蛍光回復に従うように低露光で結像されます。グラフは、この方法の再現性の高いレベルを示す別のカバーガラスからの複数のチャネルのための時間の関数としての正規化された蛍光を示します。 SAPE:DOPS:コレステロール:脳のPI(4,5)P 2:PEG2000-DOPE:NBD-PE(54.9:15:12:10:3:4.6:0.5モル%、T-SBLの脂質組成物は、POPCました)及びLP =2万。漂白面積は984.203μでしたm 3で。回復時間とフィットから算出した拡散係数は(6.1を参照)τ= 67.3秒、D =それぞれ1.99μmの2 / secであった。 この図の拡大版をご覧になるにはこちらをクリックしてください。
脂質のラベルを使用してドッキングし、核融合イベントの単一色検出
条件ごとに100から400のイベントは、良好な統計のために必要とされるようにドッキングおよび融合イベントのマニュアルカウントは面倒な作業です。 SUVが融合する前に、SBLに移動することができますので、追跡ソフトウェアが必要です。 ImageJのためのSpeckleTrackerJ 26のプラグインは、具体的には、ドッキングおよび融合事象の分析のためのSUVを追跡するために開発された機能を備えています。多くの場合、ドッキングした小胞の一部のみが融合してしまいます。ドッキング・レートを計算するために、より多くのSUVは、ヒューズまたはイベントの十分な数の信頼できる推定のために検出されるまで、映画の一部のみが分析される必要があるものよりも追跡する必要があります。主な焦点は、融合速度および/またはドッキング・ツー・融合遅れている場合、唯一の融合のSUVは、識別され追跡される必要があります。
SBLの品質を確保した後、SUVがチャネルに導入され、その蛍光は、s偏光を使用してTIR下で連続的に励起されます。最初の実験では、TIRの励起ビームの入射角を調整するために、得られたエバネッセント波の侵入深さを調整する必要があるかもしれません。代表的な単SUVドッキングおよび融合事象は、図6Aに示されています。フレームの間にドッキングされた小胞はDマークが付けられ、融合は、彼らが座って離れ融合から拡散したように、単一の脂質が認識できるになったフレームFは約1.3秒後に(フレームS)で開始しました電子。各ボックスの総蛍光強度(画素値の合計)がドッキングおよび融合を示す発生したフレームに、 図6(b)における時間の関数としてプロットされています。時間の関数をt-SNAREを使用して、五つの異なる映画のために、 図6(c)にプロットされているように融合イベントの累積数はSBLs(T-SBL、LP = 20,000平方ミクロン当たり約70トン-SNAREと)を再構成しました。各映画の融合率は、勾配に線形フィットから算出しました。 SEM±この平均融 合率は、 図6A、Bに示した例のためのドッキング・ツー・融合遅延は、0.07秒であったパネルDのバーグラフとして示されています。 158イベントの合計についても同様に計算された遅延の分布は、生存プロット、 すなわち、ドッキングされた小胞は、まだ一定の遅延によって融合していないことを確率として図6Dに示されています。約1/2の100ミリ秒以内に融合されたこれらの小胞の。コレステロールレベルを増加させる、DISTRIBとドッキング・ツー・融合遅延のutionは、脂質拡散が27を遅くしても、短くなります。
並行して対照実験を実行することが重要です。 図6B、Dのデータを取得したのと同じカバースリップ上に、制御条件は、隣接するチャネルで試験しました。 1チャンネルでは、SBLは、任意のT-SNAREとを含んでいませんでした。 60秒ずつ持続5の映画のうち、わずか7融合事象は、T-SNAREとは( 図6C)含まれていた158に比べて記録しました。他のチャンネルでは、V-SNARE VAMP2(CDV)の可溶性の細胞質ドメインが含まれていました。 CDVは、SUVの上にあり、全長VAMP2の結合を防止すること、SBL上のT-SNAREと特異的に結合します。 5 60秒のムービーでは、何の融合イベントはCDV( 図6C)の存在下で検出されませんでした。

図6。 SUV-SBLのドッキングおよび融合イベント。LR-PEの広がりを次pTIRF顕微鏡により撮像されたT-SBLとV-SUVの融合イベントの画像の(A)のシーケンス。画像は、すべて18ミリ秒、唯一の毎秒のフレームが示されているが取られています。 DおよびFはそれぞれ、ドッキングおよび融合の開始をマークします。彼らが融合部位と互いに(S)から離れて拡散するように、個々の脂質ラベルが識別可能になります。フレーム寸法:16.7ミクロンのx 16.7ミクロン。 (B)(A)に示される融合事象に対する時間の関数としての蛍光強度。 (C)別の組の実験からDIDで標識されたV-SUVのための時間の関数としての融合イベントの累積数。 (D)正規化された融合率(±SEM、SXμmの2×pMのSUVを意味する)-1、4 SUVあたり脂質および(E)の生存確率は小胞のドッキング後の一定時間、2×10を仮定。すべてのSBのための組成物SAPE:DOPS:コレステロール:脳のPI(4,5)P 2:PEG2000-DOPE:NBD-PE(54.9:15:12:10:3:4.6:0.5モル%)およびLP =2万LsがPOPCでした。 V-SUV車はPOPCで構成されていた:SAPE:DOPS:コレステロール:PEG2000-DOPE:LR-PE(57.4:15:10:12 4.6:1モル%)とLPを= CEのために、AとBのための200、V LR-PEの代わりに使用されたDID以外-SUV組成物は、同様であった。 この図の拡大版をご覧になるにはこちらをクリックしてください。
脂質拡散性、小胞サイズ、および融合細孔のプロパティ
