

Introduction
Anticorpi in grado di piegare e funzionante nell'ambiente intracellulare sono strumenti promettenti sia per la ricerca e applicazioni terapeutiche. Essi hanno la capacità di modulare l'attività della proteina legandosi a una proteina bersaglio all'interno delle cellule per evitare interazioni proteina-proteina, interrompere le interazioni degli acidi nucleici proteine, o impedire l'accesso substrato enzimi 1-5.
Sebbene gli anticorpi hanno molto potenziale per applicazioni intracellulari, li ingegneria per il corretto ripiegamento e solubilità in ambiente intracellulare pur mantenendo la capacità di legarsi ad un antigene bersaglio è impegnativo. L'ambiente citoplasmatico riducendo previene la formazione di legami disolfuro normalmente richiesti per la piegatura stabile di anticorpi integrali e frammenti anticorpali, incluse catena singola frammento variabile (scFv) anticorpi 6,7. Un certo numero di approcci evoluzione diretta sono stati impiegati per progettare anticorpi con haffinità IGH per bersaglio antigeni 8-10. Questi approcci usano comunemente phage display, visualizzazione della superficie del lievito, o la visualizzazione della superficie batterica per schermare grandi librerie di anticorpi 11-13. Questi metodi sono potenti ed efficaci per identificare gli anticorpi che si legano a bersagli, ma dipendono dalla via secretoria per trasportare proteine che verranno visualizzati 14-16. La via secretoria trasloca proteine ripiegate dal citoplasma riducendo nel lume reticolo endoplasmatico nel lievito o in periplasma nei batteri. Le proteine si ripiegano quindi in condizioni ossidanti e vengono visualizzati sulla superficie delle cellule o confezionati in particelle dei fagi per lo screening per l'affinità di legame 17,18. Come risultato, gli anticorpi isolati utilizzando queste tecniche non necessariamente piegare bene nel citoplasma, e la solubilità intracellulare spesso devono essere progettati separatamente, se verranno utilizzati gli anticorpi in applicazioni intracellulari.
Migliorarel'efficienza di anticorpi ingegneria che sono ben piegati nel citoplasma, abbiamo precedentemente riportato il successo di MAD-TRAP (display membrana ancorata riconoscimento Tat di proteine che associano), un metodo per lo screening di una libreria di anticorpi scFv usando Escherichia coli inner- Display a membrana 19. Batterica visualizzazione interna membrana si basa sul percorso twin-arginina traslocazione (Tat) per il trasporto di anticorpi esposta, a differenza di altri metodi di visualizzazione comune che utilizzano la via secretoria. Il percorso Tat contiene un meccanismo di controllo della qualità che consente solo solubili, proteine ripiegate in modo corretto per essere trasportate dal E. citoplasma coli, attraverso la membrana interna, e nel periplasma 20,21. Substrati overexpressed Tat (es., Proteine mirati al percorso Tat con una fusione N-terminale al segnale Tat peptide ssTorA) che sono ben piegati nel citoplasma formano una lunga durata traslocazione intermedio con l'N-terminale in citoplasma e il C-terminale in periplasma 19. Questo consente la visualizzazione di substrati Tat correttamente ripiegate, inclusi frammenti di anticorpi, sulla faccia periplasmic della E. coli membrana interna. Dopo aver rimosso la membrana esterna mediante digestione enzimatica per generare sferoplasti, anticorpi sono esposti allo spazio extracellulare (Figura 1). Questo permette di substrati Tat visualizzati sulla membrana interna per essere sottoposti a screening per il legame ad un target specifico. È importante sottolineare che, sfruttando il percorso Tat per la visualizzazione della superficie cellulare assicura che solo gli anticorpi nella libreria che sono ben piegati nel citoplasma saranno interrogati per la rilegatura, permettendo di ingegneria simultanea di affinità e piegatura intracellulare vincolante. In questo protocollo, si descrive come visualizzare una libreria scFv in E. coli membrana interna, pan biblioteca contro un antigene bersaglio, ed eseguire uno schermo secondario per identificare i costituenti più promettenti della biblioteca. Mentre ci concentriamo il protocollo sul scFv, il metodo potrebbe essere applicato a ingegneria proteina la cui applicazione richiede pieghevole vincolante e intracellulare.
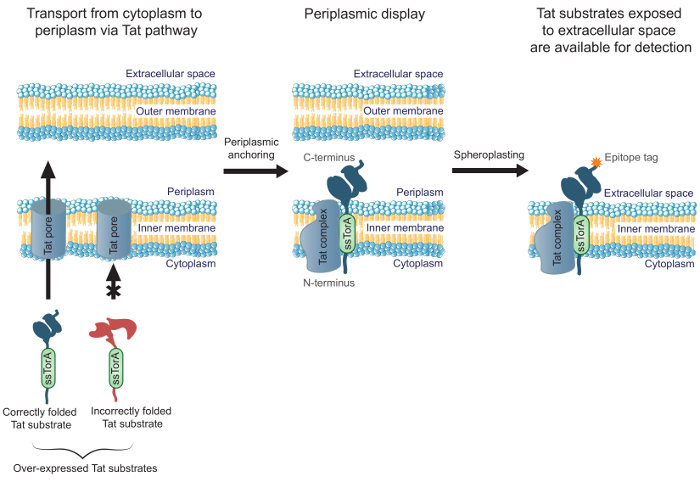
Figura 1. Display interno-membrana Tat. In E. coli, anticorpi scFv che sono espressi come una fusione alla sequenza segnale ssTorA e correttamente ripiegate nel citoplasma vengono trasportati attraverso la membrana interna. Una traslocazione forme intermedie, dove i scFv sono ancorati nella membrana interna con l'N-terminale nel citoplasma e il C-terminale in periplasma. Il E. membrana esterna coli è enzimaticamente digerito per formare sferoplasti, esponendo così gli anticorpi ancorati allo spazio extracellulare e renderli disponibili per il rilevamento utilizzando un anticorpo che si lega al tag epitopo C-terminale fuso sul anticorpi visualizzato.carico / 54583 / 54583fig1large.jpg "target =" _ blank "> Clicca qui per vedere una versione più grande di questa figura.
Protocol
1. Preparare la Libreria scFv come una fusione alla ssTorA Signal Sequence
- Ottenere una libreria di acido desossiribonucleico (DNA) contenente varianti di un gene scFv.
NOTA: La libreria può anche essere costruito utilizzando qualsiasi modalità appropriata per generare diversità sull'intera gene scFv o domini mirate 22 (ad esempio, la terza complementarità determinazione regioni, CDR3s.). - Inserire la libreria di DNA nel plasmide PIMD (figura 2) utilizzando metodi di clonazione molecolare standard 23.
NOTA: Questo plasmide esprime scFv come una fusione genetica per la sequenza di segnali ssTorA (N-terminale di scFv) e il tag FLAG epitopo (C-terminale al scFv). Il disegno del plasmide per la visualizzazione membrana interna è stato descritto in precedenza 19. Il plasmide PIMD è disponibile presso gli autori.

