

Introduction
折りたたみ及び細胞内環境内で機能することができる抗体は、研究および治療用途の両方のための有望なツールです。これらは、タンパク質-タンパク質相互作用を防止するために、細胞内で標的タンパク質に結合することによってタンパク質の活性を調節する能力を有するタンパク質-核酸相互作用を破壊する、または酵素1-5に基板へのアクセスを防ぎます。
抗体が細胞内のアプリケーションのための多くの可能性を有するが、標的抗原に結合する能力を維持しながら、細胞内環境における適切な折り畳みと溶解性のためにそれらを操作することは困難です。還元性の細胞質環境は、通常、一本鎖可変断片(scFv)で抗体6,7-含む完全長抗体および抗体フラグメントの安定なフォールディングに必要なジスルフィド結合の形成を防止します。指向進化アプローチの数は、hを有する抗体を設計するために使用されています標的抗原8-10のためのIGH親和性。これらのアプローチは、一般的に、抗体11-13の大きなライブラリーをスクリーニングするファージディスプレイ、酵母表面ディスプレイ、細菌表面ディスプレイを使用します。これらの方法は、標的に結合する抗体を同定するための強力かつ効果的で、まだ彼らは14から16に表示されるタンパク質を輸送するために分泌経路に依存しています。分泌経路は、酵母または細菌中のペリプラズムへの小胞体内腔に減少させる細胞質から展開されたタンパク質を移動します。タンパク質は、次いで、酸化条件下で折り畳むと、細胞表面上に提示または親和17,18の結合についてスクリーニングするファージ粒子中にパッケージングされます。結果として、これらの技術を用いて単離された抗体は、必ずしも細胞質内にうまく折り畳まれず、抗体は細胞内のアプリケーションで使用される場合、細胞内の溶解性は、多くの場合、個別に操作されなければなりません。
改善するために、よく細胞質内に折り畳まれる抗体を改変の効率は、我々は以前MAD-TRAP(関連付けるタンパク質のタットベースの認識のための膜固定表示)の成功、 大腸菌 inner-使用して、scFv抗体ライブラリーをスクリーニングするための方法を報告しましたメンブレンディスプレイ19。細菌の内側の膜ディスプレイは、分泌経路を使用する他の一般的な表示方法とは対照的に、表示された抗体を輸送するためのツインアルギニン転位(TAT)経路に依存しています。 Tat経路は、可溶性の正しく折り畳まれたタンパク質はE.から輸送されることを可能にする品質制御機構を含んでいます内側の膜を横切る、およびペリプラズム20,21に大腸菌細胞質、。過剰発現したTat基質( すなわち 、TatのシグナルペプチドssTorAへのN末端融合でTat経路を標的とするタンパク質)も細胞質内に折り畳まれているN末端の私との中間長寿命転座を形成します細胞質とペリプラズム19におけるC末端のn。これは、Eのペリプラズム面には、抗体フラグメントを含む、正しく折り畳まれたTat基板の表示を可能にします大腸菌内膜。スフェロプラストを生成するために酵素消化により外膜を除去した後、抗体は、細胞外空間( 図1)にさらされています。これは、内膜に表示Tatの基板は、特定の標的への結合についてスクリーニングすることを可能にします。重要なのは、細胞表面ディスプレイのためのTat経路を利用すると、結合親和性および細胞内フォールディングの同時エンジニアリングを可能にする、十分に細胞質内に折り畳まれているライブラリ内の唯一の抗体が結合するために尋問されることを保証します。このプロトコルでは、我々は、E上のscFvライブラリーを表示する方法について説明します大腸菌内膜は、標的抗原に対するライブラリをパンし、ライブラリの最も有望な成分を同定するための二次スクリーニングを行います。我々は集中しながら、 scFvのプロトコルは、この方法は、そのアプリケーションの結合および細胞内折り畳みを必要とする任意のタンパク質を操作に適用することができます。
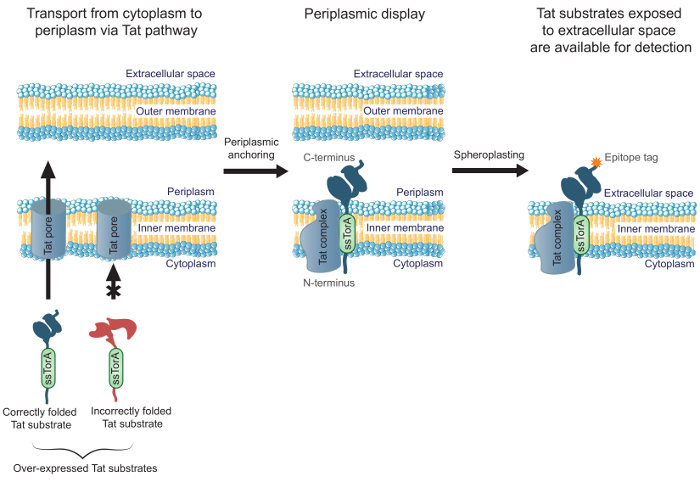
図1.タット内側の膜が表示されます。E.で大腸菌は、ssTorAシグナル配列の融合体として発現し、正常に細胞質内に折り畳まれたscFv抗体は、内膜を横切って輸送されています。 scFvをが細胞質中のN末端およびペリプラズム中のC末端と、内膜に固定されている転座の中間形態。 E.大腸菌の外膜を酵素的に、それによって細胞外空間に固定する抗体を露出し、表示された抗体のC末端に融合したエピトープタグに特異的に結合する抗体を用いて検出するためにそれらを利用可能にする、スフェロプラストを形成するために消化されます。ロード/ 54583 / 54583fig1large.jpg "ターゲット=" _空白 ">この図の拡大版をご覧になるにはこちらをクリックしてください。
Protocol
1. ssTorAシグナル配列に融合としてのscFvライブラリーを作製
- scFv遺伝子の変異体を含むデオキシリボ核酸(DNA)ライブラリーを得ます。
注:ライブラリはまた、全体のscFv遺伝子または標的ドメインにわたる多様性を生成するために、任意の適切なモードを使用して構築することができる22( 例えば 、領域の3つの相補性決定、のCDR3。)。 - 標準的な分子クローニング法23を使用してPIMDプラスミド( 図2)にDNAライブラリーを挿入します。
注:このプラスミドは(のscFvのN末端)ssTorAシグナル配列に遺伝子融合およびFLAGエピトープタグ(のscFvのC末端)としてscFvを発現します。内膜のディスプレイのためのプラスミドの設計は、これまで19に記載されています。 PIMDプラスミドは、著者から入手可能です。

図2.インナー膜ディスプレイプラスミド(PIMD)マップ(1.3を介してステップ1.2)。このプラスミドは、lacプロモーター、複製のColE1起点、およびクロラムフェニコール耐性遺伝子を含みます。挿入されたscFv遺伝子は、同じリーディングフレーム内に3つのすべてで、Tat経路およびFLAGエピトープタグへのscFvを標的とするssTorAシグナル配列に融合されます。制限酵素部位が示されています。 XbaIおよびNotI制限酵素部位の間に挿入ライブラリーのために、プラスミドの大きさは2219 bpのプラスscFvの大きさである。 この図の拡大版をご覧になるにはこちらをクリックしてください。
- MC4100 大腸菌にライブラリを含むプラスミドDNAをトランスフォームcoli細胞を 23。このライブラリの細菌の形を回復し、成長します。 RTで15分間、4000×gで遠心分離し、細胞を回収しました。上清を除去し、回収した細胞を再懸濁ルリア - ベルターニ(LB)培地中で、25%グリセロールです。必要になるまで-80℃でアリコートを保存、またはステップ2に進みます。
注:プロトコルは、他のE.ものの、MC4100細胞で確認されています大腸菌株は、プロトコルと互換性があることが予想されます。エレクトロポレーションは、高い形質転換効率による形質転換のための好ましい方法です。ライブラリーは、典型的には少なくとも10 scFvは、この段階での変異体9から構成されなければならない、と各アリコートは、ライブラリが100倍に覆われているように、十分な細胞が含まれている必要があります。
2.ライブラリを表現し、スフェロプラストを準備
- RTで(ステップ1.3から)は、細菌ライブラリーの1つのアリコートを解凍し、20μg/ mlのクロラムフェニコール(CM)と100ミリリットルのLB培地を含むフラスコにアリコートを追加します。 37°Cとインキュベートシェーカーで225rpmで3時間成長します。