アッセイの単一分子感度がドッキングおよび融合率とドッキングツー融合遅延分布に加えて、いくつかのパラメータの抽出を可能にします。彼らが融合部位( 図6A)から離れて拡散するように、単一の、蛍光標識された脂質が認識できるになります。トラッキングトンHESE直接、脂質拡散率26を提供します 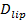 、およびSBL 27における蛍光団のための漂白率のデジタル測定。はじめに述べたように、ラベルの蛍光発光は、SBLにおける対 SUVで一般的に解くのが困難である原因の3要因を異なる:ラベルの(ⅰ)脱消光を融合すると、それらの希釈にSBLに移すとき、(ⅱ)によるTIRによって生成されたエバネッセント場、および励起光の偏光に対する蛍光団の遷移双極子の(iii)の向きの崩壊にSBLにおいてより高い励起強度を。係数の大きさは、(ii)のエバネセント場の減衰長と小胞サイズの相対的な大きさに依存し、原因SUVサイズの分散性にすべての融合イベントのために異なっています。要因は、(iii)のフルオロフォアおよび使用偏光に依存します。むしろディにしようとしてより直接融合前に、すべてのフルオロフォア後のSUVを取り囲む領域中の全画素値を比較することにより、一つの蛍光変化の大きさを測定することができ、これらの効果をsentangleする融合( 図3A以下SBLに寄託されている、(1)対( 2))。何発蛍光団が融合した後に考え回の拡散を介してそれを残していないだろうように、前作29,44とは異なり、領域が十分に大きく選択されるべきです。融合は27、通常十分である後1.6秒までの分析のために30×30画素領域(×8μmの8μm)を選択します。一瞬無視漂白、の比
、およびSBL 27における蛍光団のための漂白率のデジタル測定。はじめに述べたように、ラベルの蛍光発光は、SBLにおける対 SUVで一般的に解くのが困難である原因の3要因を異なる:ラベルの(ⅰ)脱消光を融合すると、それらの希釈にSBLに移すとき、(ⅱ)によるTIRによって生成されたエバネッセント場、および励起光の偏光に対する蛍光団の遷移双極子の(iii)の向きの崩壊にSBLにおいてより高い励起強度を。係数の大きさは、(ii)のエバネセント場の減衰長と小胞サイズの相対的な大きさに依存し、原因SUVサイズの分散性にすべての融合イベントのために異なっています。要因は、(iii)のフルオロフォアおよび使用偏光に依存します。むしろディにしようとしてより直接融合前に、すべてのフルオロフォア後のSUVを取り囲む領域中の全画素値を比較することにより、一つの蛍光変化の大きさを測定することができ、これらの効果をsentangleする融合( 図3A以下SBLに寄託されている、(1)対( 2))。何発蛍光団が融合した後に考え回の拡散を介してそれを残していないだろうように、前作29,44とは異なり、領域が十分に大きく選択されるべきです。融合は27、通常十分である後1.6秒までの分析のために30×30画素領域(×8μmの8μm)を選択します。一瞬無視漂白、の比 前後の融合は、蛍光強度の減少率を提供し、
前後の融合は、蛍光強度の減少率を提供し、 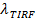 。小胞サイズは(SBL単一標識の蛍光強度を与えて推定することができますジル/ ftp_upload / 54349 / 54349eq6.jpg "/>)、それはSUV(にあった場合、その強度は減少するであろう使用される係数 )、SUVのラベルの既知の密度、および脂質の占有面積。実際には、漂白は通常、十分に速く、すべてのマーカーはSBL内に堆積している時間によって、いくつかは既に漂白していることです。このため、正確な推定のために、蛍光強度の減少率、 、および脂質の放出時間
。小胞サイズは(SBL単一標識の蛍光強度を与えて推定することができますジル/ ftp_upload / 54349 / 54349eq6.jpg "/>)、それはSUV(にあった場合、その強度は減少するであろう使用される係数 )、SUVのラベルの既知の密度、および脂質の占有面積。実際には、漂白は通常、十分に速く、すべてのマーカーはSBL内に堆積している時間によって、いくつかは既に漂白していることです。このため、正確な推定のために、蛍光強度の減少率、 、および脂質の放出時間 最高27を漂白考慮する総画素値の経時変化に適合さから抽出されます。 SBLのラベルの退色速度は、独立して、単一の脂質色素追跡から、または全強度プロファイルに指数関数を当てはめることによって推定されます
最高27を漂白考慮する総画素値の経時変化に適合さから抽出されます。 SBLのラベルの退色速度は、独立して、単一の脂質色素追跡から、または全強度プロファイルに指数関数を当てはめることによって推定されます 、長い時間に( 例えば、レジーム(3) 図3で)。定着ベシクル及び脂質拡散の大きさを知ることは、実際の脂質リリースタイムの比較を可能にします 、小胞の周りの脂質の拡散時間に
、長い時間に( 例えば、レジーム(3) 図3で)。定着ベシクル及び脂質拡散の大きさを知ることは、実際の脂質リリースタイムの比較を可能にします 、小胞の周りの脂質の拡散時間に 、VESは、小胞の面積であると脂質拡散率です。 2時間スケールが同等である場合、細孔は、脂質リリースにほとんど抵抗を提示し、リリースが拡散制限されています。対照的に、もし
、VESは、小胞の面積であると脂質拡散率です。 2時間スケールが同等である場合、細孔は、脂質リリースにほとんど抵抗を提示し、リリースが拡散制限されています。対照的に、もし 、その後細孔が大きく、脂質の流れを遅らせます。遅滞の定量的測定は、比に等しい「孔開放性」、P 0であり、
、その後細孔が大きく、脂質の流れを遅らせます。遅滞の定量的測定は、比に等しい「孔開放性」、P 0であり、  回因子Oジオメトリ27の細孔に関連するFオーダー団結。二状態、開/閉気孔については、P 0は開いた状態にある時間の割合です。そのサイズが連続的に変化するちらつき細孔について、P 0は全開半径に時間平均半径相対を表します。
回因子Oジオメトリ27の細孔に関連するFオーダー団結。二状態、開/閉気孔については、P 0は開いた状態にある時間の割合です。そのサイズが連続的に変化するちらつき細孔について、P 0は全開半径に時間平均半径相対を表します。
信号対雑音比は、単一の脂質フルオロフォアを追跡するために十分に良好である必要があります。これは、より簡単に、このようなLR-PE明るいマーカーを使用して、単色実験で達成されます。
脂質の同時検出と可溶性カーゴリリース
代表的な事象は、 図7に示されている。カプセル化のために使用される高濃度では、SRBの蛍光は、最初に自己クエンチします。そのため、ほとんどのドッキングされたV-SUVがそのDID信号27によってのみ検出可能です。急増行った蛍光は単色脂質ラベル実験のように、脂質混合をマークします。脂質放出の開始は、SRBの蛍光の増加の発症と一致しました。 SRB分子がSUVを脱出し、残りのSRBは自己消光濃度以下に希釈したように、この増加は、SRBのデクエンチングに最も可能性の高いでした。興味深いことに、SRBの蛍光は安定したプラトーに達しました。融合孔が開いて残っている場合、1は、SRB信号が原因で、残りのSRBはSUVを残したとして、またはSUVはSBLに崩壊しているときにバックグラウンドに減少が続き、脱消光を最大に高めることを期待します。安定したプラトーが最も可能性の高いすべての脂質ラベルと可溶性貨物の一部を解除した後再密封細孔を示しています。
