

Introduction
Des anticorps capables de se replier et de fonctionner dans l'environnement intracellulaire sont des outils prometteurs pour la recherche et des applications thérapeutiques. Ils ont la capacité de moduler l' activité des protéines en se liant à une protéine cible dans les cellules pour éviter des interactions protéine-protéine, les interactions perturber protéine-acide nucléique, ou d' empêcher l' accès du substrat aux enzymes 1-5.
Bien que les anticorps ont un grand potentiel pour des applications intracellulaires, les ingénierie pour le pliage correct et la solubilité dans le milieu intracellulaire, tout en maintenant la capacité à se lier à un antigène cible est difficile. L'environnement cytoplasmique réduisant empêche la formation de liaisons disulfures normalement requises pour le pliage stable d'anticorps pleine longueur et des fragments d' anticorps, y compris à chaîne unique fragment variable (scFv) des anticorps 6,7. Un certain nombre d'approches d'évolution dirigée ont été utilisées pour concevoir des anticorps avec haffinités IGH pour cible les antigènes 8-10. Ces approches utilisent couramment phage display, l' affichage de surface de levure ou d' affichage de surface bactérienne pour cribler de grandes banques d'anticorps 11-13. Ces méthodes sont puissantes et efficaces pour identifier des anticorps qui se lient à des cibles, mais ils dépendent de la voie sécrétoire de protéines de transport qui seront affichées 14-16. La voie de sécrétion de protéines dépliées translocation du cytoplasme réducteur dans la lumière du réticulum endoplasmique de la levure ou dans le périplasme dans les bactéries. Les protéines se replient alors dans des conditions d'oxydation et sont affichées sur la surface des cellules ou encapsidés dans des particules de phage pour cribler une affinité de liaison 17,18. Par conséquent, les anticorps isolés à l'aide de ces techniques ne seront pas nécessairement bien plier dans le cytoplasme, et la solubilité intracellulaire doivent souvent être conçus séparément si les anticorps sont utilisés dans des applications intracellulaires.
Améliorerl'efficacité des anticorps d'ingénierie qui sont bien pliés dans le cytoplasme, nous avons précédemment rapporté le succès de MAD-TRAP (affichage à membrane ancrée pour la reconnaissance des protéines associant à base de Tat), un procédé de criblage d' une banque d'anticorps scFv en utilisant Escherichia coli Intérieures affichage de la membrane 19. Affichage interne de la membrane bactérienne repose sur la voie de translocation à double arginine (TAT) pour le transport d'anticorps présentés, contrairement à d'autres méthodes d'affichage commun qui utilisent la voie sécrétoire. La voie Tat contient un mécanisme de contrôle de qualité qui ne permet que des protéines solubles, correctement repliées pour être transportés à partir de E. cytoplasme coli, à travers la membrane interne et dans le périplasme 20,21. Substrats surexprimé Tat (ie., Les protéines ciblées pour la voie Tat avec une fusion N-terminale du peptide signal de Tat ssTorA) qui sont bien plié dans le cytoplasme forment une translocation à long terme intermédiaire à l' extrémité N-terminale in cytoplasme et l' extrémité C-terminale 19 dans le périplasme. Ceci permet l' affichage des substrats de Tat correctement repliés, y compris des fragments d' anticorps, sur la face périplasmique de E. membrane interne coli. Après avoir enlevé la membrane externe par digestion enzymatique pour produire des sphéroplastes, des anticorps sont exposés à l'espace extracellulaire (figure 1). Ceci permet à des substrats de Tat affichés sur la membrane interne devant être criblés pour la liaison à une cible spécifique. Il est important, en exploitant la voie Tat pour l'affichage de la surface cellulaire garantit que seuls les anticorps dans la bibliothèque qui sont bien pliés dans le cytoplasme seront interrogés pour la liaison, ce qui permet l'ingénierie simultanée d'affinité et de pliage intracellulaire de liaison. Dans ce protocole, nous décrivons comment afficher une bibliothèque scFv sur la E. membrane interne coli, pan la bibliothèque contre un antigène cible, et d' effectuer un écran secondaire pour identifier les constituants les plus prometteurs de la bibliothèque. Alors que nous nous concentrons le protocole sur scFv, la méthode pourrait être appliquée à l'ingénierie toute protéine dont l'application nécessite le pliage de liaison et intracellulaire.
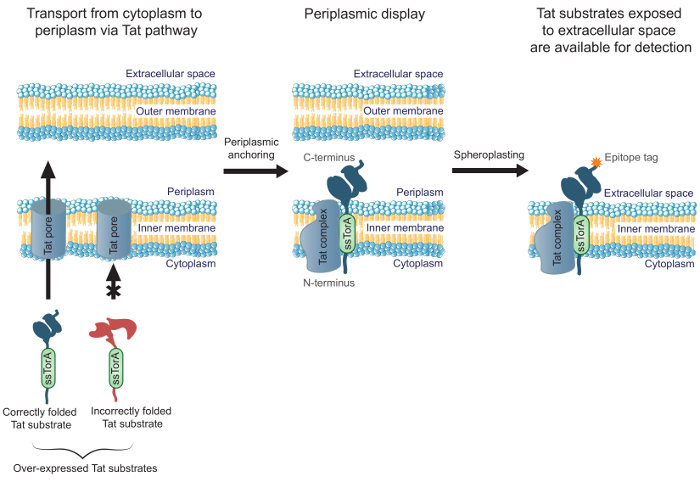
Figure 1. Affichage membrane interne Tat. Dans E. coli, des anticorps scFv exprimés sous la forme d' une fusion avec la séquence signal et ssTorA correctement replié dans le cytoplasme sont transportés à travers la membrane intérieure. Une translocation des formes intermédiaires, où les scFv sont ancrées dans la membrane interne de l'extrémité N-terminale dans le cytoplasme et l'extrémité C-terminale dans le périplasme. E. la membrane externe coli est digéré par voie enzymatique pour former des sphéroplastes, exposant ainsi les anticorps ancrés dans l'espace extracellulaire et leur mise à disposition pour la détection en utilisant un anticorps qui se lie au marqueur fusionné en C-terminal de l' épitope de l'anticorps présenté.charge / 54583 / 54583fig1large.jpg "target =" _ blank "> S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
Protocol
1. Préparer la bibliothèque scFv comme Fusion à la séquence signal ssTorA
- Obtenir une banque d'acide désoxyribonucléique (ADN) contenant des variants d'un gène scFv.
REMARQUE: La bibliothèque peut également être construit en utilisant un mode approprié pour générer de la diversité sur le gène scFv entier ou des domaines ciblés 22 (par exemple, la troisième régions déterminant la complémentarité, CDR3.). - Insérer la banque d'ADN dans le plasmide pimd (figure 2) en utilisant des procédés de clonage moléculaire standard 23.
Remarque: Ce plasmide exprime scFv comme une fusion génétique à la séquence signal ssTorA (N-terminal scFv) et le marqueur d'epitope FLAG (C-terminale du scFv). La conception du plasmide pour l' affichage de la membrane interne a été décrit précédemment 19. Le plasmide pimd est disponible auprès des auteurs.