- 3時間後、37°Cからフラスコを取り外し、シェーカーをインキュベートしました。にscFvライブラリーの発現を可能にします20℃で15〜22時間インキュベートシェーカーで225rpmでのためにO / Nを進みます。
注:PIMDプラスミドを使用した場合、プロモーターが漏れやすいようませ誘導物質は、必要ありません。 MC4100細胞はLacリプレッサーを過剰発現していない(とのLacIがプラスミド上に発見されていない)ことに注意してください。

図3. 大腸菌 細胞及びスフェロプラスト(A)E. coli細胞の形状は円筒形です。 (B) EDTA及びリゾチーム、 大腸菌の外膜を使用してスフェロプラスト後大腸菌細胞が破裂され、そして得られたスフェロプラストは、形状が球状です。微分干渉コントラスト(DIC)顕微鏡画像は、倒立顕微鏡で100倍の対物レンズを用いて得られたくださいCこの図の拡大版をご覧になるにはこちらをなめます。
- ライブラリースフェロプラストを準備します。
注:スフェロプラストは、Eの外膜を破裂させることによって形成されますcoliおよび形状が球状である( 図3)。- 必要な緩衝液を準備します。
注:すべての緩衝液は、無菌であるべきです。- 蒸留水1,000 mlの最終容量に2 O 2 PO 4 HPO 4 8gのNaCl、0.2gのKCl 1.44 G 2ナトリウムを溶解し;(pH7.4のPBS)、0.24グラムのKH 1×リン酸緩衝生理食塩水を準備します。氷の上に保管してください。
- 200 ML 1×PBS中に0.2グラムのBSAを溶解し、0.1%(w / v)のウシアルブミン血清(BSA)を含むPBSを準備します。氷の上に保管してください。
- 滅菌濾過した1 Mのショ糖、1 Mトリス緩衝液(pH8.0)、1.5mlの1 mlの蒸留H 2 O 7.5 mlと混合することによって分画バッファ(FB)を調製氷の上に保管してください。
- 30μlのOを添加することによって、1mMのエチレンジアミン四酢酸(EDTA)を調製H 2 Oを蒸留F 0.5 M EDTAを14.97ミリリットル
- H 2 Oを100mlの蒸留中に4.76グラムのMgCl 2を溶解し、0.5MのMgCl 2を準備氷の上に保管してください。
- シェーカーからフラスコを取り出し、細胞密度を決定するために、分光光度計を用いて600nmでの光学密度(OD)を測定します。誘導された培養物の容量は、スフェロプラストのために、各サンプルは、1×10 10細胞を有するように必要に応じて計算します。
注:E. 10 9細胞/ mlの濃度を示す1のOD 600の近似大腸菌は 、24を使用することができます。 - 5分間遠心室温で12,000×gで1.5ミリリットルマイクロチューブにおける誘導文化の計算されたボリューム。問題は、サンプル調製に生じた場合には、少なくとも二つのサンプルを準備します。
- 遠心分離培養物から上清を除去し、氷冷FB100μlの各細胞ペレットを再懸濁します。 12,000×で遠心分離1分間室温でグラム、その後、ピペットで上清を除去します。氷冷FB350μlの再懸濁各ペレットは、10mg / mlのリゾチームの3.5μlのを補いました。
- ゆっくりと、1 mMのEDTAの700μLを滴下しながら、各チューブをボルテックスし、ゆっくりとサンプルを混合するためにチューブ回転で回転させながら室温で20分間チューブをインキュベートします。回転子からチューブを取り外し、各チューブに氷冷した0.5 MのMgCl 2の50μlを添加し、10分間氷上でインキュベートします。遠心分離機10分間、4℃で11,000×gでチューブ。
- スフェロプラストペレットを分離します。
- ゆっくりとペレットの一部をプルアップするために1ミリリットルの先端でマイクロピペットを使用してください。直接新しい1.5 mlチューブ上部の開口部との角度でチューブを保持しながら、ゆっくりと上清のうち、ピペットチップを持ち上げ、新しいチューブにペレットをスライドさせます。
- 上清の重要なボリュームが新しいチューブに移している場合は、ピペットでそれを削除します。ペレットは、企業ENOされていない場合ぐふ2分間11,000×gで、再遠心分離を転送し、再びペレットの分離を試みます。
- 氷冷1×PBS 1 mlに各チューブ内のスフェロプラストペレットを再懸濁します。ピペッティングし、ペレットを完全に再懸濁されるまで、ゆっくりと渦ホルダーにボルテックスの間で代替。一度に複数の2分間氷のオフにサンプルを維持し、再び氷から削除する前に少なくとも5分間氷上に戻らないでください。ステップ4でパンニングに使用するまで(最大2日間)4℃でスフェロプラストを保管してください。
- 必要な緩衝液を準備します。
3.磁気ビーズ上に標的抗原を固定化
- 大腸菌中での組換え産生中にin vivoで標的抗原をビオチン化coli細胞。また、化学的結合25または既にビオチン化された購入対象抗原を使用し、3.2に進みます。
- 10×ビシンバッファを作るために50ミリリットルの水に816グラムのビシンを追加します。 BUFを希釈50°Cまでの蒸留H 2 Oと暑さの中で1×にFER。 10mMのビシン緩衝液中の5 mMのビオチンはビオチン溶液を作るために加熱された1×ビシンバッファの12ミリリットルに14.7 mgのビオチンを追加します。必要になるまで-20℃で保管してください。
- ビオチンカルボキシルキャリアタンパク質(BCCP)への融合として、標的抗原の産生を可能にするpAK400cb-BCCPプラスミド26を用いて、標的タンパク質を発現し、ビオチン化。
注:E. coli細胞ネイティブビオチニル化BCCP、精製および化学的に前ストレプトアビジンでコーティングしたビーズへの固定化に標的タンパク質をビオチン化する必要がなくなります。ネイティブE.大腸菌ビオチンリガーゼBirAを、融合タンパク質をビオチン化するために十分です。- E.を育てますで振盪しながら37℃で20 / mlのCmを用いて補充したLB培地5ml中に15〜18時間(BCCPのN末端への融合物として挿入された標的抗原を有する)ビオチン化プラスミドO / Nを含有する大腸菌 225 rpmで。
- 分光光度計を用いて600nmでODを測定し、必要な文化の体積を計算(V アド )式を用いて20μgの/ mlのCmを用いて25ミリリットル中0.05の開始OD新鮮なLB培地で継代培養する:V(= 0.05×を追加 25ミリリットル)/(OD 600 - OD 600は、O / N培養およびV アドオンの光学密度である0.05)は、新鮮なLBに追加するO / N培養の体積でありますサブカルチャーとは、37℃、225rpmでインキュベートシェーカーで0.8に0.5のODまで成長します。
- 5μMの最終濃度100μMとビオチンの最終濃度までイソプロピルβ-D-1-チオガラクトピラノシドを追加します。 20°Cおよび225 rpmで15〜22時間インキュベートシェーカーで発現を誘導します。
- 10分間、4℃で4000×gで遠心分離によって収穫細菌。上清を取り除きます。使用するまで-20℃でペレットを保管してください。
- 加えます細胞ペレットを0.2g当たり細胞溶解界面活性剤を1ml。ピペッティングにより再懸濁し、穏やかに細胞を溶解するために20分間回転させます。溶解した後、16,000×gで遠心分離し、20分間、4°C。新しい1.5 mlチューブに可溶性ライセート(上清)をピペットで。