いくつかの場合において、脂質解除信号は、任意の検出可能なコンテンツリリース信号を伴いませんでした。これは、孔がSRBの通過を可能にするには小さすぎたためのいずれか、またはいくつかの可能性どのようにSUVはすでにその可溶性貨物を失っていました。これら二つの可能性を区別するために、ドッキングされたSUV車の漂白を解析することができます。最初はSRBの一部としてSRB信号の増加で自己急冷SRBの結果は、他の製品と自己消光にフォト変換されるの漂白が緩和されるのに対し、脂質ラベルの漂白は、数秒以内にその蛍光シグナルを減少させませんでした。ほとんどのドッキングさのSUVが(58のうち48がイベントを分析した)照明の長期間にわたるSRB蛍光の予想される増加を示しているが、ごく一部(58分の10)はまったくSRB信号を示しませんでした。このように、SUV車のごく一部は、実験におけるそれらの製造と使用との間の期間中に、その可溶性のコンテンツを失う可能性があります。
SRBと信号が続くことができなかったの両方のイベント(91のうち74)の80%以上では、SRBの蛍光が増加し、安定したプラトー27のままでした。トンの〜20%で残りのSRBの蛍光が突然、数秒後までにリリースされたHESE例。これは、蓄積された光ダメージ16に1-2フレーム(18-36ミリ秒)またはSUVの破裂内の急速な、完全な放出を可能に細孔の再開が原因であるかもしれませんどちらか。何であれ、その起源、この第二は、SRBの迅速な放出は、最初のリリース後の残留SRB信号が原因でSRBはSBLとフルリリース後のガラス基板との間の空間に閉じ込められたままになる可能性を除外します。このようなメカニズムは、SBLが2 16の間の小さなスペースで、ガラス上に直接サポートされていた以前の研究で示唆されました。対照的に、このアッセイで使用されるPEG化脂質はSRB離れ融合部位から拡散することを可能にする、SBLとガラス25との間の空間の4-5 NMを提供すべきです。

図7. Simultan eous脂質およびコンテンツリリース。SUV-SBL融合イベントの脂質(青)とコンテンツ(赤)マーカーからの蛍光シグナル(詳細は本文参照します)。各チャネルからのスナップショットは、底部(4.0ミクロン幅)で示されています。漫画(中央)は、異なる段階を示します。 (1)SUVドック、DID蛍光は低いです。 SBLに行なったし、蛍光増加をしました(2)の融合は、転送を行うことができます。自己消光は、細孔を通してSRBの脱出によって緩和されるようSRB信号も増加します。 (3)SRB信号が安定してしばらく残る分子は細孔の可能な再封を示すSBL内に拡散しませんでした。 (4)SRB信号が急速なフル融合またはリポソームの破裂に、非常に急速に減少します。 SAPE:DOPS:PEG2000-DOPE:DID(67:15:12:5:1モル%)、LP 200、T-SBLsはPOPCで構成している間:SAPE:DOPS:脳のPI V-のSUVの組成はPOPCました(4,5)P 2:PEG2000-DOPE:NBD-PE(64.5:15:12:3:5:0.5モル%)とLP =2万。 = "_空白">この図の拡大版をご覧になるにはこちらをクリックしてください。
FRAP_instructions.pdf - 。FRAPシーケンスを設定するための手順は、 このファイルをダウンロードするにはこちらをクリックしてください。
ReadMe_FRAP.txt - 。提供されたMATLABコードを使用して、FRAP回復データを分析するための手順を記述したテキストファイルには、 このファイルをダウンロードするにはこちらをクリックしてください。
EK_FRAPbatch.m -光退色後のバッチ分析蛍光回復へのMATLABファイル。このファイルには、MATLABのサブルーチンEK_getFRAPvals.mとEK_SoumpassisFit.mを呼び出します。"_blank">このファイルをダウンロードするにはこちらをクリックしてください。
EK_getFRAPvals.m - 。MATLABファイル、EK_FRAPbatch.mによって呼び出されたサブルーチンは、このファイルをダウンロードするにはこちらをクリックしてください。
EK_SoumpassisFit.m - 。MATLABファイル、EK_FRAPbatch.mによって呼び出されたサブルーチンは、このファイルをダウンロードするにはこちらをクリックしてください。
JN150923c1_FRAP_list.txt - 。メインMATLABスクリプトEK_FRAPbatch.mによってバッチ分析しようとするファイル名のリストの例は、 このファイルをダウンロードするにはこちらをクリックしてください。
ザ次のファイルはTIFスタックとEK_FRAPbatch.mを実行し、ファイルリストを選択するプロンプトが表示されたら、JN150923c1_FRAP_list.txtファイルを選択することによって分析することができるFRAP配列の5例については、対応するテキストファイルです。
JN150923c1_F1_MMStack_Pos0.ome.tif - 。FRAPの画像スタックこのファイルをダウンロードするにはこちらをクリックしてください。
JN150923c1_F1_MMStack_Pos0_metadata.txt - 。メタデータと対応するテキストファイルこのファイルをダウンロードするにはこちらをクリックしてください。
JN150923c1_F2_MMStack_Pos0.ome.tif - FRAPの画像スタック。 PL容易にこのファイルをダウンロードするにはこちらをクリックしてください。
JN150923c1_F2_MMStack_Pos0_metadata.txt - 。メタデータと対応するテキストファイルこのファイルをダウンロードするにはこちらをクリックしてください。
JN150923c1_F3_MMStack_Pos0.ome.tif - 。FRAPの画像スタックこのファイルをダウンロードするにはこちらをクリックしてください。
JN150923c1_F3_MMStack_Pos0_metadata.txt - 。メタデータと対応するテキストファイルこのファイルをダウンロードするにはこちらをクリックしてください。
JN150923c1_F4_MMStack_Pos0.ome.tif - FRAP画像スタック。 このファイルをダウンロードするにはこちらをクリックしてください。