Figura 2. Inner-membrana Display plasmide (PIMD) mappa (passaggi da 1.2 a 1.3). Questo plasmide contiene un promotore lac, origine ColE1 di replica, e un gene di resistenza cloramfenicolo. Il gene scFv inserita viene fusa alla sequenza di segnali ssTorA di indirizzare il scFv al percorso Tat ed a un tag FLAG epitopo, con tutti e tre nello stesso quadro di lettura. siti enzima di restrizione sono indicati. Per una libreria inserito tra i siti di restrizione XbaI e enzimi NotI, la dimensione del plasmide è 2219 bp più la dimensione del scFv. Fare click qui per visualizzare una versione più grande di questa figura.
- Trasformare il DNA plasmide contenente la libreria in MC4100 E. coli 23. Recuperare e crescere questa forma batterica della biblioteca. Centrifugare a 4000 g per 15 min a RT per raccogliere le cellule. Rimuovere il surnatante, e risospendere le cellule raccoltenel 25% glicerolo in Luria-Bertani (LB) media. Aliquote conservare a -80 ° C fino a quando necessario, o passare al punto 2.
NOTA: Il protocollo è stato verificato con le cellule MC4100, anche se altri E. ceppi coli sono inoltre tenuti a essere compatibile con il protocollo. L'elettroporazione è il metodo preferito per la trasformazione, grazie alla sua elevata efficienza di trasformazione. La biblioteca dovrebbe tipicamente consistere di almeno 10 9 scFv varianti in questa fase, e ciascuna aliquota deve contenere abbastanza cellule tale che la libreria è coperto 100 volte.
2. Esprimere la Biblioteca e Preparare sferoplasti
- Scongelare una aliquota della libreria batterica (dal punto 1.3) a temperatura ambiente, e aggiungere la parte in una beuta contenente 100 ml mezzi LB con 20 ug / ml cloramfenicolo (Cm). Grow per 3 ore a 37 ° C e 225 rpm in un agitatore incubata.
- Dopo 3 ore, togliere il pallone da shaker incubate a 37 ° C. Consentire espressione della biblioteca scFv perprocedere O / N per 15 a 22 ore a 20 ° C e 225 rpm in un agitatore incubata.
NOTA: Non è necessario alcun induttore quando si utilizza il plasmide PIMD, come il promotore è che perde. Si noti che le cellule MC4100 non iperesprimono il repressore Lac (e LacI non si trova sul plasmide).

Figura 3. cellule di E.coli e sferoplasti. (A) E. coli sono di forma cilindrica. (B) Dopo spheroplasting usando EDTA e lisozima, la membrana esterna del E. coli è rotto, e le sferoplasti risultanti sono di forma sferica. Differenziale contrasto interferenziale (DIC) immagini al microscopio sono stati ottenuti utilizzando un obiettivo 100X su un microscopio invertito. Si prega di cleccare qui per vedere una versione più grande di questa figura.
- Preparare le sferoplasti biblioteca.
NOTA: sferoplasti sono formate dalla rottura della membrana esterna di E. coli e sono di forma sferica (Figura 3).- Preparare i buffer necessari.
NOTA: Tutti i buffer devono essere sterili.- Preparare 1 × tampone fosfato salino (PBS, pH 7,4) sciogliendo 8 g di NaCl, 0,2 g di KCl, 1,44 g di Na 2 HPO 4, e 0,24 g KH 2 PO 4 in H 2 O distillata O ad un volume finale di 1000 ml. Tenere in ghiaccio.
- Preparare PBS con 0,1% (w / v) di albumina di siero bovino (BSA) sciogliendo 0,2 g di BSA in 200 ml 1 × PBS. Tenere in ghiaccio.
- Preparare il tampone di frazionamento (FB) miscelando 7,5 ml di soluzione sterile filtrata 1 M di saccarosio, 1 ml di 1 M Tris (pH 8,0) e 1,5 ml di acqua distillata H 2 O. Tenere in ghiaccio.
- Preparare 1 mM acido etilendiamminotetraacetico (EDTA) con l'aggiunta di 30 ml of 0.5 M EDTA a 14.97 ml di acqua distillata H 2 O.
- Preparare 0,5 M MgCl 2 sciogliendo 4,76 g MgCl 2 in 100 ml di H 2 O. distillata Tenere in ghiaccio.
- Rimuovere il matraccio da shaker, e misurare la densità ottica (OD) a 600 nm utilizzando uno spettrofotometro per determinare la densità cellulare. Calcolare il volume di coltura indotta necessaria tale che ciascun campione per spheroplasting ha 1 × 10 10 cellule.
NOTA: L'approssimazione di un OD 600 di 1 indica una concentrazione di 10 9 cellule / ml per E. coli può essere utilizzato 24. - Centrifugare il volume calcolato di coltura indotta in una provetta da 1,5 ml microcentrifuga a 12.000 xg a temperatura ambiente per 5 min. Preparare almeno due campioni nel caso in cui si verificasse un problema nella preparazione del campione.
- Rimuovere il surnatante dalle culture centrifugati e risospendere ogni pellet di cellule in 100 ml di FB ghiacciata. Centrifugare a 12.000 ×g a temperatura ambiente per 1 minuto, quindi rimuovere il surnatante pipettando. Risospendere ogni pellet in 350 ml di FB ghiacciata integrati con 3,5 ml di 10 mg / ml di lisozima.
- Lentamente vortex ogni provetta aggiungendo, goccia a goccia, 700 ml di 1 mM EDTA e quindi incubare le provette a temperatura ambiente per 20 min mentre lentamente rotante su un rotatore tubo per mescolare i campioni. Rimuovere i tubi dal rotatore, aggiungere 50 ml di ghiacciata 0,5 M MgCl 2 per ciascun tubo, e incubare in ghiaccio per 10 min. Centrifugare le provette a 11.000 xg a 4 ° C per 10 min.
- Isolare il pellet sferoplasto.
- Utilizzare una micropipetta con una punta di 1 ml di tirare lentamente parte del pellet. Tenendo il tubo ad un angolo con l'apertura direttamente sopra una nuova provetta da 1,5 ml, sollevare lentamente la punta della pipetta dal surnatante e far scorrere il pellet nel nuovo tubo.
- Se un volume significativo di supernatante viene trasferito al nuovo tubetto, rimuoverlo pipettando. Se il pellet non è ferma enough di trasferire, ri-centrifugare a 11.000 g per 2 minuti e tentare l'isolamento pellet di nuovo.
- Risospendere il pellet sferoplasto in ogni tubo in 1 ml di ghiacciata 1 × PBS. Si alternano tra pipettaggio e lentamente vortex su un supporto vortice fino a quando il pellet è completamente risospeso. Non tenere campioni iniziale del ghiaccio per più di 2 min alla volta, e ritornerà sul ghiaccio per almeno 5 minuti prima di rimuovere dal ghiaccio nuovo. Mantenere le sferoplasti a 4 ° C (per un massimo di 2 giorni) fino al momento per il panning al punto 4.
- Preparare i buffer necessari.
3. immobilizzare l'antigene bersaglio su biglie magnetiche
- Biotinilato l'antigene bersaglio in vivo durante la produzione ricombinante in E. coli. In alternativa, utilizzare la coniugazione chimica di 25 o l'acquisto antigene bersaglio che è già stato biotinilato, e passare al punto 3.2.