- 未結合ビオチンを除去するために、3 kDaの分子量カットオフの列を使用します。製造元の指示に従って、20℃でカラムにライセート、および遠心分離機をピペット。溶解産物中のビオチンは、100倍に希釈され、洗浄溶解物の体積は、溶解物の元の容積に等しくなるまで1×PBSで洗浄します。新しいチューブにライセートを移します。
- ストレプトアビジン被覆磁気ビーズ上にビオチン化標的抗原を固定化します。
- ステップ2.3.1で説明したように1×PBSおよびBSA(w / v)の0.1%と1×PBSを準備します。
- 磁気ビーズを準備します。
注:これは、磁気分離ラックの使用を必要とします。- 再懸濁streptav元のバイアル中の磁性ビーズをidinがコーティングされました。または少なくとも30秒間のいずれかボルテックスで5分間回転させます。
- 1.5mlチューブに7-10×10 9ビーズを転送します。
注:必要な容量は、製造業者によって供給されたビーズ濃度に依存することになります。 - チューブの側面にビーズを収集するために2分間磁石ラックのビーズを含むチューブを置きます。まだ磁石にチューブを、慎重にビーズを中断することなく、ピペットで上清を除去します。
- 洗浄するために、磁石からチューブを削除し、気泡を発生させずにピペットで1×1mlのPBSにビーズを再懸濁します。ビーズを収集するために2分間磁石にチューブを戻し、慎重にピペットで上清を除去します。合計3回の洗浄のためのプロセスをさらに2回繰り返します。何の液体が最終洗浄後にチューブ内に残っていないことを確認してください。
- 磁気BEAにビオチン化抗原を含む溶解液を追加します。DS。
- 磁石からチューブを外し、(ステップ3.1.5から)溶解物の1ミリリットル中のビーズを再懸濁します。静かに回転させながら室温で30分間インキュベートします。
- 抗原被覆ビーズを収集するために3分間磁石上にチューブを置きます。ステップ3.2.2.4に3.2.2.3に記載したのと同じ方法で0.1%のBSAを用いて1×PBSでコーティングされたビーズを5回洗浄します。最終洗浄後、ステップ3.2.2.2で使用したのと同じ量まで0.1%BSAで1×PBS中でビーズを再懸濁します。
- 固定された標的抗原は4℃で安定している場合は、パニングのために必要とされるまで、4℃でコーティングしたビーズを格納します。それ以外の場合は、ステップ4に進みます。
4.画面のターゲット抗原に対するパニングによってscFvをライブラリ(図4)

図4パニング(ステップ4)。抗原被覆磁性ビーズは、AReは、抗体ライブラリーの変異体を発現スフェロプラストとインキュベートしました。ビーズに結合したスフェロプラストからプラスミドDNAを回収し、ELISAベースの二次スクリーニングを用いてスクリーニングされたサブライブラリを生成するために使用されます。対応するプロトコルのステップが記載されています。 この図の拡大版をご覧になるにはこちらをクリックしてください。
- スフェロプラストでコーティングしたビーズをインキュベートします。
- 約5の比率をビーズにスフェロプラストを使用してください:1。滅菌15mlチューブに4×10 9スフェロプラストと8×10 8ビーズを追加します。
注:濃度はまだ1×10 10スフェロプラスト/ mlとなるように何の細胞は、スフェロプラストの処理中に失われなかったと仮定します。 - 4ミリリットルに全量を0.1%BSAで1×PBSを追加します。 1ミリリットルそれぞれ有する4つの1.5ミリリットルチューブに小分けし。静かに回転させながら5時間4℃で反応をインキュベートします。
- 約5の比率をビーズにスフェロプラストを使用してください:1。滅菌15mlチューブに4×10 9スフェロプラストと8×10 8ビーズを追加します。
- 広報ポリメラーゼ連鎖反応(PCR)のためのビーズに結合したスフェロプラストをepare。
- 3分間磁石上にパンニング反応チューブを置きます。ピペットで上清を除去し、3.2.2.4にステップ3.2.2.3で説明したのと同様に、0.1%BSAで冷1×PBS氷でビーズに結合したスフェロプラストを4回洗浄します。蒸留H 2 Oの25μlの各管中のビーズに結合したスフェロプラストを再懸濁し-20℃でビーズを格納または4.3に進みます。
- ビーズに結合したscFvの遺伝子を含むプラスミドを増幅するために、ビーズに結合したスフェロプラストの全プラスミドPCRを行います。
- 5'CCAACTCTTTTTCCGAAGGTAACTG3 '(フォワードプライマー)と5'TAGCTCTTGATCCGGCAAACAAA3'(リバースプライマー):以下の配列を有するプライマーを取得します。
注:これらは、エンドツーエンドのPIMDプラスミドの反対の鎖に( 図2)に結合し、PIMDの共通の特徴にアニールするように設計されているので、増幅はかかわらず発生しますscFv変異体配列。 - プライマーをリン酸化します。
注:リン酸化がなければ、再ライゲーションは発生しません。プライマーはまた、むしろ、このプロトコルでは、このリン酸化方法を使用するよりも、5'-リン酸化と一緒に注文することができます。- 表1に記載されているように0.5ミリリットルのチューブでは、フォワードPCRプライマーのためのリン酸化反応を設定します。逆方向プライマーのためにこのプロセスを繰り返します。
- 37℃で1時間反応をインキュベートします。そして、T4ポリヌクレオチドキナーゼ(PNK)を無効にするには20分間65℃でそれらをインキュベートします。 -20℃でリン酸化プライマーを保管してください。
- PCRを行います。
- 表2に記載したようにPCRチューブに、PCR反応を準備します。
注:複数の反応はより高い収率のために調製することができます。未使用のビーズに結合したスフェロプラストは、-20℃で保存することができます。 - 完全な溶解を確実にするためにサーマルサイクラーで15分間、98℃でPCR反応を加熱スフェロプラストの。サーマルサイクラーからチューブを外し、それぞれに高忠実度ポリメラーゼの0.5μlを添加します。サーマルサイクラーにチューブを戻し、 表3に詳述したプログラムを使用して実行します。
- 適切なPCR産物をプール。 -20℃で保存したり、4.4に進みます。
- 表2に記載したようにPCRチューブに、PCR反応を準備します。
- 5'CCAACTCTTTTTCCGAAGGTAACTG3 '(フォワードプライマー)と5'TAGCTCTTGATCCGGCAAACAAA3'(リバースプライマー):以下の配列を有するプライマーを取得します。
表1 PNKのリン酸化反応(ステップ4.3.2.1)。
| 試薬 | 容量(μL) |
| 蒸留H 2 O | 15 |
| 10×T4 DNAリガーゼ反応バッファー | 2 |
| 100μMプライマー | 2 |
| T4ポリヌクレオチドキナーゼ(PNK) | 1 |
| 試薬 | 容量(μL) |
| 蒸留H 2 O | 28.5 |
| 5倍の高忠実度ポリメラーゼ緩衝液 | 10 |
| 10μMのリン酸化のフォワードプライマー | 2.5 |
| 10μMのリン酸化リバースプライマー | 2.5 |
| 40mMのdNTPミックス(10 mMの各dNTP) | 1 |
| ビーズに結合したスフェロプラスト | 5 |
表3のPCRプログラム(ステップ4.3.3.2)。
| ステップ | 温度(℃) | 時間(分:秒) | サイクル数 |