JN150923c1_F4_MMStack_Pos0_metadata.txt - 。メタデータと対応するテキストファイルこのファイルをダウンロードするにはこちらをクリックしてください。
JN150923c1_F5_MMStack_Pos0.ome.tif - 。FRAPの画像スタックこのファイルをダウンロードするにはこちらをクリックしてください。
JN150923c1_F5_MMStack_Pos0_metadata.txt - 。メタデータと対応するテキストファイルこのファイルをダウンロードするにはこちらをクリックしてください。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
ここで説明するSUV-SBL融合アッセイの実装を成功させるには、良質のSBLsを得、そのようなリポソームへのタンパク質の機能的再構成などのいくつかの重要なステップ、に大きく依存し、単一分子を検出するために右の撮像パラメータを選択します。それが成功するためにいくつかの時間と労力がかかることがありますが、アッセイが正常に実装されれば、それははじめに考察される任意の他のインビトロ融合アッセイからは利用できません融合プロセスに関する豊富な情報を提供します。 SUV車が上にドッキングするとSBL、ドッキング・ツー・融合時間の分布と融合する料金で、脂質の拡散、すべての融合リポソームの大きさ、および与えられた融合イベントの脂質リリースがdiffusion-または細孔制御缶であるかどうか決定されます。リリースが拡散から予想よりもはるかに遅い場合は、細孔のちらつきを前提としたモデルは、脂質リリース利回りの遅滞、P 0、時間の割合の基礎となる細孔私融合27時のオープンです。
SBLs 15-17,21,22,33,44に単一SUV-SUVの融合18-20,28またはリポソームおよびウイルス粒子の融合をプロービング前の作業は、色素分子間FRETと自己消光に依存していました。これとは対照的に、我々は、大きく2つの理由から、SBLに脂質ラベル転送の動態を調べるために、色素脱消光を避けます。まず、FRET効率は、脂質放出の非常に非線形なレポーターです。 FRET効率の大きな変化で間染料距離d結果の小さな変化が、唯一のR 0は、フェルスター距離であるD / R 0値の限られた範囲のため。つまり、実際の色素希釈と脱消光信号の観察された増加との間の遅延時間は、R 0に近い値に低下するのに必要な時間に、発生する可能性があります。色素濃度は、そのような脂質のリリース時に十分に低下したら同様に、脱消光信号にはほとんど変化があります 1.4は、染料のかなりの量は依然として小胞に残っていても。これらのプロパティは、融合事象を検出するための信号のスパイクを得るための良いツールを脱消光作るが、高い時間分解能を使用して、脂質の放出の定量的な動態解析を複雑にします。これとは対照的に、双極子再配向とSBLへのSUVからフォア転送などのエバネッセント場の効果に強度の変化は直線的にまだSUVに残っている色素の割合に関連しています。第二に、自己消光のために必要な脂質のラベルの高濃度は、融合プロセス自体に影響を与え得ます。
1.4は、染料のかなりの量は依然として小胞に残っていても。これらのプロパティは、融合事象を検出するための信号のスパイクを得るための良いツールを脱消光作るが、高い時間分解能を使用して、脂質の放出の定量的な動態解析を複雑にします。これとは対照的に、双極子再配向とSBLへのSUVからフォア転送などのエバネッセント場の効果に強度の変化は直線的にまだSUVに残っている色素の割合に関連しています。第二に、自己消光のために必要な脂質のラベルの高濃度は、融合プロセス自体に影響を与え得ます。
偏光TIRF励起のために、 図4のように自作のセットアップは、費用対効果であり、偏光特性に対して優れた制御を提供します。少なくともいくつかの商用TIRF顕微鏡は、偏光励起を使用し、既製しかし、自作のセットアップは、必要ありません2色の発光分割モジュールが用意されています。カップルに顕微鏡へのレーザーから出てくる偏励起光を偏光保持(PM)ファイバを使用する商業顕微鏡は、以前に正常に25-27を使用しました。そのような器具で、偏光顕微鏡のTIRF部にPMファイバ入力を回転させることによって簡単に回転させることができます。対照的に、いくつかの他の市販のセットアップは、偏光調整がより困難または不可能、TIRFユニットに偏光ビームスプリッタを使用することができます。残念ながら、偏光特性は、多くの場合、商用システムのために指定されていません。したがって、偏光特性を議論するために、特に商業のオプションに投資する前にデモユニットを試すために家の作成したシステムを構築したくないユーザーのために重要です。
脂質および水溶性の貨物リリースの同時モニタリングは、融合プロセスに関する更なる情報を提供します。多くの細孔はRELE後に再密封するように見えますasing SUV 27内に封入された水溶性のラベルの一部のみ。通常、すべての脂質ラベルは、細孔が再密封する時間によって放出されるので、孔の再スケーリングは、単独で、脂質の放出をモニターすることから、以前に疑われていませんでした。確かに、単独の脂質の放出を監視、キースリンクら 29は SBLへの標識された脂質の転送はSBLに全体SUV膜の非常に急速な崩壊を経由して発生した提案しました。
コンテンツの部分的な解放後の融合孔の再スケーリングは、以前リポソーム係留二層パッチ融合アッセイで観察されました。 Rawle ら 〜8 nmの長いDNAスペーサーによってカバースリップ上につながれた54準備自立型二層パッチが。そして、彼らはその融合二層パッチにリポソームとパッチ膜に固定さ相補的な一本鎖DNA - 脂質複合体のハイブリダイゼーションによって駆動されたリポソームを導入しました。すべてのイベントの12%は、一部で、その結果、一過性の融合しましたリポソーム内容物のIALリリース。 44以前のいくつかのSUV-SBL融合アッセイで見られるリポソーム破裂イベントは、非常にまれでした。著者らは、二層 - 基質相互作用を防止し、二層パッチ以下〜8 nmの空間を提案した二層の下の空間へのリポソーム内容物の非漏出性の転送を許可されている可能性があります。また、放出された可溶性の標識の拡散は著しく平面二重層とガラス基板との間の空間に妨げられていませんでした。ここに記載したアッセイでは、平面二重層は、同様に、ポリ(エチレングリコール) - 脂質コンジュゲートの存在によって約5ナノメートル離れてカバースリップから保たれます。