- Aggiungere 816 g bicine a 50 ml di acqua per fare 10 × bicine buffer. Diluire il buffer a 1 × in H 2 O distillata O e riscaldare a 50 ° C. Aggiungere 14,7 mg biotina a 12 ml della riscaldata 1 × bicine tampone per ottenere una soluzione di biotina che è 5 mM biotina in 10 mM tampone bicine. Conservare a -20 ° C fino al momento dell'uso.
- Express e biotinilato proteina bersaglio utilizzando il plasmide pAK400cb-BCCP 26, che consente la produzione dell'antigene bersaglio come una fusione alla proteina carrier biotina carbossilico (BCCP).
NOTA: E. coli nativamente biotinilato BCCP, eliminando la necessità di purificare e chimicamente biotinilato la proteina bersaglio prima di immobilizzazione su perle rivestite di streptavidina. Il nativo E. coli biotina ligasi Bira è sufficiente per biotinylating la proteina di fusione.- Grow E. coli contenenti il plasmide biotinilazione (con l'antigene bersaglio inserita come fusione al N-terminale di BCCP) O / N per 15 a 18 ore in 5 ml di mezzi LB addizionato con 20 ug / ml Cm a 37 ° C agitando a 225 rpm.
- Misurare la densità ottica a 600 nm utilizzando uno spettrofotometro e calcolare il volume di coltura necessario (V add) per subcoltura in un OD di partenza di 0,05 a 25 ml fresca mezzi LB con 20 ug / ml Cm usando l'equazione: V aggiungere = (0,05 × 25 ml) / (OD 600 - 0,05), dove OD 600 è la densità ottica della O / N cultura e V è il volume aggiuntivo del / culture O N da aggiungere al LB. fresco Subculture e crescere ad un OD di 0.5 e 0.8 in un agitatore incubate a 37 ° C e 225 rpm.
- Aggiungere isopropil β-D-1-thiogalactopyranoside ad una concentrazione finale di 100 mM e biotina ad una concentrazione finale di 5 mM. Indurre l'espressione in un agitatore incubate per 15 a 22 ore a 20 ° C e 225 rpm.
- batteri Harvest per centrifugazione a 4.000 xg a 4 ° C per 10 min. Rimuovere il surnatante. Conservare il pellet a -20 ° C fino al momento dell'uso.
- Aggiungere1 ml di un detergente lisi cellulare per 0,2 g di pellet cellulare. Risospendere pipettando e ruotare delicatamente per 20 minuti per lisare le cellule. Dopo lisi, centrifugare a 16.000 xg e 4 ° C per 20 min. Pipettare il lisato solubile (surnatante) in una nuova provetta da 1,5 ml.
- Utilizzare una colonna di peso molecolare di taglio 3 kDa per rimuovere la biotina non legato. Pipettare il lisato nella colonna e centrifugare a 20 ° C secondo le istruzioni del produttore. Lavare con 1 × PBS fino alla biotina nel lisato è stato diluito 100 volte e il volume del lisato lavato è uguale al volume originale del lisato. Trasferire il lisato in una nuova provetta.
- Immobilizzare l'antigene bersaglio biotinilato su sfere magnetiche rivestite di streptavidina.
- Preparare 1 × PBS e 1 × PBS con 0,1% (w / v) BSA come descritto al punto 2.3.1.
- Preparare le sfere magnetiche.
NOTA: Ciò richiede l'uso di un rack di separazione magnetica.- Risospendere streptavIdin-rivestito sfere magnetiche nella loro flacone originale. In entrambi i casi vortex per almeno 30 sec o ruotare per 5 min.
- Trasferire 7-10 × 10 9 perline in una provetta da 1,5 ml.
NOTA: Il volume richiesto sarà dipendente dalla concentrazione tallone fornito dal produttore. - Posizionare il tubo contenente le perline sulla cremagliera magnete per 2 minuti per raccogliere le perline sul lato del tubo. Con il tubo ancora sul magnete, rimuovere con attenzione il surnatante pipettando senza interrompere le perline.
- Per lavare, rimuovere il tubo dal magnete, e risospendere le sfere in 1 ml di 1 × PBS pipettando senza generare bolle. Riportare il tubo al magnete per 2 minuti per raccogliere le perline, e rimuovere con attenzione il surnatante pipettando. Ripetere la procedura altre due volte per un totale di tre lavaggi. Assicurarsi che nessun liquido viene lasciato nel tubo dopo il lavaggio finale.
- Aggiungere il lisato contenente l'antigene biotinilato al bea magneticads.
- Rimuovere il tubo dal magnete e risospendere le sfere in 1 ml di lisato (dal punto 3.1.5). Incubare a temperatura ambiente per 30 min mentre delicatamente rotazione.
- Posizionare il tubo sul magnete per 3 minuti per raccogliere le perline rivestite antigene. Lavare le perline rivestite cinque volte con 1 × PBS con 0,1% BSA nello stesso modo come descritto nei passi 3.2.2.3 a 3.2.2.4. Dopo l'ultimo lavaggio, risospendere le sfere in 1 × PBS con 0,1% BSA fino allo stesso volume utilizzato nel passaggio 3.2.2.2.
- Se l'antigene bersaglio immobilizzato è stabile a 4 ° C, conservare le perline rivestite a 4 ° C fino al momento della panning. In caso contrario, passare al punto 4.
4. Schermo del scFv Library di Panoramica contro l'antigene bersaglio (figura 4)

Figura 4. Panoramica (fase 4). Antigen-rivestito sfere magnetiche are incubato con sferoplasti esprimono varianti di libreria di anticorpi. Plasmidi DNA da sferoplasti bead-bound viene recuperato e utilizzato per generare un sublibrary, che viene proiettato utilizzando lo schermo secondario ELISA-based. Corrispondenti operazioni di protocollo sono noti. Cliccate qui per vedere una versione più grande di questa figura.
- Incubare le perline rivestite con sferoplasti.
- Utilizzare un sferoplasto da bordare rapporto di circa 5: 1. Aggiungere 4 × 10 9 sferoplasti e 8 × 10 8 perle di una provetta sterile 15 ml.
NOTA: Si supponga che non le cellule sono stati persi durante il processo di spheroplasting, quindi la concentrazione è ancora 1 × 10 10 sferoplasti / ml. - Aggiungere 1 × PBS con 0,1% BSA per portare il volume totale a 4 ml. Aliquota in quattro provette da 1,5 ml con 1 ml ciascuno. Incubare le reazioni a 4 ° C per 5 ore mentre delicatamente rotazione.
- Utilizzare un sferoplasto da bordare rapporto di circa 5: 1. Aggiungere 4 × 10 9 sferoplasti e 8 × 10 8 perle di una provetta sterile 15 ml.
- Prepare le sferoplasti tallone-bound per la reazione a catena della polimerasi (PCR).
- Mettere le provette di reazione panning sul magnete per 3 min. Rimuovere il surnatante pipettando e lavare i sferoplasti stallonatori vincolati quattro volte con ghiaccio freddo 1 × PBS con 0,1% BSA nello stesso modo come descritto nei passi 3.2.2.3 a 3.2.2.4. Risospendere le sferoplasti tallone-bound in ogni tubo in 25 ml di H 2 O. distillata Conservare le perline a -20 ° C o passare al punto 4.3.