Figure 2. Inner-membrane affichage plasmide (PIMD) carte (étapes 1.2 à 1.3). Ce plasmide contient un promoteur lac, l' origine ColE1 de réplication, et un gène de résistance au chloramphénicol. Le gène inséré scFv est fusionné à la séquence signal pour cibler ssTorA scFv à la voie Tat et à un marqueur d'epitope FLAG, avec toutes les trois dans le même cadre de lecture. les sites d'enzymes de restriction sont indiqués. Pour une bibliothèque insérée entre les sites d'enzymes de restriction Xbal et Notl, la taille du plasmide est 2219 pb plus la taille du scFv. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
- Transformer l'ADN plasmidique contenant la banque dans MC4100 de E. 23 cellules de E. coli. Récupérer et développer cette forme bactérienne de la bibliothèque. Centrifuger à 4000 xg pendant 15 min à température ambiante pour recueillir les cellules. Retirer le surnageant et remettre en suspension les cellules recueilliesdans 25% de glycérol dans Luria-Bertani (LB) médias. aliquotes Conserver à -80 ° C jusqu'à ce que nécessaire, ou passez à l'étape 2.
NOTE: Le protocole a été vérifié avec des cellules MC4100, bien que d' autres E. souches coli devraient également être compatibles avec le protocole. L'électroporation est la méthode préférée pour la transformation, en raison de sa grande efficacité de transformation. La bibliothèque devrait typiquement être composé d'au moins 10 9 variantes scFv à ce stade, et chaque aliquote doit contenir suffisamment de cellules de telle sorte que la bibliothèque est recouverte de 100 fois.
2. Exprimer la Bibliothèque et Préparer sphéroplastes
- Décongeler une aliquote de la banque bactérienne (étape 1.3) à la température ambiante, et à l'aliquote à un flacon contenant 100 ml de milieu LB avec 20 ug / ml de chloramphenicol (Cm). Développer pendant 3 heures à 37 ° C et à 225 tours par minute dans un agitateur incubé.
- Après 3 heures, enlever le ballon de l'agitateur incubé 37 ° C. Permettre l'expression de la bibliothèque de scFvprocéder O / N pendant 15-22 heures à 20 ° C et à 225 tours par minute dans un agitateur incubé.
REMARQUE: Aucun inducteur est nécessaire lors de l'utilisation du plasmide pimd, comme le promoteur est percé. Notez que les cellules MC4100 ne surexpriment le répresseur Lac (et Lad est introuvable sur le plasmide).

Figure 3. cellules de E. coli et sphéroplastes. (A) E. cellules coli sont de forme cylindrique. (B) Après sphéroplastes à l' aide de l' EDTA et du lysozyme, la membrane externe de E. cellules coli est rompu, et les sphéroplastes résultants sont de forme sphérique. Contraste interférentiel différentiel (DIC) des images de microscopie ont été obtenues en utilisant un objectif 100X sur un microscope inversé. S'il vous plaît clécher ici pour voir une version plus grande de cette figure.
- Préparer les sphéroplastes de la bibliothèque.
REMARQUE: Les sphéroplastes sont formés par la rupture de la membrane externe de E. coli et sont de forme sphérique (Figure 3).- Préparer les tampons nécessaires.
REMARQUE: Tous les tampons doivent être stériles.- Préparer 1 x tampon phosphate salin (PBS; pH 7,4) en dissolvant 8 g de NaCl, 0,2 g de KCl, 1,44 g de Na 2 HPO 4 et 0,24 g de KH 2 PO 4 dans H2O distillée jusqu'à un volume final de 1000 ml. Gardez sur la glace.
- Préparer PBS avec 0,1% (p / v) de sérum-albumine bovine (BSA) en dissolvant 0,2 g de BSA dans 200 ml de 1 x PBS. Gardez sur la glace.
- Préparer le tampon de fractionnement (FB) en mélangeant 7,5 ml d' eau filtrée stérilement 1 M de saccharose, 1 ml de tampon Tris 1 M (pH 8,0) et 1,5 ml d' eau distillée 2 O. Gardez sur la glace.
- Préparer 1 mM d'acide éthylènediaminetétraacétique (EDTA) en ajoutant 30 ul of EDTA 0,5 M à 14,97 ml d'eau distillée H 2 O.
- Préparer 0,5 M MgCl2 par dissolution de 4,76 g de MgCl 2 dans 100 ml de H 2 O distillée Gardez sur la glace.
- Enlever le ballon de l'agitateur, et à mesurer la densité optique (DO) à 600 nm en utilisant un spectrophotomètre pour déterminer la densité cellulaire. Calculer le volume de la culture induite nécessaire de telle sorte que chaque échantillon de sphéroplastes a 1 × 10 10 cellules.
NOTE: L'approximation d'une DO600 de 1 indique une concentration de 10 9 cellules / ml pour E. coli 24 peut être utilisée. - Centrifugeuse le volume calculé de la culture induite dans un tube de 1,5 ml à 12000 × g à température ambiante pendant 5 min. Préparer au moins deux échantillons dans le cas où un problème se pose dans la préparation des échantillons.
- Retirer le surnageant de la culture centrifugée et remise en suspension chaque culot de cellules dans 100 ul de FB glacée. Centrifugeuse à 12.000 ×g à température ambiante pendant 1 min, puis éliminer le surnageant par pipetage. Remettre en suspension chaque culot dans 350 ul de FB glacé supplémenté avec 3,5 ul de 10 mg / ml de lysozyme.
- Chaque tube vortex lentement tout en ajoutant goutte à goutte 700 pi d'EDTA 1 mM, et ensuite incuber les tubes à température ambiante pendant 20 minutes tout en faisant tourner lentement sur un dispositif de rotation de tube pour mélanger les échantillons. Enlever les tubes du dispositif de rotation, ajouter 50 ul de glace froid 0,5 M MgCl2 à chaque tube, et on incube sur la glace pendant 10 minutes. Centrifuger les tubes à 11 000 x g à 4 ° C pendant 10 min.
- Isoler le culot de sphéroplastes.
- Utilisez un micropipette avec une pointe de 1 ml pour tirer lentement partie du culot. Tout en maintenant le tube à un angle avec l'ouverture directement au-dessus d'un nouveau tube de 1,5 ml, soulevez lentement la pointe de la pipette sur le surnageant et faites glisser la pastille dans le nouveau tube.
- Si un volume important de surnageant est transféré dans le nouveau tube, retirez-le par pipetage. Si le culot est pas ferme enough pour transférer, re-centrifuger à 11000 × g pendant 2 min et tentative d'isolement de granulés à nouveau.
- Remettre en suspension le culot de sphéroplastes dans chaque tube 1 ml de glace froid 1 x PBS. Alternez entre pipetage et tourbillonner lentement sur un support de vortex jusqu'à ce que le culot est totalement remis en suspension. Ne pas conserver les échantillons hors de la glace pendant plus de 2 minutes à la fois, et revenir à la glace pendant au moins 5 minutes avant de retirer de la glace à nouveau. Gardez les sphéroplastes à 4 ° C (jusqu'à 2 jours) jusqu'à son utilisation pour le panoramique à l'étape 4.
- Préparer les tampons nécessaires.
3. Immobiliser l'antigène cible sur des billes magnétiques
- Biotinyler l'antigène cible in vivo au cours de la production recombinante dans E. cellules de E. coli. Vous pouvez également utiliser la conjugaison chimique 25 ou antigène cible d'achat qui a déjà été biotinylé et passez à l' étape 3.2.