| 初期変性 | 98 | 0時30分 | 1 |
| 変性 | 98 | 0時10分 | 35 |
| アニーリング | 69 | 0時30分 | |
| 拡張 | 72 | キロバイトあたりの夜12時30分 | |
| 最終エクステンション | 72 | 6時 | 1 |
| ホールド | 12 | 無限 | 1 |
- 再環状化全プラスミドPCR産物を、およびMC4100 大腸菌を形質転換するためにライゲーションされた製品を使用coli細胞。
- アガロースゲル23のPCR反応を実行してゲル23にDNAを染色し、使用して、PCR産物を精製し製造業者によって供給された指示に従うことによって線状化プラスミドを精製するためのゲルクリーンアップキット。 260nmで分光光度計を用いて濃度を測定します。必要になるまで-20℃で精製した断片を保存、または4.4.2に進みます。
- PCR産物からのプラスミドを再環状化。
- PCR産物の分子間ライゲーションを防止するために、PCR産物の1ng /μLの低濃度27との連結反応を行います。この濃度で800μlのライゲーション反応を調製するために必要な体積を計算します。
- 氷上でライゲーション反応を準備します。チューブでは、800μlのまでのPCR産物のステップ4.4.2.1で計算されたボリューム、10×DNAリガーゼ緩衝液80μlの、そして蒸留H 2 Oを追加します。 T4 DNAリガーゼの4μlを添加し、直ちに水浴またはサーマルサイクラー中で16℃でチューブを配置します。 14〜18時間、16°CO / Nでインキュベートします。 -20°Cで完了連結反応を保管してください必要になるまで、または4.4.3に進みます。
- DNAリガーゼを加熱、不活性化するために15分間65℃でヒートブロック上のライゲーション反応を置きます。その後、連結したDNAを、塩を解除するために微小透析膜やDNAクリーンアップキットを使用します。 20℃で保存するか、または4.4.4に進みます。
- MC4100 大腸菌を形質転換するために全体の熱不活性化、脱塩漬け連結産物を使用してくださいcoli細胞を 23。 -80℃で、得られたパンニングサブライブラリを含む細胞、および店舗のアリコートを、ステップ1.3で説明したように、グリセロールストックを準備します。
- サブライブラリ上の第二のパンニングを行うために、ステップ4.4.4からのアリコートを使用してその全体を繰り返し、ステップ4。
注:第二のパンは、標的抗原19に良好に結合するライブラリーの成分を濃縮することができます。
5.さらなる特徴付けのために有望なクローンを同定するために酵素結合免疫吸着アッセイ法を用いた二次スクリーニングを行います(図5)</ P>

図5 ELISAに基づく二次スクリーニング(ステップ5)。パンニング中富化サブライブラリから(A)ライブラリの変異体は、増殖および発現のための培養プレートの個々のウェルに接種します。 (B)ELISAプレートを、標的抗原で被覆されています。 (C)ライブラリの変異体は、プロトコルに記載のELISAベースの二次スクリーニングを用いてスクリーニングされます。二次スクリーニングから得られたデータの分析に、興味のある変異体が選択され、さらに特徴付けられます。対応するプロトコルのステップが記載されています。 この図の拡大版をご覧になるにはこちらをクリックしてください。
- LB寒天プレート上にプレート(ステップ4.4.4から)パンニングサブライブラリーの1のチューブを解凍します。プレートいくつかの希釈( - 10〜6倍希釈液を例えば 、10 2)個々のコロニーを確保するのに十分低い濃度で。 37℃で15〜18時間、プレートをインキュベートします。 4℃でプレートを保管または5.2に進みます。
- パンニングサブライブラリーからコロニーを培養し、誘導します。無菌条件下でのすべての手順を実行します。 96ウェルプレートを伴う工程のためのマルチチャンネルピペッターを使用してください。
- 丸底96ウェル培養プレートの各ウェルに20 / mlのCmを含むLBの200μLを加えます。
- 、ピペットチップで寒天プレートからの個々のコロニーを採取し、96ウェルプレートの最初のウェルにチップを置き、静かに接種するためにかき混ぜます。各ウェルのための新しいチップを使用してください。各ウェルに1つのコロニーを接種します。対照として、接種していないコロニーとよく少なくとも一つの無菌性のコントロールが含まれています。
- いくつかの96ウェルプレートに接種し、ステップ5.2.1と5.2.2を繰り返します。
- 310 rpmでマイクロプレートシェーカー上で96ウェルプレートを置きます。 37インキュベート6; 20〜24時間のためのCは、scFvを発現させること。
- ステップ5.2で調製した各培養プレート、標的抗原でコート1、96ウェルELISAプレートのために。
- コーティング溶液を作るために1×PBSで適切な濃度(4 / mlのに例えば、1μgの/ mlの)に精製された標的抗原を希釈します。各96ウェルプレートのためのコーティング溶液5mlを加えます。
注:適切な濃度は、使用されている特定の抗原に依存し、調整する必要があります。 - 96ウェル高結合クリアポリスチレンELISAプレートの各ウェルにコーティング溶液50μlを加えます。静かに、各ウェルの全表面がコーティングされていることを確認するために、ベンチトップ面にプレートをタップします。各プレートについて、この手順を繰り返します。 4°CO / Nでプレートをインキュベートします。
- コーティング溶液を作るために1×PBSで適切な濃度(4 / mlのに例えば、1μgの/ mlの)に精製された標的抗原を希釈します。各96ウェルプレートのためのコーティング溶液5mlを加えます。
- 寒天プレート上に96ウェル培養プレートからコロニーを複製します。
- liquの少量を収集するために培養プレートのウェルに滅菌ポリスチレンレプリケータを配置ID。慎重にレプリケータを上げ、すべての先端が板に触れているように、15cmのLB寒天プレートに移します。液体が移転した後は、レプリケータをまっすぐに持ち上げます。各培養プレートのために繰り返します。
- さらなる特性が所望されるならば、96ウェルプレート中で二次スクリーニングからの結果は、プレート上の正しい複製されたコロニーを一致させることができるように、正しい向きで寒天プレートにラベルを付けます。 15〜18時間、37℃で成長し、その後、必要になるまで4℃で保存します。
- ELISA二次スクリーニングを行います。
- 1×PBS中のドライミルク(w / v)の2%にすることによってブロッキング溶液を準備します。 ELISAプレートからのコーティング液を空にします。各ウェルにブロッキング溶液100μlを加えます。 4℃で少なくとも2時間、またはブロックO / NをRTでインキュベートします。
- 1×PBS中の0.05%の最終濃度にポリソルベート20を添加することにより、洗浄バッファーを準備します。 ELISAプレート当たり250ミリリットルを加えます。
- 20μlのを追加します。丸底培養プレートの各ウェルに細胞溶解界面活性剤を濃縮し、15〜20分間、室温でマイクロプレートシェーカー上で培養プレートをインキュベートします。溶解および洗浄工程5.5.4を同時に行うことができるようにELISAプレートのブロッキングが完了すると同時に溶解を始めます。