可溶性および脂質貨物の2色の監視によって提供される追加情報は、いくつかのトレードオフが付属しています。まず、2つのラベルを持つSUV車の製造は、単独の脂質のラベルを含めたよりもやや複雑です。第二に、いくつかの必然的な損失があり、信号対雑音tにおける脂質の標識からの信号を監視します単色の脂質の放出実験のように、彼と同じ取得レート(フレーム当たり15-20ミリ秒)。この損失の一部は、DIDに起因する可溶性SRBマーカーと共に脂質標識として使用される、LR-PEは、単一色の実験で使用されるように明るくありません。追加の損失は、光路における同時2レーザー励起と追加コンポーネント( 図4)に高いバックグラウンドから生じます。これまでのところ、低い信号対雑音は単一の信頼性の追跡から私たちを防ぐ二色実験でラベルをしました。これにより、単色LR-PE測定値とすることができるすべての情報を抽出するために徹底的な分析を妨げました。この課題は、取得条件を最適化し、改善された特性と互換性標識の他の組をテストすることによって、将来的に克服することができます。
可溶性および脂質貨物リリースの同時モニタリングもhemifusion、近位リーフレットが融合した状態が、先端を検出することができます。リーフレットはそうではありません。実際、このような二色実験は、 インビトロの融合アッセイ18,20 の他の内hemifusionの存在だけで決定的な評価を提供することができます。しかし、hemifusion確実脂質放出単独22,44を監視する単色SUV-SBL実験で検出することができます。このような実験では、SUVの約1/2の初期脂質標識強度は、融合スポットのまま。スポットはまだ漂白されていない場合は、遠位リーフレットは、後でいくつかの時間を融合したとき、残りの強度が消えることがあります。
PEG化脂質の使用は、SBLとガラス支持体との間に柔らかいクッションがエキソサイトーシスのSNAP25要件を再生する際に鍵となっている導入します。同様の流路に面したSBLの側を覆うPEGブラシのプロトコルの結果。これは、はじめに説明したように、SUVとSBLとの間の非特異的相互作用を低減するようにいくつかの利点を有しています。のリン脂質と相互作用するタンパク質の場合SBLは、アッセイに導入される、しかし、PEGブラシは、そのような相互作用に対して立体障害を提示します。したがって、いくつかの用途のために、それだけSBLとカバーガラスとの間に、非対称PEG層を含むことが望ましいです。流路に面しSBLの側は、その後、シナプトタグミンなどのリン脂質結合タンパク質と相互作用することができるであろう。これは、共有結合で一方の端部にカバーガラスに結合させることができる二官能性PEG鎖を使用して他に脂質アンカーを搬送することによって達成することができます。ラングミュア-ブロジェット法は、その後、PEG層55上の脂質単分子層を堆積させるために使用することができます。リポソームは、二重層56を形成し 、それをこの疎水性の表面ヒューズの上に導入しました。このアプローチは、将来のマイクロフルイディクスと組み合わされることが期待されます。
ここで提示SUV-SBLアッセイはのSNAREと相互作用する追加のタンパク質の役割を探求するのに役立つことが期待されますこのようなMunc18 / SEC1ファミリー、カルシウムセンサーシナプトタグミン-1、およびcomplexinなど。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
著者は、彼らが競合する金融利害関係を持たないことを宣言します。
Materials
| Name | Company | Catalog Number | Comments |
| Reagents | |||
| Milli-Q (MQ) water | Millipore | ||
| KOH | J.T. Baker | 3040-05 | |
| Ethanol 190 Proof | Decon | ||
| Isopropanol | Fisher Chemical | A416P4 | |
| HEPES | AmericanBio | AB00892 | |
| Sodium Cholride (KCl) | AmericanBio | AB01915 | |
| Dithiothreitol | AmericanBio | AB00490 | |
| N-[2-hydroxyethyl] piperazine-N'-[2-ethanesulfonic acid] (HEPES) | AmericanBio | AB00892 | |
| EGTA | Acros Organics | 409911000 | |
| Buffers | |||
| HEPES-KOH buffer (pH 7.4) | 25 mM HEPES-KOH, 140 mM KCl, 100 μM EGTA, 1 mM DTT | ||
| Solvents | |||
| Chloroform | J.T. Baker | 9180-01 | in glass bottle, CAUTION, wear PPE |
| Methanol | J.T. Baker | 9070-03 | in glass bottle, CAUTION, wear PPE |
| Liposome preparation | |||
| Gastight Hamilton syringe | Hamilton | var. sizes | only use glass sringe with solents (Chlorophorm/ Methanon, 2:1, v/v) |
| Glass tubes Pyrex Vista 11 ml, 16x100 mm screw cap culture tube | Pyrex | 70825-16 | clean thoroughly, rinse with chloroform |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, 16:0-18:1 PC (POPC) | Avanti Polar Lipids | 850457 | Lipids come dissolved in CHCl3 or as lyphilized powder in sealed vials. Aliquot upon opening. Store extra as dried lipid films under inert atmosphere at -20 °C. Keep stocks in CHCl3/MeOH (2:1, v/v) at -20 °C. let come to RT before opening |