- Ajouter 816 g bicine à 50 ml d'eau pour faire 10 × bicine tampon. Diluer le buffer à 1 x H 2 O distillée et de la chaleur à 50 ° C. Ajouter 14,7 mg de biotine pour 12 ml de tampon 1 x bicine chauffé pour former une solution de biotine qui est la biotine 5 mM dans 10 mM de tampon bicine. Conserver à -20 ° C jusqu'à leur utilisation.
- Et exprimer biotinyler la protéine cible en utilisant le plasmide pAK400cb-BCCP 26, ce qui permet la production de l'antigène cible en tant que fusion de la protéine porteuse de biotine carboxyle (BCCP).
NOTE: E. cellules de E. coli natif biotinyler BCCP, ce qui élimine la nécessité de purifier et chimiquement biotinyler la protéine cible avant immobilisation sur des billes revêtues de streptavidine. Le natif E. coli , la biotine - ligase BirA est suffisante pour biotinyler la protéine de fusion.- Cultivez E. coli contenant le plasmide de biotinylation (avec l'antigène cible inséré en tant que fusion à l' extrémité N-terminale de la BCCP) O / N pendant 15 à 18 heures dans 5 ml de milieu LB additionné de 20 pg / ml de Cm à 37 ° C tout en agitant à 225 tours par minute.
- Mesurer la DO à 600 nm en utilisant un spectrophotomètre et calculer le volume de la culture nécessaire (V add) à la sous - culture à une DO de départ de 0,05 à 25 ml de milieu LB frais avec 20 pg / ml cm en utilisant l'équation: V ajouter = (0,05 × 25 ml) / (DO 600 à 0,05), où OD 600 est la densité optique de la culture O / N et V add est le volume de la culture O / N à ajouter à la LB. frais La sous-culture et de croître jusqu'à une DO de 0,5 à 0,8 dans un agitateur mis à incuber à 37 ° C et à 225 tours par minute.
- Ajouter β-D-thiogalactopyranoside d'isopropyle 1 à une concentration finale de 100 pM et de la biotine à une concentration finale de 5 uM. Induire l'expression dans un agitateur à incuber pendant 15-22 heures à 20 ° C et à 225 tours par minute.
- Récolte des bactéries par centrifugation à 4000 x g à 4 ° C pendant 10 min. Retirer le surnageant. Conserver le culot à -20 ° C jusqu'à utilisation.
- Ajouter1 ml d'un détergent de lyse cellulaire par 0,2 g de culot cellulaire. Remettre en suspension par pipetage et faire tourner doucement pendant 20 minutes pour lyser les cellules. Après la lyse, centrifuger à 16 000 x g et 4 ° C pendant 20 min. Pipeter le lysat soluble (surnageant) dans un nouveau tube de 1,5 ml.
- Utilisez une colonne de poids moléculaire de coupure de 3 kDa pour éliminer la biotine non liée. Pipeter le lysat dans la colonne, et centrifuger à 20 ° C selon les instructions du fabricant. Laver avec du PBS 1 x jusqu'à ce que la biotine dans le lysat a été dilué 100 fois, et le volume du lysat lavé est égal au volume initial du lysat. Transférer le lysat dans un nouveau tube.
- Immobiliser l'antigène cible biotinylé sur des billes magnétiques revêtues de streptavidine.
- Préparer de 1 x PBS et 1 x PBS avec 0,1% (p / v) de BSA comme décrit dans l'étape 2.3.1.
- Préparer les billes magnétiques.
REMARQUE: Ceci nécessite l'utilisation d'une grille de séparation magnétique.- Resuspendre streptavidin revêtu des billes magnétiques dans leur flacon d'origine. Soit vortex pendant au moins 30 secondes ou tourner pendant 5 min.
- Transférer 7 à 10 × 10 9 billes dans un tube de 1,5 ml.
REMARQUE: le volume requis dépendra de la concentration de billes fourni par le fabricant. - Placer le tube contenant les perles sur le porte-aimant pendant 2 minutes pour collecter les perles sur le côté du tube. Avec le tube toujours sur l'aimant, retirez délicatement le surnageant par pipetage sans perturber les perles.
- Pour laver, retirer le tube de l'aimant, et remettre en suspension les billes dans 1 ml de 1 x PBS par pipetage sans générer des bulles. Retour le tube à l'aimant pendant 2 minutes pour recueillir les perles, et retirer délicatement le surnageant par pipetage. Répétez le processus deux fois de plus pour un total de trois lavages. Veiller à ce qu'aucun liquide est laissé dans le tube après le lavage final.
- Ajouter le lysat contenant l'antigène biotinylé au bea magnétiqueds.
- Retirer le tube de l'aimant et remettre en suspension les billes dans 1 ml de lysat (étape 3.1.5). Incuber à température ambiante pendant 30 minutes tout en faisant tourner doucement.
- Placer le tube sur l'aimant pendant 3 min pour recueillir les billes revêtues d'antigène. Laver les billes revêtues cinq fois avec 1 x PBS avec 0,1% de BSA de la même manière que celle décrite dans les étapes 3.2.2.3 à 3.2.2.4. Après le lavage final, la remise en suspension des billes dans 1 x PBS avec 0,1% de BSA au même volume utilisé dans l'étape 3.2.2.2.
- Si l'antigène cible immobilisé est stable à 4 ° C, de stocker les billes revêtues à 4 ° C jusqu'à ce que nécessaire pour un balayage panoramique. Sinon, passez à l'étape 4.
4. Écran de la Bibliothèque scFv par Panning contre l'antigène cible (Figure 4)

Figure 4. Panning (étape 4). Antigen-billes magnétiques recouvertes are incubée avec des sphéroplastes exprimant des variants de la bibliothèque d'anticorps. L'ADN plasmidique de sphéroplastes liées aux billes est récupérée et utilisée pour générer une sous-banque qui est criblée en utilisant l'écran secondaire à base d'ELISA. Correspondantes étapes de protocole sont notées. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
- Incuber les billes revêtues avec sphéroplastes.
- Utiliser un sphéroplastes à perler rapport d'environ 5: 1. Ajouter 4 × 10 9 sphéroplastes et 8 × 10 8 perles dans un tube de 15 ml stérile.
NOTE: Supposons que pas de cellules ont été perdus au cours du processus de sphéroplastes, de sorte que la concentration est encore 1 × 10 10 sphéroplastes / ml. - Ajouter 1 x PBS avec 0,1% de SAB pour porter le volume total à 4 ml. Aliquoter en quatre tubes de 1,5 ml avec 1 ml chacun. Incuber les réactions à 4 ° C pendant 5 heures tout en tournant doucement.
- Utiliser un sphéroplastes à perler rapport d'environ 5: 1. Ajouter 4 × 10 9 sphéroplastes et 8 × 10 8 perles dans un tube de 15 ml stérile.
- Prepare sphéroplastes liées aux billes pour une réaction en chaîne par polymérase (PCR).
- Placer les tubes de réaction panning sur l'aimant pendant 3 min. Retirer le surnageant par pipetage, et laver les sphéroplastes liées aux billes quatre fois avec de la glace froide 1 × PBS avec 0,1% de BSA de la même manière que celle décrite dans les étapes 3.2.2.3 à 3.2.2.4. Resuspendre les sphéroplastes liées aux billes dans chaque tube dans 25 pi d'eau distillée 2 O. Stocker les perles à -20 ° C ou passez à l'étape 4.3.