- ELISAプレートからブロッキング溶液を空にします。 1回の洗浄につきウェル当たり洗浄緩衝液200μlでブロックしたELISAプレートを4回洗浄します。ウェルから洗浄バッファーを空にします。
- 各ウェルについて新しいチップを使用して、ELISAプレートの対応するウェルに細胞溶解プレートの各ウェルからの転送を50μl。 1〜2時間、室温でELISAプレートをインキュベートします。
- バインドされたscFvを検出するために、抗体溶液を準備します。
- 西洋ワサビペルオキシダーゼ(HRP)は、ライブラリのscFvに融合したFLAGエピトープタグに結合する一次抗体を抱合使用。
- ELISA(参照サプライヤー&#で使用するために適切な希釈に抗体を希釈39; 1×PBS中の0.05%ポリソルベート20で2%での推奨)(w / v)のドライミルク。各プレートのための5ミリリットルを準備します。
- ステップ5.5.4で説明したようにELISAプレートを洗浄緩衝液で4回洗浄します。
- ELISAプレートの各ウェルに、抗体溶液50μlを加えます。室温で1〜2時間インキュベートします。
- 光を避けながら、製造業者のプロトコルごとに蒸留H 2 O中のOフェニレンジアミン二塩酸塩(OPD)錠を溶解することにより、HRP基質を準備します。 ELISAプレート当たり20ミリリットルを準備します。
- 必要に応じて蒸留H 2 Oで濃H 2 SO 4を希釈することによって3 MH 2 SO 4を準備します。 ELISAプレートあたり5ミリリットルを準備します。
注意:H 2 SO 4を強い酸です。適切な個人保護具を着用してください。 - ステップ5.5.4で説明したように、洗浄緩衝液でELISAプレートを4回洗浄します。
- HRPのsubstraでELISAプレートをインキュベートTE。
- 各ウェルにHRP基質200μlのを追加します。 、露光を最小限に一度に一つのELISAプレートに基板を追加し、次のプレートに進む前に、アルミホイルでラップします。暗所で室温で30〜60分間、プレートをインキュベートします。
- 最初の30分後、基板の黒ずみのためにプレートをチェックし、必要に応じて色を開発して可視化するために長くインキュベートします。
- 反応をクエンチするために各ウェルに3 MH 2 SO 4の50μlのを追加します。優しく泡立てずに上下にピペッティングすることにより、ウェル中の溶液を混ぜて、各ウェルに異なるチップを用いて。一貫性を保つためと飽和を防ぐために、各プレートのための溶液とを混合する前に、ELISAプレートのすべてにSO 4迅速かつ慎重にH 2を追加します。
- プレートリーダーを用いて492nmで各プレートのウェル内の溶液の吸光度を測定します。
- scFvはそのexhibバリアント識別するために、吸光度データを分析それが結合シグナルを約束し、これらの有望なscFvを特徴づけます。各プレートの平均信号よりもバックグラウンドシグナルよりも高く、高い吸光度信号を示すscFvを選択します。
注:吸光度レベルは、スクリーニングにおいて単離されたのscFv変異体の強さと一緒に、使用される抗原と抗FLAG抗体の特性に依存します。
Representative Results
E.におけるTat経路の細胞内のタンパク質フォールディング品質管理機構コリがよく、還元細胞質環境に折り畳まれるタンパク質に内部細胞膜を横切る輸送を制限します。 ssTorAシグナル配列(天然のTat経路20により搬送されたトラタンパク質からのシグナル配列)へのscFvの融合体を過剰発現することにより、転座は内膜19上のscFvの表示をもたらす、ストールします。外膜の酵素破壊した後、表示された抗体は、抗原結合活性をスクリーニングするために利用できるようになります。 scFvをディスプレイするためのTat経路を利用する能力は、カールソンらによって示された。19( 図6)。 scFv抗体scFv13とscFv13.R4はによって認識アルギニン - アルギニン残基のペアを欠いているネイティブssTorA配列または修正ssTorAのいずれかに融合させましたTat経路。 scFv13.R4は、指向進化の4ラウンドを通じてscFv13から。マーティらによって設計され、細胞質9でよく折り畳むことが知られています。このscFvは、内膜上に表示したが、ネイティブssTorAシグナル配列への融合物として発現のみ( 図6)。逆に、scFv13はよく細胞質9が折り畳まれていないので、関係なく、シグナル配列は、それが融合された、内膜にも表示されません。また、場合のscFvは、ディスプレイが内側の膜の表示およびTat経路間の重要なリンクを示す、観察されなかった、TATCタンパク質、タット機械20,28の重要なコンポーネントを欠いていた細胞で発現させました。これらの結果は、Tat経路を介した輸送は、細胞内FOL用スクリーンとして機能するように、正確に細胞質内に折り畳まれているだけTatシグナルペプチドを含むタンパク質とは、内膜上に表示されていることを示しています鼎。

内膜上に表示されたscFvの図6.検出。フローサイトメトリー分析は、内膜上の不十分折り畳まscFv13、よく折り畳まれたscFv13.R4の表示を検出しました。 scFvはssTorAシーケンス内のArg-ArgのペアがLys-Lysのに変更されたネイティブssTorAまたはssTorA(KK)に融合させました。 scFvのC末端FLAGエピトープタグは、フルオレセインイソチオシアネート(FITC)で検出した抗FLAG抗体を抱合。 FLAGタグなしTATCタンパク質(ΔtatC)とssTorA-scFv13ない細胞を対照として試験しました。 Mは、メジアン蛍光値を示しています。許可を得て、基準19から転載。 この図の拡大版をご覧になるにはこちらをクリックしてください。
19を向上させます。この、βガラクトシダーゼに対する結合親和性の低いレベルを有するscFv13に基づくエラープローンPCRライブラリーを実証するために、ディスプレイを使用して、標的抗原のβガラクトシダーゼに対してパニングプロトコルに記載の方法をパンニングしました。 scFvを1-4は、突然変異誘発の1ラウンドの後に単離され、パン、scFv13よりも( 図7A)および細胞質溶解性( 図7B)、より高いレベルのβ-ガラクトシダーゼするために、より高い結合親和性を示しました。
scFvを1-4に基づいて、新しいライブラリは、エラープローンPCRを使用して作られ、そしてに対するこの第二世代のライブラリーのパニングましたβガラクトシダーゼを記載したプロトコルの変形を使用して行きました。進化の第二ラウンドのためのβガラクトシダーゼに対するパニングは、scFv 1-4よりも高い親和性を有するクローンを単離する可能性を向上させるために競争相手として精製し、可溶性scFv 14の存在下で行いました。突然変異誘発とパンのこの第二ラウンドの後、scFvを2-1およびscFv 2-3は、ELISAに基づく二次スクリーニングを用いて単離しました。これらのscFvはscFv13よりβガラクトシダーゼに対する高い結合親和性を示しただけでなく、最初のラウンドのクローンのscFv 1-4よりも良好な結合を示しただけではなく。 scFvを2-1 scFv13.R4( 図7A)に匹敵する結合βガラクトシダーゼを示しました。 scFvを2-3また、溶解性および抗原結合の同時エンジニアリングを強調し、scFvを14に比べて細胞質溶解性のさらなる増加を示しています。親和性のscFvの可溶性の発現を同時にについてスクリーニングされるので、選択されたscFvは、MODを有することが可能ですERATEの溶解度が、高い結合、またはその逆。例えば、scFvを2-1は、scFv 2-3よりも低い水溶性発現を有するが、それはβ-ガラクトシダーゼする高い結合親和性を示します。