- Eseguire tutto il plasmide PCR su sferoplasti tallone-bound per amplificare i plasmidi contenenti i geni per scFv bead-bound.
- Ottenere primer con le seguenti sequenze: 5'CCAACTCTTTTTCCGAAGGTAACTG3 '(forward primer) e 5'TAGCTCTTGATCCGGCAAACAAA3' (primer reverse).
NOTA: Questi si legherà end-to-end su filoni opposti del plasmide PIMD (Figura 2) e sono progettati per temprare a una caratteristica comune di PIMD, quindi l'amplificazione si verifica indipendentemente dalscFv sequenza variante. - Fosforilare i primer.
NOTA: Senza la fosforilazione, non si verificano ri-legatura. Primer possono anche essere ordinati con 5'-fosforilazione, piuttosto che utilizzare questo metodo fosforilazione in questo protocollo.- In una provetta da 0,5 ml, impostare una reazione di fosforilazione per il primer PCR in avanti, come descritto nella tabella 1. Ripetere questo processo per il primer inverso.
- Incubare le reazioni a 37 ° C per 1 ora. Poi incubare a 65 ° C per 20 min per disattivare chinasi T4 polinucleotide (PNK). Conservare i primer fosforilata a -20 ° C.
- Eseguire la PCR.
- In un tubo di PCR, preparare la reazione di PCR come descritto nella Tabella 2.
NOTA: Le reazioni multiple possono essere preparati per rendimento più elevato. I sferoplasti bead-bound non utilizzati possono essere conservati a -20 ° C. - Riscaldare le reazioni PCR a 98 ° C per 15 min in un ciclatore termico per garantire la piena lisi dei sferoplasti. Rimuovere i tubi dal termociclatore e aggiungere 0,5 ml di un alto polimerasi fedeltà a ciascuno. Rientro tubi al termociclatore ed eseguire utilizzando il programma dettagliato nella tabella 3.
- Pool prodotti di PCR a seconda dei casi. Conservare a -20 ° C o passare al punto 4.4.
- In un tubo di PCR, preparare la reazione di PCR come descritto nella Tabella 2.
- Ottenere primer con le seguenti sequenze: 5'CCAACTCTTTTTCCGAAGGTAACTG3 '(forward primer) e 5'TAGCTCTTGATCCGGCAAACAAA3' (primer reverse).
Tabella 1. reazione di fosforilazione PNK (Step 4.3.2.1).
| Reagente | Volume (ml) |
| H 2 O distillata | 15 |
| tampone di reazione 10x ligasi T4 DNA | 2 |
| 100 micron di primer | 2 |
| T4 polinucleotide chinasi (PNK) | 1 |
| Reagente | Volume (ml) |
| H 2 O distillata | 28.5 |
| 5x Alta fedeltà tampone polimerasi | 10 |
| 10 micron fosforilato primer forward | 2.5 |
| 10 micron fosforilato primer reverse | 2.5 |
| 40 mM dNTP mix (10 millimetri ogni dNTP) | 1 |
| sferoplasti Bead-bound | 5 |
Tabella 3. programma PCR (Step 4.3.3.2).
| Passo | Temperatura (° C) | Tempo (min: sec) | Numero di cicli |
| denaturare iniziale | 98 | 00:30 | 1 |
| Denaturare | 98 | 00:10 | 35 |
| ricottura | 69 | 00:30 | |
| Estensione | 72 | 00:30 per kb | |
| estensione finale | 72 | 06:00 | 1 |
| tenere | 12 | Infinito | 1 |
- Re-circolari ai prodotti integrali-plasmide PCR, e utilizzare il prodotto legatura di trasformare MC4100 E. coli.
- Purificare il prodotto di PCR eseguendo la reazione di PCR su un gel di agarosio 23, macchiando il DNA nel gel 23, e utilizzando unkit di pulizia gel per purificare il plasmide linearizzato seguendo le istruzioni fornite dal produttore. Misurare la concentrazione utilizzando uno spettrofotometro a 260 nm. Conservare il frammento purificato a -20 ° C fino al momento, o continuare al passo 4.4.2.
- Re-circolare la plasmide dal prodotto di PCR.
- Per evitare legatura intermolecolare del prodotto PCR, effettuare la reazione legatura con una bassa concentrazione di 27 1 ng / ml di prodotto della PCR. Calcolare il volume necessario per la preparazione di una reazione di ligasi 800 microlitri a questa concentrazione.
- Preparare la reazione legatura sul ghiaccio. In un tubo, aggiungere il volume calcolato al punto 4.4.2.1 del prodotto della PCR, 80 ml di tampone ligasi 10 × DNA, e H 2 O distillata fino a 800 ml. Aggiungere 4 ml di T4 DNA ligasi, e collocare immediatamente provette a 16 ° C in un bagno d'acqua o termociclatore. Incubare a 16 ° CO / N per 14 a 18 ore. Conservare le reazioni legatura completati a -20 ° Cfino a quando necessario, o passare al punto 4.4.3.
- Posizionare la reazione legatura su un blocco di calore a 65 ° C per 15 min per riscaldare-inattivare il DNA ligasi. Quindi utilizzare un kit di microdialisi membrana o la pulizia del DNA di de-sale il DNA legatura. Conservare a 20 ° C o passare al punto 4.4.4.
- Utilizzare l'intero, prodotto inattivato al calore de-salata legatura per trasformare MC4100 E. coli 23. Preparare le scorte glicerolo, come descritto al punto 1.3, delle cellule che contengono il sublibrary stroncato risultante, e aliquote conservare a -80 ° C.
- Ripetere il punto 4 nella sua interezza con una aliquota dal punto 4.4.4 di fare un secondo panoramica sul sublibrary.
NOTA: Una seconda panning contribuisce ad arricchire di componenti di libreria che si legano bene alla antigene bersaglio 19.
5. Eseguire una schermata secondaria L'utilizzo di un metodo di analisi Immunoenzimatico Enzyme-linked per identificare cloni promettenti per un'ulteriore caratterizzazione (Figura 5) </ P>

Lo screening Figura 5. ELISA a base secondaria (punto 5). Varianti (A) libreria dal sublibrary arricchito durante il panning vengono inoculate in singoli pozzetti di una piastra di coltura per la crescita e l'espressione. (B) Un piastra ELISA è rivestita con antigene bersaglio. (C) Le varianti di libreria sono proiettati utilizzando lo schermo secondario ELISA-based descritto nel protocollo. Dopo analisi dei dati ottenuti dallo schermo secondario, varianti di interesse sono selezionati e caratterizzati ulteriormente. Corrispondenti operazioni di protocollo sono noti. Cliccate qui per vedere una versione più grande di questa figura.
- Scongelare un tubo del sublibrary panoramica (dal punto 4.4.4) e la piastra su piastre di agar LB. Piastra diverse diluizionia concentrazioni sufficientemente bassa per garantire singole colonie (ad esempio, 10 2 - 10 6 diluizioni fold). Incubare le piastre per 15 a 18 ore a 37 ° C. Conservare le piastre a 4 ° C o passare al punto 5.2.
- Cultura e indurre colonie dalla sublibrary stroncato. Eseguire tutte le operazioni in condizioni sterili. Utilizzare una pipetta multicanale per i passaggi che coinvolgono piastre a 96 pozzetti.