| 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (sodium salt), 18:1 PS (DOPS) | Avanti Polar Lipids | 840035 | Same as above. |
| 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine, 18:0-20:4 PE (SAPE) | Avanti Polar Lipids | 850804 | Same as above. |
| L-α-phosphatidylinositol-4,5-bisphosphate (Brain, Porcine) (ammonium salt), Brain PI(4,5)P2 | Avanti Polar Lipids | 840046 | Same as above. |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt), 18:1 NBD PE | Avanti Polar Lipids | 810145 | Same as above. |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt), 18:1 PEG2000 PE | Avanti Polar Lipids | 880130 | Same as above. |
| cholesterol (ovine wool, >98%) | Avanti Polar Lipids | 700000 | Same as above. |
| DiD' oil; DiIC18(5) oil (1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindodicarbocyanine Perchlorate) | Molecular Probes | D-307 | |
| Rotavapor R-210 | Buchi | R-210 | heat bath above Tm of lipids used |
| OG n-Octyl-β-D-Glucopyranoside | Affymetrix | 0311 | store at -20 °C, let come to RT before opening |
| Shaker - Eppendorf Thermomixer R | Eppendorf | ||
| Slide-A-Lyze Dialysis Cassettes, 20k MWCO, 3 ml | life technologies | 66003 | |
| Bio-Beads SM-2 Adsorbents | Bio-Rad | 1523920 | |
| OptiPrep Density Gradient Medium | Sigma-Aldrich | D1556 | |
| Ultracentrifugation tube, Thinwall, Ultra-Clear, 13.2 ml, 14 x 89 mm | Beckman Coulter | 41121703 | |
| Beckman SW41 Ti rotor | |||
| SuflorhodamineB | Molecular Probes | S-1307 | |
| Econo-Column Chromatography Columns, 2.5 × 10 cm | Bio-Rad | 7372512 | |
| Sepharose CL-4B | GE Healthcare | 17-0150-01 | |
| SYPRO Orange Protein Gel Stain | Molecular Probes | S-6650 | 5,000x Concentrate in DMSO |
| PDMS block | |||
| Sylgard 184 Silicone elastomer kit, PDMS | Dow Corning | 3097358-1004 | |
| Pyrex glass petri dish, 150 x 20 mm, complete with cover | Corning | 3160-152 | |
| Hole puncher - Reusable Biopsy Punch, 0.75 mm | World Precision Instruments | 504529 | |
| Manual Hole Punching Machine | SYNEO | MHPM-UNV | |
| Drill .035 x .026 x 1.5 304 SS TiN coated round punch | SYNEO | CR0350265N20R4 | drill diameter: 0.9 mm, punch bore size: 0.74 mm |
| Tygon Microbore tubing, 0.25 mm ID, 0.76 mm OD | Cole-Parmer | 06419-00 | 0.010" ID, 0.030" OD |
| Silicone Tubing (0.51 mm ID, 2.1 mm OD | Cole-Parmer | 95802-00 | 0.020" ID, 0.083" OD |
| Cover glass - cleanroom cleaned | |||
| Schott Nexterion cover slip glass D | Schott | 1472305 | |
| plasma cleaner | Harrick | PDC-32G | |
| pTIRF setup and accessories | |||
| IX81 microscope body | Olympus | IX81 | |
| EM CCD camera | Andor | ixon-ultra-897 | |
| Thermo Plate, heated microscope stage | Tokai Hit | MATS-U52RA26 | |
| 1 ml hamilton glass syringes (4x) | Hamilton | 81365 | |
| syringe pump | kd Scientific | KDS-230 | |
References
- Sudhof, T. C., Rothman, J. E. Membrane fusion: grappling with SNARE and SM proteins. Science. 323, 474-477 (2009).