- Effectuer l'ensemble du plasmide PCR sur sphéroplastes liées aux billes pour amplifier les plasmides contenant les gènes de scFv liées aux billes.
- Obtenir des amorces avec les séquences suivantes: 5'CCAACTCTTTTTCCGAAGGTAACTG3 '(amorce directe) et 5'TAGCTCTTGATCCGGCAAACAAA3' (amorce inverse).
NOTE: Ce sera lier bout à bout sur des brins opposés du plasmide pimd (figure 2) et sont conçus pour recuire à une caractéristique commune de pimd, donc l' amplification se produit quel que soit levariante séquence scFv. - Phosphoryler les amorces.
Remarque: l'absence de phosphorylation, re-ligature ne se produit. Les amorces peuvent également être commandés avec 5'-phosphorylation, plutôt que d'utiliser cette méthode de phosphorylation dans ce protocole.- Dans un tube de 0,5 ml, mettre en place une réaction de phosphorylation pour l'amorce de PCR vers l' avant décrite dans le tableau 1. Répétez ce processus pour l'amorce inverse.
- Incuber les réactions à 37 ° C pendant 1 heure. Puis incuber à 65 ° C pendant 20 minutes pour désactiver le polynucleotide T4 kinase (PNK). Stocker les amorces phosphorylées à -20 ° C.
- Effectuer PCR.
- Dans un tube de PCR, préparer la réaction de PCR telle que décrite dans le tableau 2.
NOTE: Les réactions multiples peuvent être préparés pour un rendement plus élevé. Les sphéroplastes liées aux billes non utilisés peuvent être conservés à -20 ° C. - Chauffer les réactions de PCR à 98 ° C pendant 15 minutes dans un cycleur thermique pour assurer la pleine lyse des sphéroplastes. Retirer les tubes du cycleur thermique, et d'ajouter 0,5 ul d'une polymerase haute fidélité à chacun. Tubes Retour au cycleur thermique et exécuter en utilisant le programme détaillé dans le tableau 3.
- Réunir les produits de PCR, le cas échéant. Conserver à -20 ° C ou passez à l'étape 4.4.
- Dans un tube de PCR, préparer la réaction de PCR telle que décrite dans le tableau 2.
- Obtenir des amorces avec les séquences suivantes: 5'CCAACTCTTTTTCCGAAGGTAACTG3 '(amorce directe) et 5'TAGCTCTTGATCCGGCAAACAAA3' (amorce inverse).
Tableau 1. PNK réaction de phosphorylation (étape 4.3.2.1).
| Réactif | Volume (ul) |
| H2O distillée | 15 |
| un tampon de réaction de ligase T4 ADN 10x | 2 |
| 100 pM amorce | 2 |
| T4 Polynucleotide kinase (PNK) | 1 |
| Réactif | Volume (ul) |
| H2O distillée | 28,5 |
| tampon 5x haute fidélité de la polymérase | dix |
| amorce sens 10 uM phosphorylée | 2.5 |
| amorce antisens 10 uM phosphorylée | 2.5 |
| 40 mM dNTP mix (10 mM de chaque dNTP) | 1 |
| sphéroplastes de perles assorties | 5 |
Programme Tableau 3. PCR (étape 4.3.3.2).
| Étape | Température (° C) | Temps (min: sec) | Nombre de cycles |
| denature initiale | 98 | 00h30 | 1 |
| Dénaturer | 98 | 00h10 | 35 |
| recuit | 69 | 00h30 | |
| Extension | 72 | 0:30 par kb | |
| L'extension finale | 72 | 06h00 | 1 |
| Tenir | 12 | Infini | 1 |
- Re-circulariser les produits entiers plasmidique PCR, et utiliser le produit ligaturé pour transformer MC4100 E. cellules de E. coli.
- On purifie le produit PCR en exécutant la réaction de PCR sur un gel d'agarose à 23, la coloration de l'ADN dans le gel 23, et en utilisant unkit de nettoyage de gel pour purifier le plasmide linéarisé en suivant les instructions fournies par le fabricant. Mesurer la concentration à l'aide d'un spectrophotomètre à 260 nm. Stocker le fragment purifié à -20 ° C jusqu'à ce que nécessaire, ou passez à l'étape 4.4.2.
- Re-circulariser le plasmide à partir du produit de PCR.
- Pour empêcher la ligature intermoléculaire du produit de PCR, effectuer la réaction de ligature avec une faible concentration 27 de 1 ng / pl de produit PCR. Calculer le volume nécessaire pour la préparation d'une réaction de ligature 800 ul à cette concentration.
- Préparer la réaction de ligature sur la glace. Dans un tube, ajouter le volume calculé à l' étape 4.4.2.1 du produit de PCR, 80 ul de tampon de ligase 10X de l' ADN et H2O distillée jusqu'à 800 ul. Ajouter 4 ul d'ADN ligase de T4, et placer immédiatement les tubes à 16 ° C dans un bain d'eau ou thermocycleur. Incuber à 16 ° CO / N pour 14 à 18 ans h. Stocker les réactions de ligature terminé à -20 ° Cjusqu'à ce que nécessaire, ou passer à l'étape 4.4.3.
- Placer la réaction de ligature sur un bloc chauffant à 65 ° C pendant 15 min à la chaleur inactiver l'ADN ligase. Ensuite, utilisez un kit de membrane de microdialyse ou de l'ADN de nettoyage pour dessaler l'ADN ligaturé. Conserver à 20 ° C ou passez à l'étape 4.4.4.
- Utilisez le, produit toute inactivé à la chaleur de-salé ligature pour transformer MC4100 E. 23 cellules de E. coli. Préparer les stocks de glycérol, tel que décrit à l'étape 1.3, des cellules contenant le sous-banque batée résultant, et aliquotes stocker à -80 ° C.
- Répétez l'étape 4 dans son intégralité à l'aide d'une aliquote de l'étape 4.4.4 pour faire une deuxième panoramique sur la sous-banque.
REMARQUE: Un deuxième panoramique permet d' enrichir pour les constituants de bibliothèque qui se lient bien à l'antigène cible 19.
5. Effectuer un écran secondaire à l'aide d'un dosage immunoenzymatique Méthode enzymatique liée à l'identification des clones prometteurs pour une caractérisation (Figure 5) </ P>

Dépistage ELISA de Figure 5. secondaire (étape 5). Variantes (A) de la bibliothèque de la sous - bibliothèque enrichie pendant le panoramique sont inoculées dans des puits individuels d'une plaque de culture pour la croissance et l' expression. (B) Une plaque ELISA revêtue d' un antigène cible. (C) Les variantes de la bibliothèque sont criblées en utilisant l'écran secondaire à base d'ELISA décrit dans le protocole. Lors de l'analyse des données obtenues à partir de l'écran secondaire, des variantes d'intérêt sont sélectionnés et caractérisés davantage. Correspondantes étapes de protocole sont notées. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
- Décongeler un tube de la sous-banque batée (de l'étape 4.4.4) et de la plaque sur des plaques d'agar LB. Plate plusieurs dilutionsà des concentrations suffisamment faibles pour assurer des colonies individuelles (par exemple, 10 2 - 10 6 dilutions -fold). Incuber les plaques pendant 15 à 18 heures à 37 ° C. Stocker les plaques à 4 ° C ou passez à l'étape 5.2.