- Aggiungere 200 ml di LB con 20 ug / ml Cm in ciascun pozzetto di una piastra a fondo rotondo coltura da 96 pozzetti.
- Scegliere una colonia individuo dalla piastra di agar con un puntale, posizionare la punta nel primo pozzetto della piastra a 96 pozzetti, e mescolare delicatamente per inoculare. Utilizzare una nuova punta per ogni bene. Seminare una colonia in tutti i pozzetti. Come controllo, includere almeno un controllo di sterilità e senza colonia inoculato.
- Ripetere i punti 5.2.1 e 5.2.2 per inoculare numerose piastre a 96 pozzetti.
- Posizionare le piastre a 96 pozzetti, un vortex a 310 rpm. Incubare a 376; C per 20 a 24 ore per esprimere la scFv.
- Per ogni piastra di coltura preparato al punto 5.2, mano a 96 pozzetti ELISA con l'antigene bersaglio.
- Diluire purificato antigene bersaglio a concentrazione appropriata (ad esempio, 1 ug / ml a 4 mg / ml) in 1 × PBS per rendere la soluzione di rivestimento. Effettuare 5 ml di soluzione di rivestimento per ciascuna piastra da 96 pozzetti.
NOTA: La concentrazione appropriata dipende l'antigene specifico che viene utilizzato e può essere necessario un aggiustamento. - Aggiungere 50 ml di soluzione di rivestimento a ciascun pozzetto di una piastra di polistirene ELISA chiaro 96 pozzetti alta vincolante. Agitare gentilmente la piastra sulla superficie banco per assicurare che l'intera superficie di ogni pozzetto è rivestito. Ripetere per ogni piatto. Incubare le piastre a 4 ° CO / N.
- Diluire purificato antigene bersaglio a concentrazione appropriata (ad esempio, 1 ug / ml a 4 mg / ml) in 1 × PBS per rendere la soluzione di rivestimento. Effettuare 5 ml di soluzione di rivestimento per ciascuna piastra da 96 pozzetti.
- Replicare le colonie delle piastre di coltura a 96 pozzetti su piastre di agar.
- Inserire un replicatore polistirene sterile nei pozzetti di una piastra di coltura per raccogliere una piccola quantità di sapone liquidid. Sollevare con cautela il replicatore e trasferimento in una piastra di agar LB 15 centimetri in modo tale che tutte le punte toccano il piatto. Una volta che il liquido ha trasferito, sollevare il replicatore verso l'alto. Ripetere per ogni piastra di coltura.
- Etichettare la piastra di agar con l'orientamento corretto in modo che i risultati dallo schermo secondario nella piastra a 96 pozzetti possono essere abbinati con la corretta colonia replicato sul piatto, se ulteriore caratterizzazione è desiderata. Crescere a 37 ° C per 15 a 18 ore, e quindi memorizzare a 4 ° C fino al momento dell'uso.
- Eseguire lo schermo secondario ELISA.
- Preparare la soluzione di saturazione rendendo 2% (w / v) di latte secco in 1 × PBS. Svuotare la soluzione di rivestimento dalle piastre ELISA. Aggiungere 100 microlitri della soluzione bloccante a ciascun pozzetto. Incubare a temperatura ambiente per almeno 2 ore, o il blocco O / N a 4 ° C.
- Preparare il tampone di lavaggio aggiungendo polisorbato 20 ad una concentrazione finale di 0,05% in 1 × PBS. Fare 250 ml per piastra ELISA.
- Aggiungere 20 ml di unaconcentrato lisi cellulare detergente a ciascun pozzetto della piastra a fondo tondo coltura e incubare la piastra di coltura, un vortex a temperatura ambiente per 15 a 20 min. Iniziare la lisi allo stesso tempo che il blocco delle piastre ELISA è completo in modo che la lisi e lavaggio Fase 5.5.4 possono essere eseguite contemporaneamente.
- Svuotare la soluzione di saturazione dalle piastre ELISA. Lavare le piastre ELISA bloccati per quattro volte con 200 ml di tampone di lavaggio per pozzetto a lavaggio. Svuotare il tampone di lavaggio dai pozzetti.
- Trasferimento 50 microlitri di ciascun pozzetto della piastra di lisi cellulare al corrispondente pozzetto della piastra ELISA, utilizzando una nuova punta per ogni pozzetto. Incubare la piastra ELISA a temperatura ambiente per 1 a 2 ore.
- Preparare la soluzione di anticorpi per rilevare scFv rilegati.
- Utilizzare un perossidasi di rafano (HRP) coniugata anticorpo primario che si lega al tag FLAG epitopo fuso alle scFv biblioteca.
- Diluire l'anticorpo per la diluizione appropriata da utilizzare in un test ELISA (vedi il fornitore & #39; s consigli) in 2% (w / v) di latte secco 0,05% polisorbato 20 in 1 × PBS. Preparare 5 ml per ogni piatto.
- Lavare le piastre ELISA quattro volte con tampone di lavaggio come descritto al punto 5.5.4.
- Aggiungere 50 ml di soluzione di anticorpi a ciascun pozzetto della piastra ELISA. Incubare per 1 a 2 ore a RT.
- Preparare il substrato HRP sciogliendo o fenilendiamina dicloridrato compresse (OPD) in H 2 O distillata per protocollo del produttore, evitando luce. Preparare 20 ml per piastra ELISA.
- Preparare 3 MH 2 SO 4 concentrato diluendo H 2 SO 4 con H 2 O distillata O come necessario. Preparare 5 ml per piastra ELISA.
Attenzione: H 2 SO 4 è un acido forte. Assicurarsi di indossare dispositivi di protezione adeguati. - Lavare le piastre ELISA quattro volte con tampone di lavaggio, come descritto al punto 5.5.4.
- Incubare le piastre ELISA con la substra HRPTE.
- Aggiungere 200 ml di substrato HRP in ogni pozzetto. Per ridurre al minimo l'esposizione alla luce, aggiungere il substrato da una piastra ELISA alla volta, e avvolgere con un foglio di alluminio prima di procedere alla piastra successiva. Incubare le piastre per 30 a 60 minuti a temperatura ambiente al buio.
- Dopo i primi 30 min, controlla le piastre per scurire del substrato e incubare più a lungo se necessario visualizzare di sviluppo colore.
- Aggiungere 50 ml di 3 MH 2 SO 4 a ciascun pozzetto per placare la reazione. Utilizzando una punta diversa per ogni bene, mescolare la soluzione nei pozzetti pipettando gentilmente su e giù senza schiumare. Per coerenza e per evitare la saturazione, aggiungere l'H 2 SO 4 rapidamente e con attenzione a tutte le piastre ELISA prima della miscelazione la soluzione per ogni piastra.
- Misurare l'assorbanza della soluzione nei pozzetti di ciascuna piastra a 492 nm usando un lettore di piastre.
- Analizzare i dati di assorbanza per identificare scFv varianti che exhibsi promette segnali vincolanti e caratterizzare questi scFv promettenti. Selezionare scFv che presentano segnali assorbanza superiore al segnale di fondo e superiori del segnale medio su ciascuna piastra.