- Wickner, W., Schekman, R. Membrane fusion. Nat Struct Mol Biol. 15, 658-664 (2008).
- Harrison, S. C. Viral membrane fusion. Nat Struct Mol Biol. 15, 690-698 (2008).
- Jahn, R., Scheller, R. H. SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol. 7, 631-643 (2006).
- Lindau, M., Alvarez de Toledo, G. The fusion pore. Biochim Biophys Acta. 1641, 167-173 (2003).
- Staal, R. G., Mosharov, E. V., Sulzer, D. Dopamine neurons release transmitter via a flickering fusion pore. Nat Neurosci. 7, 341-346 (2004).
- Wu, Z., et al. Nanodisc-cell fusion: Control of fusion pore nucleation and lifetimes by SNARE protein transmembrane domains. Sci. Rep. 6, 27287 (2016).
- Alabi, A. A., Tsien, R. W. Perspectives on kiss-and-run: role in exocytosis, endocytosis, and neurotransmission. Ann Rev Physiol. 75, 393-422 (2013).
- Rossetto, O., Pirazzini, M., Montecucco, C. Botulinum neurotoxins: genetic, structural and mechanistic insights. Nature Rev Microbiol. 12, 535-549 (2014).
- Weber, T., et al. SNAREpins: minimal machinery for membrane fusion. Cell. 92, 759-772 (1998).
- Nickel, W., et al. Content mixing and membrane integrity during membrane fusion driven by pairing of isolated v-SNAREs and t-SNAREs. Proc Natl Acad Sci U S A. 96, 12571-12576 (1999).
- McNew, J. A., et al. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature. 407, 153-159 (2000).
- Melia, T. J., You, D. Q., Tareste, D. C., Rothman, J. E. Lipidic antagonists to SNARE-mediated fusion. J Biol Chem. 281, 29597-29605 (2006).
- Hernandez, J. M., et al. Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science. 336, 1581-1584 (2012).
- Fix, M., et al. Imaging single membrane fusion events mediated by SNARE proteins. Proc Natl Acad Sci U S A. 101, 7311-7316 (2004).
- Bowen, M. E., Weninger, K., Brunger, A. T., Chu, S. Single molecule observation of liposome-bilayer fusion thermally induced by soluble N-ethyl maleimide sensitive-factor attachment protein receptors (SNAREs). Biophys J. 87, 3569-3584 (2004).
- Liu, T., Tucker, W. C., Bhalla, A., Chapman, E. R., Weisshaar, J. C. SNARE-driven, 25-millisecond vesicle fusion in vitro. Biophys J. 89, 2458-2472 (2005).
- Yoon, T. Y., Okumus, B., Zhang, F., Shin, Y. K., Ha, T. Multiple intermediates in SNARE-induced membrane fusion. Proc Natl Acad Sci U S A. 103, 19731-19736 (2006).
- Diao, J., et al. A single-vesicle content mixing assay for SNARE-mediated membrane fusion. Nat Commun. 1, 1-6 (2010).
- Kyoung, M., et al. In vitro system capable of differentiating fast Ca2+-triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release. Proc Natl Acad Sci U S A. 108, E304-E313 (2011).
- Domanska, M. K., Kiessling, V., Stein, A., Fasshauer, D., Tamm, L. K. Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J Biol Chem. 284, 32158-32166 (2009).
- Kreutzberger, A. J., Kiessling, V., Tamm, L. K. High Cholesterol Obviates a Prolonged Hemifusion Intermediate in Fast SNARE-Mediated Membrane Fusion. Biophys J. 109, 319-329 (2015).
- Schwenen, L. L., et al. Resolving single membrane fusion events on planar pore-spanning membranes. Sci Rep. 5, 12006 (2015).
- Karatekin, E., et al. A fast, single-vesicle fusion assay mimics physiological SNARE requirements. Proc Natl Acad Sci U S A. 107, 3517-3521 (2010).