図7.ターゲット結合およびscFvの細胞質発現は、内側の膜ディスプレイを用いて単離し変種。(A)のscFvは、 大腸菌の細胞質で発現させました大腸菌細胞( 例えば 、ssTorAシグナル配列なし)ヘキサヒスチジン(6×-His)タグおよびニッケル-ニトリロ三酢酸のスピンカラムを用いて精製とは。 β-ガラクトシダーゼ、精製scFvの結合は、ELISAで測定しました。精製されたscFvはβガラクトシダーゼでコーティングしたELISAプレートにロードし、結合したscFvを抗6×-His抗体を用いて検出しました。データは、6つの反復試験の平均であり、エラーバーは平均の標準誤差を示します。(B)細胞質抗6×-His抗体でプローブしたウェスタンブロットによって分析したscFvを発現する細胞からの細胞溶解物の可溶性画分および不溶性画分。総タンパク質濃度を、サンプルのローディングを標準化するために使用しました。 (A)転載及び権限を持つ参照19からの(B)に適合される。 この図の拡大版をご覧になるにはこちらをクリックしてください。
Discussion
細胞質活性のために抗体を操作することは、ジスルフィド結合6,7の安定化の形成を妨げる細胞質、の還元環境に困難な作業です。これは、それらが細胞質中の安定性と溶解性のために設計されていない限り、ほとんどの抗体が結合親和性のために設計されることに加えて、細胞質に不活性であることが原因となります。ファージディスプレイ、細菌表面ディスプレイ、酵母表面ディスプレイ法の既存の方法は、すべての操作された抗体の表示のために分泌経路14-16を使用しますが、これらの方法は、細胞内の折り畳みを操作する手段がありません。 Tat経路の折り品質管理が不十分に折りたたまれ、細胞質内の不安定される抗体の移行を防止するため、内側膜ディスプレイを使用して操作された抗体は、細胞質の安定性と溶解性が改善されています。この方法は、親和性aの工学、細胞内抗体の反復プロセスを簡素化目の溶解度、二つの特性は、1ステップで操作されているよう。この方法は、還元細胞内環境中での溶解度を有する抗体を改変するために設計されたが、この方法を使用して操作されたタンパク質は、ペリプラズムの酸化性環境中でそれらのフォールディングを維持するので、また、非還元条件下で機能する技術的抗体に適用することができます。
この技術は、高い親和性と高い細胞質溶解性を有する抗体を設計するプロセスを簡素化しますが、いくつかの制限は、このプロトコルを使用する際に考慮することが重要です。有望なscFvは変異体を識別するために、二次スクリーニングELISAシグナルを分析する場合、適切な抗原結合を示さないかもしれない潜在的に興味深い変異体およびそれらの間の目の肥えたためのしきい値は、いくつかのクローンをさらに特徴付けられている後まで明らかになることはまずありません。改善された親抗体を介して結合を探すことが重要です。しかしながら、異常に高い信号は、内側の膜ディスプレイスクリーニングアプローチに固有のものではありません挑戦アビディティ29または凝集効果30の指標である可能性があります。それらは非生存(未発表データ)であるため、このプロトコルを使用するときに覚えておくべき重要な制限は、パニングの後にスフェロプラストを回復することができないことです。これは、抗体をコードするプラスミドを回収するためにDNA増幅および変換ステップを必要とします。
プロトコルのいくつかの重要なステップは、折り畳みおよび抗体の結合の同時エンジニアリングを可能にします。スクリーニングを成功させるには、スクリーニングされるscFvライブラリーはssTorAシグナルペプチドとの融合として発現されなければなりません。このシーケンスがなければ、抗体は、Tat経路に導かれることはありませんので、ペリプラズム19に移行されることはありません。加えて、C末端エピトープタグがビンに表示された抗体の検出を可能にするために、抗体に融合されることが不可欠です鼎アッセイ。明らかに、E。 coli株も必要Tat経路機構を有していなければならないscFvを発現するために使用されるが、これは一般的に使用されるEの真でありますcoli株。
このプロトコルへの改変は、所望の特性を有する抗体を単離する可能性を向上させることが可能です。減法パニングステップは、前の非所望成分のscFvライブラリーを枯渇するために標的抗原に対してパニングを完了することができます。ライブラリーのスフェロプラストは、BCCP単独で、または非所望のタンパク質でコーティングされてコーティングした磁気ビーズとともにインキュベートすることができ、それらのビーズに結合したスフェロプラストは、所望の標的に結合するために、残りの未結合のスフェロプラストをスクリーニングする前に廃棄することができます。代表的な結果で述べたように、分離されたscFvの親和性を改善するための方法は、スフェロプラスト上に表示されたscFvと競合するパンニング反応中の可溶性競合他社を含むことです。可溶性コンプため、etitorにはDNAは、スフェロプラスト上に表示されたscFvのように配列のみでは、PCR反応で回収され、それから増幅されていない、精製されたタンパク質です。さらに、この方法は、抗体の他のタイプの工学または非抗体結合タンパク質に拡張することができます。
大腸菌内側の膜ディスプレイは、高親和性および細胞内溶解性の高レベルの抗体を改変するための強力なプラットフォームです。この方法は、細胞内環境内で機能するように設計された抗体の効率的な操作のために特に適しています。これらの細胞内抗体は、すでに神経変性疾患、癌、およびウイルス感染31を含むフィールドの数の潜在的な治療薬として検討されています。この技術は、研究と医療これらの分野でその場でタンパク質標的を研究が望まれる任意の他のフィールドのためのツールとしての細胞内抗体のより広範な使用を可能にします。
Materials
| Name | Company | Catalog Number | Comments |
| scFv library | Varies | A suitable scFv library should be obtained from a commercial or academic source. | |
| MC4100 E. coli cells | Coli Genetic Stock Center | 6152 | Cells need to be chemically competent or electrocompetent, depending on the selected transformation method. |
| Glycerol | Fisher Scientific | BP229-4 | |
| Difco dehydrated culture media LB Broth, Miller (Luria-Bertani) | BD | 244610 | |
| Chloramphenicol (Cm) | Fisher Scientific | BP904-100 | |
| Sodium chloride (NaCl) | Fisher Scientific | BP358-1 | |
| Potassium chloride (KCl) | Fisher Scientific | BP366-500 | |