- Culture et induire des colonies de la sous-banque batée. Effectuer toutes les étapes dans des conditions stériles. Utilisez une pipette multicanaux pour les étapes impliquant des plaques à 96 puits.
- Ajouter 200 pi de LB avec 20 ug / ml cm dans chaque puits d'une plaque à 96 puits de culture à fond rond.
- Choisissez une colonie individuelle de la plaque de gélose avec une pointe de pipette, placer la pointe dans le premier puits de la plaque de 96 puits, et remuer doucement pour inoculer. Utiliser un nouvel embout pour chaque puits. Inoculer une colonie dans chaque puits. En tant que témoin, comprennent au moins un contrôle de stérilité bien avec aucune colonie inoculées.
- Répétez les étapes 5.2.1 et 5.2.2 pour inoculer plusieurs plaques à 96 puits.
- Placer les plaques à 96 puits sur un agitateur de microplaque à 310 tours par minute. Incuber à 376; C pendant 20 à 24 heures pour exprimer les scFv.
- Pour chaque plaque de culture préparé à l'étape 5.2, manteau une plaque à 96 puits ELISA avec l'antigène cible.
- Diluer l' antigène cible purifié à une concentration appropriée (par exemple, 1 pg / ml à 4 mg / ml) dans 1 x PBS pour rendre la solution de revêtement. Faire 5 ml d'une solution de revêtement pour chaque plaque à 96 puits.
NOTE: La concentration appropriée dépend de l'antigène spécifique utilisé et peut être nécessaire d'ajuster. - Ajouter 50 ul de la solution de revêtement pour chaque puits d'une plaque de polystyrène clair 96 puits à haute liaison ELISA. Tapoter doucement la plaque sur la surface de la paillasse pour assurer que toute la surface de chaque puits est revêtue. Répétez l'opération pour chaque plaque. Incuber les plaques à 4 ° CO / N.
- Diluer l' antigène cible purifié à une concentration appropriée (par exemple, 1 pg / ml à 4 mg / ml) dans 1 x PBS pour rendre la solution de revêtement. Faire 5 ml d'une solution de revêtement pour chaque plaque à 96 puits.
- Répliquer les colonies provenant des plaques de culture à 96 puits sur des plaques d'agar-agar.
- Placez un polystyrène réplicateur stérile dans les puits d'une plaque de culture pour recueillir une petite quantité de savon mouid. Soulevez avec précaution le réplicateur et le transfert à une plaque de gélose LB 15 cm de telle sorte que tous les conseils sont en contact avec la plaque. Une fois que le liquide a transféré, soulevez le réplicateur directement vers le haut. Répétez l'opération pour chaque plaque de culture.
- Étiquette de la plaque de gélose avec l'orientation correcte de sorte que les résultats de l'écran secondaire dans la plaque de 96 puits peuvent être jumelés à la colonie répliquée correcte sur la plaque, si une caractérisation plus poussée est souhaitée. Cultivez à 37 ° C pendant 15 à 18 heures, puis stocker à 4 ° C jusqu'à ce que nécessaire.
- Effectuer l'écran secondaire ELISA.
- Préparer la solution de blocage en faisant 2% (p / v) de lait en poudre dans 1 x PBS. Vider la solution de revêtement des plaques ELISA. Ajouter 100 ul de la solution de blocage dans chaque puits. Incuber à température ambiante pendant au moins 2 heures, ou le bloc O / N à 4 ° C.
- Préparer le tampon de lavage en ajoutant le polysorbate 20 à une concentration finale de 0,05% dans 1 x PBS. Faire 250 ml par plaque ELISA.
- Ajouter 20 ul d'uneconcentré détergent lyse cellulaire à chaque puits de la plaque de culture à fond rond et incuber la plaque de culture sur un agitateur de microplaque à température ambiante pendant 15 à 20 min. Commencer la lyse en même temps que le blocage des plaques ELISA est terminée de sorte que la lyse et le lavage à l'étape 5.5.4 peuvent être effectuées simultanément.
- Vider la solution de blocage des plaques ELISA. Laver les plaques ELISA bloqués quatre fois avec 200 pi de tampon de lavage par puits par lavage. Vider le tampon de lavage dans les puits.
- Transfert de 50 pi de chaque puits de la plaque de lyse cellulaire au puits correspondant de la plaque ELISA, en utilisant une nouvelle astuce pour chaque puits. Incuber la plaque ELISA à température ambiante pendant 1 ou 2 heures.
- Préparer la solution d'anticorps pour détecter les scFv liés.
- Utiliser une peroxydase de raifort (HRP) conjugué à un anticorps primaire qui se lie au marqueur épitopique FLAG fusionné au scFv de la bibliothèque.
- Diluer l'anticorps à la dilution appropriée à utiliser dans un test ELISA (voir le fournisseur & #39; s recommandations) à 2% (p / v) de lait sec dans 0,05% de polysorbate 20 dans 1 x PBS. Préparer 5 ml pour chaque plaque.
- Laver les plaques ELISA quatre fois avec le tampon de lavage tel que décrit à l'étape 5.5.4.
- Ajouter 50 ul de la solution d'anticorps dans chaque puits de la plaque ELISA. Incuber pendant 2 heures à 1 à la température ambiante.
- Préparer le substrat HRP en dissolvant o phénylènediamine dichlorhydrate (OPD) comprimés dans H 2 O distillée selon le protocole du fabricant tout en évitant la lumière. Préparer 20 ml par plaque ELISA.
- Préparer 3 MH 2 SO 4 par dilution de H 2 SO 4 concentré avec H2O distillée, si nécessaire. Préparer 5 ml par plaque ELISA.
Attention: H 2 SO 4 est un acide fort. Assurez-vous de porter un équipement de protection individuelle approprié. - Laver les plaques ELISA à quatre reprises avec du tampon de lavage, comme décrit dans l'étape 5.5.4.
- Incuber les plaques ELISA avec le SUBSTRA HRPte.
- Ajouter 200 pi de substrat de HRP dans chaque puits. Pour réduire au minimum l'exposition lumineuse, ajouter le substrat à une plaque ELISA à la fois, et l'envelopper de papier d'aluminium avant de passer à la plaque suivante. Incuber les plaques pendant 30 à 60 min à température ambiante dans l'obscurité.
- Après les 30 premières minutes, vérifier les plaques d'assombrissement du substrat, et incuber plus si nécessaire pour visualiser le développement de la couleur.
- Ajouter 50 ul de 3 MH 2 SO 4 à chaque puits pour arrêter la réaction. En utilisant une pointe différente pour chaque puits, mélanger la solution dans les puits par pipetage de haut en bas, sans faire mousser. Par souci de cohérence et d'éviter la saturation, ajouter le H 2 SO 4 rapidement et soigneusement à l' ensemble des plaques ELISA avant de mélanger la solution pour chaque plaque.
- Mesurer l'absorbance de la solution dans les puits de chaque plaque à 492 nm en utilisant un lecteur de plaque.
- Analyser les données d'absorbance pour identifier scFv variantes que exhibil promet des signaux de liaison et de caractériser ces scFv prometteurs. Sélectionnez scFv qui présentent des signaux d'absorbance plus élevées que le signal de fond et plus élevé que le signal moyen sur chaque plaque.