NOTA: Il livello assorbanza sarà dipendente dalle proprietà della antigene e anticorpo anti-FLAG usati, insieme con la forza delle varianti scFv che sono stati isolati nello screening.
Representative Results
Il ripiegamento delle proteine meccanismo di controllo della qualità intracellulare del percorso Tat in E. coli limita trasporto attraverso la membrana cellulare interna di proteine che sono ben piegati in ambiente citoplasmatico riducente. Con iperespressione una fusione di un scFv alla sequenza segnale di ssTorA (sequenza segnale dalla proteina TORA, che è naturalmente trasportato attraverso la via Tat 20), traslocazione è bloccato, con conseguente esposizione del scFv sulla membrana interna 19. Dopo interruzione enzimatica della membrana esterna, gli anticorpi visualizzati vengono resi disponibili per lo screening per l'attività antigene-legante. La possibilità di usufruire del percorso Tat per la visualizzazione scFv è stato dimostrato da Karlsson et al. 19 (Figura 6). Gli anticorpi scFv scFv13 e scFv13.R4 sono stati fusi per la sequenza di ssTorA nativo o un ssTorA modificato che manca la coppia residuo di arginina-arginina riconosciuto dail percorso Tat. scFv13.R4 è stato progettato da Martineau et al. da scFv13 in quattro manche di evoluzione diretta ed è noto per piegare bene nel citoplasma 9. Questo scFv è stato visualizzato sulla membrana interna, ma solo quando espressa come una fusione alla sequenza segnale nativa ssTorA (Figura 6). Al contrario, scFv13 non è ben piegato cytoplasmically 9, quindi non viene visualizzata bene sulla membrana interna, indipendentemente dalla sequenza segnale al quale è fuso. Inoltre, se le scFv sono stati espressi nelle cellule che mancava la proteina TATC, una componente vitale della macchina Tat 20,28, il display non è stato osservato, che mostra l'importante legame tra il display interno-membrana e il percorso Tat. Questi risultati dimostrano che solo proteine che contengono il segnale peptide Tat e che siano correttamente ripiegate nel citoplasma vengono visualizzati sulla membrana interna, permettendo il trasporto attraverso la via Tat di funzionare come schermo per fol intracellulareding.

Figura 6. Rilevamento di scFv visualizzato sulla membrana interna. Citometria a flusso analisi è stata effettuata per rilevare la visualizzazione di mal ripiegata scFv13 e ben piegata scFv13.R4 sulla membrana interna. scFv sono state fuse al nativo ssTorA o ssTorA (KK), dove la coppia Arg-Arg nella sequenza ssTorA stato modificato per Lys-Lys. I tag FLAG epitopi C-terminale sul scFv sono stati rilevati con un isotiocianato di fluoresceina (FITC) coniugata anticorpo anti-FLAG. Le cellule senza la proteina TATC (ΔtatC) e ssTorA-scFv13 senza il cartellino del FLAG sono stati testati come controlli. M indica il valore mediano di fluorescenza. Ristampato da riferimento 19 con il permesso. Cliccate qui per vedere una versione più grande di questa figura.
19. Per dimostrare questo, una libreria PCR incline all'errore basato su scFv13, che ha un basso livello di affinità di legame per β-galattosidasi, fu stroncato contro l'antigene bersaglio β-galattosidasi utilizzando il display e panning metodo descritto nel protocollo. scFv 1-4 è stato isolato dopo un round di mutagenesi e panning, ed espone più alta affinità di legame per i beta-galattosidasi di scFv13 (Figura 7A) e un più alto livello di solubilità citoplasmatica (Figura 7B).
Una nuova libreria, sulla base di scFv 1-4, è stato realizzato utilizzando soggetto a errori PCR, e panoramica di questa libreria di seconda generazione controβ-galattosidasi è stato fatto usando una modifica del protocollo descritto. Il panning contro β-galattosidasi per il secondo turno di evoluzione è stata fatta in presenza di purificato, solubile scFv 14 come un concorrente per migliorare la probabilità di isolare cloni con maggiore affinità rispetto scFv 1-4. Dopo questo secondo round di mutagenesi e panning, scFv 2-1 e 2-3 scFv sono stati isolati mediante lo screening secondario ELISA-based. Questi scFv non solo hanno mostrato una maggiore affinità di legame per β-galattosidasi di scFv13, ma anche esposti meglio vincolante che il clone primo turno scFv 1-4. scFv 2-1 esposto β-galattosidasi legame paragonabile a quella di scFv13.R4 (Figura 7A). scFv 2-3 mostra anche un ulteriore aumento di solubilità citoplasmatica rispetto al scFv 14, evidenziando l'ingegneria simultanea di solubilità e antigene-legante. Poiché affinità e espressione solubile dei scFv sono schermati per simultaneamente, è possibile che un scFv selezionato è modrano solubilità ma alta versa vincolante o vice. Ad esempio, scFv 2-1 trovi espressione solubile inferiore scFv 2-3, ma presenta una maggiore affinità di legame al ß-galattosidasi.

Figura 7. target vincolante e l'espressione citoplasmatica di scFv varianti isolata usando Display interno-membrana. (A) scFv sono state espresse nel citoplasma di E. coli (ad es., senza la sequenza di segnali ssTorA) con un hexahistidine tag (6 × -Il suo) e purificate utilizzando acido spin-colonne di nichel-nitrilotriacetico. Il legame dei scFv purificati di beta-galattosidasi è stata misurata con un test ELISA. scFv purificati sono stati caricati su piastre ELISA β-galattosidasi-rivestito ed i scFv legati sono stati rilevati con un anticorpo anti-6 × -Il suo. I dati sono una media di sei repliche, e la barra di errore indica l'errore standard della media.(B) Le frazioni solubili e insolubili dei lisati cellulari di cellule che esprimono i scFv cytoplasmically sono stati analizzati da una macchia occidentale sondati con un anticorpo anti-6 × -Il suo. concentrazione proteica totale è stato utilizzato per normalizzare il caricamento dei campioni. Ristampato (A) e adattato (B) dal riferimento 19 con il permesso. Cliccate qui per vedere una versione più grande di questa figura.
Discussion
Ingegneria anticorpi per l'attività citoplasmatica è un compito difficile a causa l'ambiente riducente del citoplasma, che impedisce la formazione di legami disolfuro stabilizzante 6,7. Questo fa sì che la maggior parte degli anticorpi di essere cytoplasmically inattiva meno che non siano progettati per la stabilità e la solubilità nel citoplasma, oltre ad essere progettato per affinità di legame. I metodi esistenti di phage display, visualizzazione di superficie batterica, e metodi di visualizzazione superficiali lievito utilizzare tutti la via secretoria 14-16 per la visualizzazione di anticorpi ingegnerizzati, ma questi metodi non hanno mezzi per progettare piegatura intracellulare. Anticorpi ingegnerizzati utilizzando visualizzazione interna membrana hanno migliorato la stabilità citoplasmatica e la solubilità in quanto il controllo di qualità di piegatura del percorso Tat impedisce traslocazione di anticorpi che sono mal ripiegate e instabili nel citoplasma. Questo metodo semplifica il processo iterativo di anticorpi intracellulari engineering per affinità unnd solubilità, come i due sono costruiti in un unico passaggio. Anche se questo metodo è stato progettato per ingegneria anticorpi con solubilità in ambiente intracellulare riduzione, può essere applicato anche ad anticorpi ingegneria di funzionare in condizioni non riducenti, poiché le proteine progettati utilizzando questo metodo mantenere il loro ripiegamento nell'ambiente ossidante periplasma.