| Sodium phosphate, dibasic (Na2HPO4) | Fisher Scientific | BP332-500 | |
| Potassium phosphate, monobasic (KH2PO4) | Fisher Scientific | BP362-500 | |
| Bovine serum albumin (BSA) | Fisher Scientific | BP9706-100 | |
| Sucrose | Fisher Scientific | BP220-1 | |
| Tris base | Fisher Scientific | BP1521 | |
| Ethylenediaminetetraacetic acid (EDTA), 0.5 M | Fisher Scientific | BP2482-500 | |
| Magnesium chloride (MgCl2) | Fisher Scientific | BP214-500 | |
| Lysozyme | Sigma Aldrich | L3790-10X1ML | |
| Vortex mixer | VWR | 97043-564 | |
| Bicine | Fisher Scientific | BP2646100 | |
| D-Biotin | Fisher Scientific | BP232-1 | |
| Isopropyl β-D-1-thiogalactopyranoside | Fisher Scientific | BP1755-1 | |
| BugBuster Master Mix (cell lysis detergent) | EMD Millipore | 71456 | |
| Vivaspin 2 MWCO, 3,000 daltons | GE Healthcare Sciences | 28932240 | |
| Target antigen | Varies | N/A | Purified target antigen may be purchased or produced/purified. |
| Dynabeads MyOne Streptavidin T1 | Invitrogen | 65601 | |
| Dynamag-2 magnet | Invitrogen | 12321D | |
| Tube rotator | VWR | 13916-822 | |
| PCR primers | IDT | N/A | Primer sequences are as described in the protocol. |
| 10x T4 DNA ligase reaction buffer | New England BioLabs | B0202S | |
| T4 Polynucelotide kinase (PNK) | New England BioLabs | M0201S | Make sure the T4 ligase buffer used in the primer phosphorylation reaction contains 1 mM ATP. |
| 5x Phusion HF buffer pack | New England BioLabs | B0518S | |
| Deoxynucleotide (dNTP) solution mix, 10 mM each dNTP | New England BioLabs | N0447L | |
| Phusion DNA polymerase | New England BioLabs | M0530S | Other high-fidelity polymerases may be used as an alternative, but the annealing temperature in Table 3 must be adjusted. |
| C1000 Touch thermal cycler with dual 48/48 fast reaction module | Bio-Rad | 185-1148 | |
| Agarose | Promega | V3121 | |
| SYBR Safe DNA gel stain | Invitrogen | S33102 | |
| Wizard SV gel and PCR clean-up system | Promega | A9281 | |
| T4 DNA ligase | New England BioLabs | M0202S | |
| Microdialysis membrane filter | EMD Millipore | VSWP04700 | |
| Agar | BD | 214030 | |
| 96-well polystyrene round-bottom cell culture plates | VWR | 10062-902 | |
| Costar general polystyrene assay plate lids | Corning | 3931 | |
| Microtitre plate shaker | VWR | 12620-926 | |
| Costar 96 well EIA/RIA Easy Wash clear flat bottom polystyrene high bind microplate | Corning | 3369 | |
| Bel-blotter polycarbonate 96-well replicating tool | Bel-Art Products | 378760002 | |
| Instant nonfat dry milk | Quality Biological | A614-1000 | |
| Tween 20 (polysorbate 20) | Fisher Scientific | BP337-500 | |
| PopCulture reagent (concentrated cell lysis detergent) | EMD Millipore | 71092-3 | |
| Monoclonal ANTI-FLAG M2-Peroxidase(HRP) antibody produced in mouse | Sigma Aldrich | A8592 | |
| SigmaFast OPD | Sigma Aldrich | P9187-50SET | |
| Sulfuric acid (H2SO4), 10 N solution | Fisher Scientific | SA200-1 | |
| Reynolds Wrap aluminum foil | VWR | 89079-075 | |
| BioTek Epoch microplate spectrophotometer | Fisher Scientific | 11120570 |
References
- Biocca, S., Pierandrei-Amaldi, P., Campioni, N., Cattaneo, A. Intracellular immunization with cytosolic recombinant antibodies. Biotechnology (NY). 12 (4), 396-399 (1994).