NOTE: Le niveau d'absorption dépendra des propriétés de l'antigène et de l'anticorps anti-FLAG utilisés, ainsi que la force des variantes scFv qui ont été isolées dans le criblage.
Representative Results
Le pliage de la protéine mécanisme de contrôle de la qualité intracellulaire de la voie Tat chez E. coli limite le transport à travers la membrane cellulaire interne à des protéines qui sont bien pliées dans l'environnement cytoplasmique réducteur. En surexprimant une fusion d'un scFv à la séquence de signal ssTorA (la séquence signal de la protéine TorA, qui est naturellement transportées par la voie Tat 20), la translocation est bloqué, ce qui entraîne l' affichage de scFv sur la membrane interne 19. Après rupture enzymatique de la membrane externe, les anticorps affichés sont mis à disposition pour le criblage pour l'activité de liaison à l'antigène. La capacité à tirer parti de la voie Tat pour l' affichage scFv a été montré par Karlsson et al. 19 (Figure 6). Les anticorps scFv scFv13 et scFv13.R4 ont été fusionnés soit la séquence ssTorA native ou un ssTorA modifié qui n'a pas la paire de résidus arginine-arginine reconnu parla voie Tat. scFv13.R4 a été conçu par Martineau et al. de scFv13 grâce à quatre tours de l' évolution dirigée et est connu pour bien plier dans le cytoplasme 9. Ce scFv a été affiché sur la membrane intérieure, mais seulement lorsqu'il est exprimé en tant que fusion à la séquence native de signal ssTorA (figure 6). Au contraire, scFv13 est pas bien plié cytoplasme 9, donc il est pas bien affiché sur la membrane interne, quelle que soit la séquence de signal auquel il est fusionné. En outre, si les scFv ont été exprimés dans des cellules qui ne disposaient pas de la protéine TATC, une composante essentielle de la machine Tat 20,28, l' affichage n'a pas été observée, montrant le lien important entre l' affichage membrane interne et la voie Tat. Ces résultats montrent que seules les protéines qui contiennent le peptide signal Tat et qui sont correctement repliées dans le cytoplasme sont affichés sur la membrane intérieure, ce qui permet le transport à travers la voie Tat pour fonctionner comme un écran pour fol intracellulaireding.

Figure 6. Détection de scFv affichés sur la membrane interne. Cytométrie de flux analyse a été effectuée pour détecter l'affichage de mal plié scFv13 et bien plié scFv13.R4 sur la membrane interne. scFv ont été fusionnés à ssTorA native ou ssTorA (KK), où la paire Arg-Arg dans la séquence a été modifiée pour ssTorA Lys-Lys. Les balises C-terminal épitope FLAG sur les scFv ont été détectés avec un isothiocyanate de fluorescéine (FITC) anticorps anti-FLAG. Les cellules sans la protéine TATC (ΔtatC) et ssTorA-scFv13 sans l'étiquette FLAG ont été testés comme témoins. M indique la valeur médiane de la fluorescence. Reproduit de référence 19 avec la permission. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
19. Pour le démontrer, une bibliothèque PCR sujette aux erreurs sur la base de scFv13, qui a un niveau d'affinité de liaison pour les β-galactosidase faible, a été éreinté contre l'antigène cible β-galactosidase en utilisant l'affichage et de panoramique méthode décrite dans le protocole. scFv 1-4 a été isolé après un tour de mutagenèse et de panoramique, et présentait une affinité de liaison supérieure à la ß-galactosidase que scFv13 (figure 7A) et un niveau plus élevé de solubilité cytoplasmique (figure 7B).
Une nouvelle bibliothèque, sur la base de scFv 1-4, a été faite par PCR sujette à l'erreur, et le panoramique de cette bibliothèque de deuxième génération contreβ-galactosidase a été effectuée en utilisant une modification du protocole décrit. Le panoramique contre β-galactosidase pour le second tour de l'évolution a été faite en présence de purifiée, soluble scFv 14 comme un concurrent pour améliorer la probabilité d'isoler des clones avec une affinité supérieure scFv 1-4. Après ce deuxième cycle de mutagenèse et de panoramique, scFv 2-1 et 2-3 scFv ont été isolés en utilisant le criblage secondaire à base d'ELISA. Ces scFv non seulement présentent une affinité de liaison plus élevée pour les β-galactosidase que scFv13, mais aussi présenté une meilleure liaison que le clone de première ronde scFv 1-4. scFv 01/02 présentait β-galactosidase de liaison comparable à celle de scFv13.R4 (figure 7A). scFv 2-3 montre également une nouvelle augmentation de la solubilité cytoplasmique par rapport à scFv 14, mettant en évidence l'ingénierie simultanée de solubilité et de liaison d'antigène. Etant donné que l'affinité et l'expression soluble des scFv sont criblés pour en même temps, il est possible qu'un scFv sélectionné a modnérer solubilité, mais haute versa liaison ou vice. Par exemple, scFv 01/02 a l'expression soluble inférieure à celle scFv 2-3, mais il présente une affinité de liaison supérieure à la ß-galactosidase.

Figure 7. Cible de liaison et d' expression cytoplasmique de scFv variants isolés en utilisant l' affichage membrane interne. (A) scFv ont été exprimées dans le cytoplasme de E. coli (par exemple des cellules., sans la séquence signal ssTorA) avec une hexahistidine (6 × -Son) tag et purifiée en utilisant l' acide spin-colonnes de nickel-nitrilotriacétique. La liaison du scFv purifiées à la ß-galactosidase a été mesurée par un test ELISA. scFv purifiées ont été chargés sur des plaques ELISA β-galactosidase revêtues et les scFv liés ont été détectés avec un anticorps anti-6 x His. Les données sont une moyenne de six répétitions, et la barre d'erreur indique l'erreur standard de la moyenne.(B) Les fractions solubles et insolubles des lysats cellulaires provenant de cellules exprimant scFv cytoplasmique ont été analysés par Western blot sondé avec un anticorps anti-6 x His. La concentration totale des protéines a été utilisée pour normaliser le chargement des échantillons. Reproduit (A) et adapté (B) de référence 19 avec la permission. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
Discussion
Ingénierie des anticorps pour l' activité cytoplasmique est une tâche difficile en raison du milieu réducteur du cytoplasme, ce qui empêche la formation de liaisons disulfure stabilisant à 6,7. Cela provoque la plupart des anticorps pour être inactif dans le cytoplasme, sauf si elles sont conçues pour assurer la stabilité et la solubilité dans le cytoplasme, en plus d'être conçu pour une affinité de liaison. Les méthodes de phage display, affichage de la surface bactérienne, et les méthodes d'affichage de surface de levure existantes tout utiliser la voie sécrétoire 14-16 pour l'affichage des anticorps génétiquement modifiés , mais ces méthodes ont aucun moyen de pliage ingénieur intracellulaire. Anticorps modifiés génétiquement en utilisant l'affichage membrane interne ont permis d'améliorer la stabilité et la solubilité dans cytoplasmique parce que le contrôle de la qualité du repliement de la voie Tat empêche la translocation des anticorps qui sont mal repliées et instables dans le cytoplasme. Cette méthode simplifie le processus itératif d'anticorps intracellulaires d'ingénierie pour l'affinité d'unnd solubilité, comme les deux propriétés sont conçues en une seule étape. Bien que ce procédé a été conçu pour l'ingénierie des anticorps ayant une solubilité dans l'environnement intracellulaire réducteur, il pourrait également être appliquée à des anticorps d'ingénierie pour fonctionner dans des conditions non réductrices, étant donné que les protéines conçues en utilisant cette méthode conservent leur pliage dans l'environnement oxydant du périplasme.