Sebbene questa tecnica semplifica il processo di ingegneria anticorpi con alta affinità e alta solubilità citoplasmatica, diverse limitazioni sono importanti da considerare quando si utilizza questo protocollo. Quando si analizzano i segnali ELISA schermo secondario da identificare promettenti scFv varianti, la soglia per discernere tra le varianti potenzialmente interessanti e quelli che non possono esibire adeguata antigene vincolante non è probabile che sia evidente solo dopo parecchi cloni sono stati ulteriormente caratterizzati. E 'importante cercare migliorata legame sopra l'anticorpo genitore; però,un segnale anormalmente elevato potrebbe essere indicativo di avidità 29 o effetti di aggregazione 30, una sfida che non riguardano solo l'approccio di screening del display interno-membrana. Una limitazione chiave da ricordare quando si utilizza questo protocollo è l'incapacità di recuperare sferoplasti dopo panning, in quanto sono non vitali (dati non pubblicati). Ciò richiede le fasi di amplificazione del DNA e trasformazione di recuperare i plasmidi codificanti anticorpi.
Parecchi punti critici del protocollo consentono dell'ingegneria simultanea di piegatura e legame di anticorpi. Per lo screening per avere successo, la libreria di scFv viene proiettato deve essere espressa come una fusione al segnale peptide ssTorA. Senza questa sequenza, gli anticorpi non saranno indirizzati al percorso Tat e quindi non saranno traslocati al periplasma 19. Inoltre, è imperativo che un epitopo C-terminale è fuso agli anticorpi per consentire il rilevamento degli anticorpi visualizzati nel bidonesaggi ding. Chiaramente, la E. coli ceppo utilizzato per esprimere i scFv deve avere anche la necessaria macchinari percorso Tat, ma questo è vero per la E. comunemente utilizzati ceppi coli.
Modifiche questo protocollo è possibile migliorare il suo potenziale di isolare anticorpi con le caratteristiche desiderate. Un passo panning sottrattiva può essere completata prima di panning contro l'antigene bersaglio per esaurire la biblioteca scFv dei costituenti non desiderato. I sferoplasti biblioteca possono essere incubate con perline magnetiche rivestite con BCCP da solo o rivestita con una proteina non desiderato, ei sferoplasti che si legano a quelle sfere possono essere eliminati prima dello screening i restanti sferoplasti non legati per il legame con il target desiderato. Come indicato nelle Risultati rappresentativi, un metodo per migliorare l'affinità di un isolato scFv è quello di includere un concorrente solubili nella reazione panning di competere con gli scFv visualizzati sulle sferoplasti. Perché il comp solubileetitor è una proteina purificata, nessun DNA viene amplificato da esso, in modo che solo le sequenze di scFv esposti sulle sferoplasti saranno recuperati nella reazione PCR. Inoltre, questo metodo potrebbe essere estesa ad altri tipi di ingegneria anticorpi o alla non-anticorpi proteine leganti.
E. coli Display interno-membrana è una potente piattaforma per gli anticorpi di ingegneria con alta affinità e alti livelli di solubilità intracellulare. Questo metodo è particolarmente adatto per engineering efficiente di anticorpi progettati per funzionare in ambiente intracellulare. Questi anticorpi intracellulari sono già in fase di studio come potenziali terapie in una serie di settori, tra cui le malattie neurodegenerative, cancro e infezioni virali 31. Questa tecnica permetterà un uso più diffuso di anticorpi intracellulari come strumenti per la ricerca e la medicina in questi campi e qualsiasi altro campo in cui lo studio di una proteina bersaglio in situ è desiderato.
Materials
| Name | Company | Catalog Number | Comments |
| scFv library | Varies | A suitable scFv library should be obtained from a commercial or academic source. | |
| MC4100 E. coli cells | Coli Genetic Stock Center | 6152 | Cells need to be chemically competent or electrocompetent, depending on the selected transformation method. |
| Glycerol | Fisher Scientific | BP229-4 | |
| Difco dehydrated culture media LB Broth, Miller (Luria-Bertani) | BD | 244610 | |
| Chloramphenicol (Cm) | Fisher Scientific | BP904-100 | |
| Sodium chloride (NaCl) | Fisher Scientific | BP358-1 | |
| Potassium chloride (KCl) | Fisher Scientific | BP366-500 | |
| Sodium phosphate, dibasic (Na2HPO4) | Fisher Scientific | BP332-500 | |
| Potassium phosphate, monobasic (KH2PO4) | Fisher Scientific | BP362-500 | |
| Bovine serum albumin (BSA) | Fisher Scientific | BP9706-100 | |
| Sucrose | Fisher Scientific | BP220-1 | |
| Tris base | Fisher Scientific | BP1521 | |
| Ethylenediaminetetraacetic acid (EDTA), 0.5 M | Fisher Scientific | BP2482-500 | |
| Magnesium chloride (MgCl2) | Fisher Scientific | BP214-500 | |
| Lysozyme | Sigma Aldrich | L3790-10X1ML | |
| Vortex mixer | VWR | 97043-564 | |
| Bicine | Fisher Scientific | BP2646100 | |
| D-Biotin | Fisher Scientific | BP232-1 | |
| Isopropyl β-D-1-thiogalactopyranoside | Fisher Scientific | BP1755-1 | |
| BugBuster Master Mix (cell lysis detergent) | EMD Millipore | 71456 | |
| Vivaspin 2 MWCO, 3,000 daltons | GE Healthcare Sciences | 28932240 | |
| Target antigen | Varies | N/A | Purified target antigen may be purchased or produced/purified. |
| Dynabeads MyOne Streptavidin T1 | Invitrogen | 65601 | |
| Dynamag-2 magnet | Invitrogen | 12321D | |
| Tube rotator | VWR | 13916-822 | |
| PCR primers | IDT | N/A | Primer sequences are as described in the protocol. |
| 10x T4 DNA ligase reaction buffer | New England BioLabs | B0202S | |
| T4 Polynucelotide kinase (PNK) | New England BioLabs | M0201S | Make sure the T4 ligase buffer used in the primer phosphorylation reaction contains 1 mM ATP. |
| 5x Phusion HF buffer pack | New England BioLabs | B0518S | |
| Deoxynucleotide (dNTP) solution mix, 10 mM each dNTP | New England BioLabs | N0447L | |
| Phusion DNA polymerase | New England BioLabs | M0530S | Other high-fidelity polymerases may be used as an alternative, but the annealing temperature in Table 3 must be adjusted. |
| C1000 Touch thermal cycler with dual 48/48 fast reaction module | Bio-Rad | 185-1148 | |
| Agarose | Promega | V3121 | |
| SYBR Safe DNA gel stain | Invitrogen | S33102 | |
| Wizard SV gel and PCR clean-up system | Promega | A9281 | |
| T4 DNA ligase | New England BioLabs | M0202S | |
| Microdialysis membrane filter | EMD Millipore | VSWP04700 | |
| Agar | BD | 214030 | |
| 96-well polystyrene round-bottom cell culture plates | VWR | 10062-902 | |
| Costar general polystyrene assay plate lids | Corning | 3931 | |
| Microtitre plate shaker | VWR | 12620-926 | |
| Costar 96 well EIA/RIA Easy Wash clear flat bottom polystyrene high bind microplate | Corning | 3369 | |
| Bel-blotter polycarbonate 96-well replicating tool | Bel-Art Products | 378760002 | |
| Instant nonfat dry milk | Quality Biological | A614-1000 | |
| Tween 20 (polysorbate 20) | Fisher Scientific | BP337-500 | |
| PopCulture reagent (concentrated cell lysis detergent) | EMD Millipore | 71092-3 | |
| Monoclonal ANTI-FLAG M2-Peroxidase(HRP) antibody produced in mouse | Sigma Aldrich | A8592 | |
| SigmaFast OPD | Sigma Aldrich | P9187-50SET | |
| Sulfuric acid (H2SO4), 10 N solution | Fisher Scientific | SA200-1 | |
| Reynolds Wrap aluminum foil | VWR | 89079-075 | |
| BioTek Epoch microplate spectrophotometer | Fisher Scientific | 11120570 |
References
- Biocca, S., Pierandrei-Amaldi, P., Campioni, N., Cattaneo, A. Intracellular immunization with cytosolic recombinant antibodies. Biotechnology (NY). 12 (4), 396-399 (1994).