- Karatekin, E., Rothman, J. E. Fusion of single proteoliposomes with planar, cushioned bilayers in microfluidic flow cells. Nat Protoc. 7, 903-920 (2012).
- Smith, M. B., et al. Interactive, computer-assisted tracking of speckle trajectories in fluorescence microscopy: application to actin polymerization and membrane fusion. Biophys J. 101, 1794-1804 (2011).
- Stratton, B. S., et al. Cholesterol Increases the Openness of SNARE-mediated Flickering Fusion Pores. Biophysical journal. 110, (2016).
- Diao, J., et al. Synaptic proteins promote calcium-triggered fast transition from point contact to full fusion. Elife. 1, e00109 (2012).
- Kiessling, V., Domanska, M. K., Tamm, L. K. Single SNARE-mediated vesicle fusion observed in vitro by polarized TIRFM. Biophys J. 99, 4047-4055 (2010).
- Blasi, J., et al. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature. 365, 160-163 (1993).
- Washbourne, P., et al. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci. 5, 19-26 (2002).
- Diaz, A. J., Albertorio, F., Daniel, S., Cremer, P. S. Double cushions preserve transmembrane protein mobility in supported bilayer systems. Langmuir. 24, 6820-6826 (2008).
- Floyd, D. L., Ragains, J. R., Skehel, J. J., Harrison, S. C., van Oijen, A. M. Single-particle kinetics of influenza virus membrane fusion. Proc Natl Acad Sci U S A. 105, 15382-15387 (2008).
- Albertorio, F., et al. Fluid and air-stable lipopolymer membranes for biosensor applications. Langmuir. 21, 7476-7482 (2005).
- Daniel, S., Albertorio, F., Cremer, P. S. Making lipid membranes rough, tough, and ready to hit the road. Mrs Bulletin. 31, 536-540 (2006).
- Gao, Y., et al. Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science. 337, 1340-1343 (2012).
- Kenworthy, A. K., Hristova, K., Needham, D., Mcintosh, T. J. Range and Magnitude of the Steric Pressure between Bilayers Containing Phospholipids with Covalently Attached Poly(Ethylene Glycol). Biophys J. 68, 1921-1936 (1995).
- Knoll, W., et al. Solid supported lipid membranes: New concepts for the biomimetic functionalization of solid surfaces. Biointerphases. 3, Fa125-Fa135 (2008).
- Quinn, P., Griffiths, G., Warren, G. Density of newly synthesized plasma membrane proteins in intracellular membranes II. Biochemical studies. J Cell Biol. 98, 2142-2147 (1984).
- Sund, S. E., Swanson, J. A., Axelrod, D. Cell membrane orientation visualized by polarized total internal reflection fluorescence. Biophys J. 77, 2266-2283 (1999).
- Johnson, D. S., Toledo-Crow, R., Mattheyses, A. L., Simon, S. M. Polarization-controlled TIRFM with focal drift and spatial field intensity correction. Biophys J. 106, 1008-1019 (2014).
- Anantharam, A., Onoa, B., Edwards, R. H., Holz, R. W., Axelrod, D. Localized topological changes of the plasma membrane upon exocytosis visualized by polarized TIRFM. J Cell Biol. 188, 415-428 (2010).
- Axelrod, D. Carbocyanine dye orientation in red cell membrane studied by microscopic fluorescence polarization. Biophys J. 26, 557-573 (1979).
- Wang, T., Smith, E. A., Chapman, E. R., Weisshaar, J. C. Lipid mixing and content release in single-vesicle, SNARE-driven fusion assay with 1-5 msec resolution. Biophys J. 96, 4122-4131 (2009).
- Chernomordik, L. V., Frolov, V. A., Leikina, E., Bronk, P., Zimmerberg, J. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J Cell Biol. 140, 1369-1382 (1998).
- Takamori, S., et al. Molecular anatomy of a trafficking organelle. Cell. 127, 831-846 (2006).
- Wilhelm, B. G., et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science. 344, 1023-1028 (2014).
- Scott, B. L., et al. Liposome fusion assay to monitor intracellular membrane fusion machines. Methods Enzymol. 372, 274-300 (2003).
- Linkert, M., et al. Metadata matters: access to image data in the real world. J Cell Biol. 189, 777-782 (2010).
- Soumpasis, D. M. Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys J. 41, 95-97 (1983).
- Ohki, S. A mechanism of divalent ion-induced phosphatidylserine membrane fusion. Biochim Biophys Acta. 689, 1-11 (1982).
- Berquand, A., et al. Two-step formation of streptavidin-supported lipid bilayers by PEG-triggered vesicle fusion. Fluorescence and atomic force microscopy characterization. Langmuir. 19, 1700-1707 (2003).
- Tamm, L. K., McConnell, H. M. Supported phospholipid bilayers. Biophys J. 47, 105-113 (1985).
- Rawle, R. J., van Lengerich, B., Chung, M., Bendix, P. M., Boxer, S. G. Vesicle fusion observed by content transfer across a tethered lipid bilayer. Biophys J. 101, L37-L39 (2011).
- Wagner, M. L., Tamm, L. K. Reconstituted syntaxin1a/SNAP25 interacts with negatively charged lipids as measured by lateral diffusion in planar supported bilayers. Biophys J. 81, 266-275 (2001).
- Kalb, E., Frey, S., Tamm, L. K. Formation of Supported Planar Bilayers by Fusion of Vesicles to Supported Phospholipid Monolayers. Biochimica Et Biophysica Acta. 1103, 307-316 (1992).