Bien que cette technique simplifie le processus d'ingénierie des anticorps avec une affinité élevée et une haute solubilité cytoplasmique, plusieurs limitations sont importants à considérer lors de l'utilisation de ce protocole. Lors de l'analyse des signaux ELISA écran secondaire pour identifier des variants prometteurs scFv, le seuil pour discerner entre les variantes potentiellement intéressantes et celles qui ne peuvent pas exposer adéquate liaison à l'antigène ne sont pas susceptibles d'être apparent qu'après plusieurs clones ont été caractérisés davantage. Il est important de rechercher une liaison améliorée sur l'anticorps parent; cependant,un signal anormalement élevé pourrait être le signe de l' avidité 29 ou des effets d'agrégation 30, un défi qui est pas unique à l'approche de criblage d'affichage membrane interne. Une limitation clé à retenir lors de l'utilisation de ce protocole est l'incapacité de récupérer sphéroplastes après le panoramique, car ils sont non-viables (données non publiées). Cela nécessite l'amplification de l'ADN et de transformation des mesures pour récupérer les plasmides codant pour les anticorps.
Plusieurs étapes critiques du protocole permettent l'ingénierie simultanée de pliage et de liaison des anticorps. Pour le dépistage, pour réussir, la bibliothèque scFv projeté doit être exprimé comme une fusion au signal peptide ssTorA. Sans cette séquence, les anticorps ne seront pas dirigés vers la voie Tat et ne seront donc pas translocation vers le périplasme 19. En outre, il est impératif qu'un marqueur d'épitope C-terminale est fusionnée à des anticorps pour permettre la détection des anticorps présentés dans le bacdosages ding. Il est clair que E. coli utilisé pour exprimer les scFv doit également avoir la machinerie de la voie Tat nécessaire, mais cela est vrai du E. couramment utilisé souches de E. coli.
Les modifications apportées à ce protocole sont possibles pour améliorer son potentiel pour isoler des anticorps avec les caractéristiques souhaitées. Une étape de balayage panoramique soustractive peut être complété avant lavage contre l'antigène cible pour épuiser la bibliothèque scFv des constituants non désirés. Les sphéroplastes de la bibliothèque peuvent être incubées avec des billes magnétiques revêtues d'BCCP seul ou enduit d'une protéine non désirée et les sphéroplastes qui se lient à ces billes peuvent être éliminés avant le criblage des sphéroplastes non liés restants pour se lier à la cible souhaitée. Comme indiqué dans les résultats représentatifs, un procédé pour améliorer l'affinité d'un scFv isolé est d'inclure un concurrent soluble dans la réaction de panning pour rivaliser avec les scFv affichées sur les sphéroplastes. Parce que l'échantillon solubleetitor est une protéine purifiée, aucun ADN est amplifié à partir d'elle, de sorte que seules des séquences de scFv présentées sur les sphéroplastes seront récupérés dans la réaction PCR. En outre, cette méthode pourrait être étendue à d'autres types d'ingénierie d'anticorps ou de protéines de liaison non-anticorps.
E. affichage membrane interne coli est une plate - forme puissante pour les anticorps d'ingénierie avec une affinité élevée et des niveaux élevés de solubilité intracellulaire. Cette méthode est particulièrement adaptée pour l'ingénierie efficace des anticorps conçus pour fonctionner dans l'environnement intracellulaire. Ces anticorps intracellulaires sont déjà explorés en tant qu'agents thérapeutiques potentiels dans un certain nombre de domaines, y compris les maladies neurodégénératives, le cancer et les infections virales 31. Cette technique permettra une utilisation plus répandue des anticorps intracellulaires comme outils de recherche et de la médecine dans ces domaines et dans tout autre domaine où l' étude d' une protéine cible in situ est souhaitée.
Materials
| Name | Company | Catalog Number | Comments |
| scFv library | Varies | A suitable scFv library should be obtained from a commercial or academic source. | |
| MC4100 E. coli cells | Coli Genetic Stock Center | 6152 | Cells need to be chemically competent or electrocompetent, depending on the selected transformation method. |
| Glycerol | Fisher Scientific | BP229-4 | |
| Difco dehydrated culture media LB Broth, Miller (Luria-Bertani) | BD | 244610 | |
| Chloramphenicol (Cm) | Fisher Scientific | BP904-100 | |
| Sodium chloride (NaCl) | Fisher Scientific | BP358-1 | |
| Potassium chloride (KCl) | Fisher Scientific | BP366-500 | |
| Sodium phosphate, dibasic (Na2HPO4) | Fisher Scientific | BP332-500 | |
| Potassium phosphate, monobasic (KH2PO4) | Fisher Scientific | BP362-500 | |
| Bovine serum albumin (BSA) | Fisher Scientific | BP9706-100 | |
| Sucrose | Fisher Scientific | BP220-1 | |
| Tris base | Fisher Scientific | BP1521 | |
| Ethylenediaminetetraacetic acid (EDTA), 0.5 M | Fisher Scientific | BP2482-500 | |
| Magnesium chloride (MgCl2) | Fisher Scientific | BP214-500 | |
| Lysozyme | Sigma Aldrich | L3790-10X1ML | |
| Vortex mixer | VWR | 97043-564 | |
| Bicine | Fisher Scientific | BP2646100 | |
| D-Biotin | Fisher Scientific | BP232-1 | |
| Isopropyl β-D-1-thiogalactopyranoside | Fisher Scientific | BP1755-1 | |
| BugBuster Master Mix (cell lysis detergent) | EMD Millipore | 71456 | |
| Vivaspin 2 MWCO, 3,000 daltons | GE Healthcare Sciences | 28932240 | |
| Target antigen | Varies | N/A | Purified target antigen may be purchased or produced/purified. |
| Dynabeads MyOne Streptavidin T1 | Invitrogen | 65601 | |
| Dynamag-2 magnet | Invitrogen | 12321D | |
| Tube rotator | VWR | 13916-822 | |
| PCR primers | IDT | N/A | Primer sequences are as described in the protocol. |
| 10x T4 DNA ligase reaction buffer | New England BioLabs | B0202S | |
| T4 Polynucelotide kinase (PNK) | New England BioLabs | M0201S | Make sure the T4 ligase buffer used in the primer phosphorylation reaction contains 1 mM ATP. |
| 5x Phusion HF buffer pack | New England BioLabs | B0518S | |
| Deoxynucleotide (dNTP) solution mix, 10 mM each dNTP | New England BioLabs | N0447L | |
| Phusion DNA polymerase | New England BioLabs | M0530S | Other high-fidelity polymerases may be used as an alternative, but the annealing temperature in Table 3 must be adjusted. |
| C1000 Touch thermal cycler with dual 48/48 fast reaction module | Bio-Rad | 185-1148 | |
| Agarose | Promega | V3121 | |
| SYBR Safe DNA gel stain | Invitrogen | S33102 | |
| Wizard SV gel and PCR clean-up system | Promega | A9281 | |
| T4 DNA ligase | New England BioLabs | M0202S | |
| Microdialysis membrane filter | EMD Millipore | VSWP04700 | |
| Agar | BD | 214030 | |
| 96-well polystyrene round-bottom cell culture plates | VWR | 10062-902 | |
| Costar general polystyrene assay plate lids | Corning | 3931 | |
| Microtitre plate shaker | VWR | 12620-926 | |
| Costar 96 well EIA/RIA Easy Wash clear flat bottom polystyrene high bind microplate | Corning | 3369 | |
| Bel-blotter polycarbonate 96-well replicating tool | Bel-Art Products | 378760002 | |
| Instant nonfat dry milk | Quality Biological | A614-1000 | |
| Tween 20 (polysorbate 20) | Fisher Scientific | BP337-500 | |
| PopCulture reagent (concentrated cell lysis detergent) | EMD Millipore | 71092-3 | |
| Monoclonal ANTI-FLAG M2-Peroxidase(HRP) antibody produced in mouse | Sigma Aldrich | A8592 | |
| SigmaFast OPD | Sigma Aldrich | P9187-50SET | |
| Sulfuric acid (H2SO4), 10 N solution | Fisher Scientific | SA200-1 | |
| Reynolds Wrap aluminum foil | VWR | 89079-075 | |
| BioTek Epoch microplate spectrophotometer | Fisher Scientific | 11120570 |
References
- Biocca, S., Pierandrei-Amaldi, P., Campioni, N., Cattaneo, A. Intracellular immunization with cytosolic recombinant antibodies. Biotechnology (NY). 12 (4), 396-399 (1994).