- Chen, S. Y., Bagley, J., Marasco, W. A. Intracellular antibodies as a new class of therapeutic molecules for gene-therapy. Hum. Gene Ther. 5 (5), 595-601 (2008).
- Gargano, N., Biocca, S., Bradbury, A., Cattaneo, A. Human recombinant antibody fragments neutralizing human immunodeficiency virus type 1 reverse transcriptase provide an experimental basis for the structural classification of the DNA polymerase family. J Virol. 70 (11), 7706-7712 (1996).
- Mhashilkar, A. M., et al. Inhibition of HIV-1 Tat-mediated LTR transactivation and HIV-1 infection by anti-Tat single chain intrabodies. Embo J. 14 (7), 1542-1551 (1995).
- Strube, R. W., Chen, S. Y. Characterization of anti-cyclin E single-chain Fv antibodies and intrabodies in breast cancer cells: enhanced intracellular stability of novel sFv-F-c intrabodies. J. Immunol. Meth. 263 (1-2), 149-197 (2002).
- Mössner, E., Koch, H., Plückthun, A. Fast selection of antibodies without antigen purification: adaptation of the protein fragment complementation assay to select antigen-antibody pairs. J. Mol. Biol. 308 (2), 115-122 (2001).
- Wörn, A., et al. Correlation between in vitro stability and in vivo performance of anti-GCN4 intrabodies as cytoplasmic inhibitors. J Biol Chem. 275 (4), 2795-2803 (2000).
- Knappik, A., Plückthun, A. Engineered turns of a recombinant antibody improve its in vivo folding. Protein Eng. 8 (1), 81-89 (1995).
- Martineau, P., Jones, P., Winter, G. Expression of an antibody fragment at high levels in the bacterial cytoplasm. J Mol Biol. 280 (1), 117-127 (1998).
- Steipe, B., Schiller, B., Plückthun, A., Steinbacher, S. Sequence statistics reliably predict stabilizing mutations in a protein domain. J. Mol. Biol. 240 (3), 188-192 (1994).
- Daugherty, P. S. Protein engineering with bacterial display. Curr Opin Struct Biol. 17 (4), 474-480 (2007).
- Lener, M., et al. Diverting a protein from its cellular location by intracellular antibodies. Eur. J. Biochem. 267 (4), 1196-1205 (2000).
- Lynch, S. M., Zhou, C., Messer, A. An scFv intrabody against the nonamyloid component of α-synuclein reduces intracellular aggregation and toxicity. J. Mol. Biol. 377 (1), 136-147 (2008).
- Gai, S. A., Wittrup, K. D. Yeast surface display for protein engineering and characterization. Curr. Opin. Struc. Biol. 17 (4), 467-473 (2007).
- Kieke, M. C., et al. Selection of functional T cell receptor mutants from a yeast surface-display library. Proc. Natl. Acad. Sci. USA. 96 (10), 5651-5656 (1999).
- Steiner, D., Forrer, P., Stumpp, M. T., Pluckthun, A. Signal sequences directing cotranslational translocation expand the range of proteins amenable to phage display. Nat. Biotechnol. 24, 823-831 (2006).
- Pugsley, A. P. The complete general secretory pathway in gram-negative bacteria. Microbiol. Rev. 57 (1), 50-108 (1993).
- Rapoza, M. P., Webster, R. E. The filamentous bacteriophage assembly proteins require the bacterial SecA protein for correct localization to the membrane. J. Bacteriol. 175 (6), 1856-1859 (1993).
- Karlsson, A. J., et al. Engineering antibody fitness and function using membrane-anchored display of correctly folded proteins. J. Molec. Biol. 416 (1), 94-107 (2012).
- DeLisa, M. P., Tullman, D., Georgiou, G. Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc Natl Acad Sci U S A. 100 (10), 6115-6120 (2003).
- Fisher, A. C., Kim, W., DeLisa, M. P. Genetic selection for protein solubility enabled by the folding quality control feature of the twin-arginine translocation pathway. Protein Sci. 15 (3), 449-458 (2006).
- Maynard, J., Georgiou, G. Antibody engineering. Annu Rev Biomed Eng. 2, 339-376 (2000).
- Green, M. R., Sambrook, J. Molecular Cloning: A Laboratory Manual. 1, Fourth, Cold Spring Harbor Laboratory Press. (2012).
- Milo, R., Jorgensen, P., Moran, U., Weber, G., Springer, M. BioNumbers--the database of key numbers in molecular and cell biology. Nucleic Acids Res. 38, D750-D753 (2010).
- Hermanson, G. T. Bioconjugate Techniques. , Third, Elsevier/Academic Press. (2013).
- Tayapiwatana, C., Chotpadiwetkul, R., Kasinrerk, W. A novel approach using streptavidin magnetic bead-sorted in vivo biotinylated survivin for monoclonal antibody production. J Immunol Methods. 317 (1-2), 1-11 (2006).
- Zhu, G., Song, L., Lippard, S. J. Visualizing inhibition of nucleosome mobility and transcription by cisplatin-DNA interstrand crosslinks in live mammalian cells. Cancer Res. 73 (14), 4451-4460 (2013).
- Bogsch, E. G., et al. An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J. Biol. Chem. 273, 18003-18006 (1998).
- Julian, M. C., et al. Co-evolution of affinity and stability of grafted amyloid-motif domain antibodies. Protein Eng. Des. Sel. 28 (10), 339-350 (2015).
- Garber, K. Bispecific antibodies rise again. Nat. Rev. Drug. Discov. 13 (11), 799-801 (2014).
- Marschall, A. L., Dübel, S., Böldicke, T. Specific in vivo knockdown of protein function by intrabodies. MAbs. 7 (6), 1010-1035 (2015).