- Chen, S. Y., Bagley, J., Marasco, W. A. Intracellular antibodies as a new class of therapeutic molecules for gene-therapy. Hum. Gene Ther. 5 (5), 595-601 (2008).
- Gargano, N., Biocca, S., Bradbury, A., Cattaneo, A. Human recombinant antibody fragments neutralizing human immunodeficiency virus type 1 reverse transcriptase provide an experimental basis for the structural classification of the DNA polymerase family. J Virol. 70 (11), 7706-7712 (1996).
- Mhashilkar, A. M., et al. Inhibition of HIV-1 Tat-mediated LTR transactivation and HIV-1 infection by anti-Tat single chain intrabodies. Embo J. 14 (7), 1542-1551 (1995).
- Strube, R. W., Chen, S. Y. Characterization of anti-cyclin E single-chain Fv antibodies and intrabodies in breast cancer cells: enhanced intracellular stability of novel sFv-F-c intrabodies. J. Immunol. Meth. 263 (1-2), 149-197 (2002).
- Mössner, E., Koch, H., Plückthun, A. Fast selection of antibodies without antigen purification: adaptation of the protein fragment complementation assay to select antigen-antibody pairs. J. Mol. Biol. 308 (2), 115-122 (2001).
- Wörn, A., et al. Correlation between in vitro stability and in vivo performance of anti-GCN4 intrabodies as cytoplasmic inhibitors. J Biol Chem. 275 (4), 2795-2803 (2000).
- Knappik, A., Plückthun, A. Engineered turns of a recombinant antibody improve its in vivo folding. Protein Eng. 8 (1), 81-89 (1995).
- Martineau, P., Jones, P., Winter, G. Expression of an antibody fragment at high levels in the bacterial cytoplasm. J Mol Biol. 280 (1), 117-127 (1998).
- Steipe, B., Schiller, B., Plückthun, A., Steinbacher, S. Sequence statistics reliably predict stabilizing mutations in a protein domain. J. Mol. Biol. 240 (3), 188-192 (1994).
- Daugherty, P. S. Protein engineering with bacterial display. Curr Opin Struct Biol. 17 (4), 474-480 (2007).
- Lener, M., et al. Diverting a protein from its cellular location by intracellular antibodies. Eur. J. Biochem. 267 (4), 1196-1205 (2000).
- Lynch, S. M., Zhou, C., Messer, A. An scFv intrabody against the nonamyloid component of α-synuclein reduces intracellular aggregation and toxicity. J. Mol. Biol. 377 (1), 136-147 (2008).
- Gai, S. A., Wittrup, K. D. Yeast surface display for protein engineering and characterization. Curr. Opin. Struc. Biol. 17 (4), 467-473 (2007).
- Kieke, M. C., et al. Selection of functional T cell receptor mutants from a yeast surface-display library. Proc. Natl. Acad. Sci. USA. 96 (10), 5651-5656 (1999).
- Steiner, D., Forrer, P., Stumpp, M. T., Pluckthun, A. Signal sequences directing cotranslational translocation expand the range of proteins amenable to phage display. Nat. Biotechnol. 24, 823-831 (2006).
- Pugsley, A. P. The complete general secretory pathway in gram-negative bacteria. Microbiol. Rev. 57 (1), 50-108 (1993).
- Rapoza, M. P., Webster, R. E. The filamentous bacteriophage assembly proteins require the bacterial SecA protein for correct localization to the membrane. J. Bacteriol. 175 (6), 1856-1859 (1993).
- Karlsson, A. J., et al. Engineering antibody fitness and function using membrane-anchored display of correctly folded proteins. J. Molec. Biol. 416 (1), 94-107 (2012).
- DeLisa, M. P., Tullman, D., Georgiou, G. Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc Natl Acad Sci U S A. 100 (10), 6115-6120 (2003).
- Fisher, A. C., Kim, W., DeLisa, M. P. Genetic selection for protein solubility enabled by the folding quality control feature of the twin-arginine translocation pathway. Protein Sci. 15 (3), 449-458 (2006).
- Maynard, J., Georgiou, G. Antibody engineering. Annu Rev Biomed Eng. 2, 339-376 (2000).
- Green, M. R., Sambrook, J. Molecular Cloning: A Laboratory Manual. 1, Fourth, Cold Spring Harbor Laboratory Press. (2012).
- Milo, R., Jorgensen, P., Moran, U., Weber, G., Springer, M. BioNumbers--the database of key numbers in molecular and cell biology. Nucleic Acids Res. 38, D750-D753 (2010).
- Hermanson, G. T. Bioconjugate Techniques. , Third, Elsevier/Academic Press. (2013).
- Tayapiwatana, C., Chotpadiwetkul, R., Kasinrerk, W. A novel approach using streptavidin magnetic bead-sorted in vivo biotinylated survivin for monoclonal antibody production. J Immunol Methods. 317 (1-2), 1-11 (2006).
- Zhu, G., Song, L., Lippard, S. J. Visualizing inhibition of nucleosome mobility and transcription by cisplatin-DNA interstrand crosslinks in live mammalian cells. Cancer Res. 73 (14), 4451-4460 (2013).
- Bogsch, E. G., et al. An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J. Biol. Chem. 273, 18003-18006 (1998).
- Julian, M. C., et al. Co-evolution of affinity and stability of grafted amyloid-motif domain antibodies. Protein Eng. Des. Sel. 28 (10), 339-350 (2015).
- Garber, K. Bispecific antibodies rise again. Nat. Rev. Drug. Discov. 13 (11), 799-801 (2014).
- Marschall, A. L., Dübel, S., Böldicke, T. Specific in vivo knockdown of protein function by intrabodies. MAbs. 7 (6), 1010-1035 (2015).