- Chen, S. Y., Bagley, J., Marasco, W. A. Intracellular antibodies as a new class of therapeutic molecules for gene-therapy. Hum. Gene Ther. 5 (5), 595-601 (2008).
- Gargano, N., Biocca, S., Bradbury, A., Cattaneo, A. Human recombinant antibody fragments neutralizing human immunodeficiency virus type 1 reverse transcriptase provide an experimental basis for the structural classification of the DNA polymerase family. J Virol. 70 (11), 7706-7712 (1996).
- Mhashilkar, A. M., et al. Inhibition of HIV-1 Tat-mediated LTR transactivation and HIV-1 infection by anti-Tat single chain intrabodies. Embo J. 14 (7), 1542-1551 (1995).
- Strube, R. W., Chen, S. Y. Characterization of anti-cyclin E single-chain Fv antibodies and intrabodies in breast cancer cells: enhanced intracellular stability of novel sFv-F-c intrabodies. J. Immunol. Meth. 263 (1-2), 149-197 (2002).
- Mössner, E., Koch, H., Plückthun, A. Fast selection of antibodies without antigen purification: adaptation of the protein fragment complementation assay to select antigen-antibody pairs. J. Mol. Biol. 308 (2), 115-122 (2001).
- Wörn, A., et al. Correlation between in vitro stability and in vivo performance of anti-GCN4 intrabodies as cytoplasmic inhibitors. J Biol Chem. 275 (4), 2795-2803 (2000).
- Knappik, A., Plückthun, A. Engineered turns of a recombinant antibody improve its in vivo folding. Protein Eng. 8 (1), 81-89 (1995).
- Martineau, P., Jones, P., Winter, G. Expression of an antibody fragment at high levels in the bacterial cytoplasm. J Mol Biol. 280 (1), 117-127 (1998).
- Steipe, B., Schiller, B., Plückthun, A., Steinbacher, S. Sequence statistics reliably predict stabilizing mutations in a protein domain. J. Mol. Biol. 240 (3), 188-192 (1994).
- Daugherty, P. S. Protein engineering with bacterial display. Curr Opin Struct Biol. 17 (4), 474-480 (2007).
- Lener, M., et al. Diverting a protein from its cellular location by intracellular antibodies. Eur. J. Biochem. 267 (4), 1196-1205 (2000).
- Lynch, S. M., Zhou, C., Messer, A. An scFv intrabody against the nonamyloid component of α-synuclein reduces intracellular aggregation and toxicity. J. Mol. Biol. 377 (1), 136-147 (2008).
- Gai, S. A., Wittrup, K. D. Yeast surface display for protein engineering and characterization. Curr. Opin. Struc. Biol. 17 (4), 467-473 (2007).
- Kieke, M. C., et al. Selection of functional T cell receptor mutants from a yeast surface-display library. Proc. Natl. Acad. Sci. USA. 96 (10), 5651-5656 (1999).
- Steiner, D., Forrer, P., Stumpp, M. T., Pluckthun, A. Signal sequences directing cotranslational translocation expand the range of proteins amenable to phage display. Nat. Biotechnol. 24, 823-831 (2006).
- Pugsley, A. P. The complete general secretory pathway in gram-negative bacteria. Microbiol. Rev. 57 (1), 50-108 (1993).
- Rapoza, M. P., Webster, R. E. The filamentous bacteriophage assembly proteins require the bacterial SecA protein for correct localization to the membrane. J. Bacteriol. 175 (6), 1856-1859 (1993).
- Karlsson, A. J., et al. Engineering antibody fitness and function using membrane-anchored display of correctly folded proteins. J. Molec. Biol. 416 (1), 94-107 (2012).
- DeLisa, M. P., Tullman, D., Georgiou, G. Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc Natl Acad Sci U S A. 100 (10), 6115-6120 (2003).
- Fisher, A. C., Kim, W., DeLisa, M. P. Genetic selection for protein solubility enabled by the folding quality control feature of the twin-arginine translocation pathway. Protein Sci. 15 (3), 449-458 (2006).
- Maynard, J., Georgiou, G. Antibody engineering. Annu Rev Biomed Eng. 2, 339-376 (2000).
- Green, M. R., Sambrook, J. Molecular Cloning: A Laboratory Manual. 1, Fourth, Cold Spring Harbor Laboratory Press. (2012).
- Milo, R., Jorgensen, P., Moran, U., Weber, G., Springer, M. BioNumbers--the database of key numbers in molecular and cell biology. Nucleic Acids Res. 38, D750-D753 (2010).
- Hermanson, G. T. Bioconjugate Techniques. , Third, Elsevier/Academic Press. (2013).
- Tayapiwatana, C., Chotpadiwetkul, R., Kasinrerk, W. A novel approach using streptavidin magnetic bead-sorted in vivo biotinylated survivin for monoclonal antibody production. J Immunol Methods. 317 (1-2), 1-11 (2006).
- Zhu, G., Song, L., Lippard, S. J. Visualizing inhibition of nucleosome mobility and transcription by cisplatin-DNA interstrand crosslinks in live mammalian cells. Cancer Res. 73 (14), 4451-4460 (2013).
- Bogsch, E. G., et al. An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J. Biol. Chem. 273, 18003-18006 (1998).
- Julian, M. C., et al. Co-evolution of affinity and stability of grafted amyloid-motif domain antibodies. Protein Eng. Des. Sel. 28 (10), 339-350 (2015).
- Garber, K. Bispecific antibodies rise again. Nat. Rev. Drug. Discov. 13 (11), 799-801 (2014).
- Marschall, A. L., Dübel, S., Böldicke, T. Specific in vivo knockdown of protein function by intrabodies. MAbs. 7 (6), 1010-1035 (2015).
