

Introduction
能够折叠,并在细胞内环境中起作用的抗体可用于研究和治疗应用中有希望的工具。它们具有通过在细胞内结合到靶蛋白,以防止蛋白质-蛋白质相互作用,破坏蛋白质-核酸相互作用,或阻止对酶1-5基底入口调节蛋白活性的能力。
尽管抗体具有细胞内的应用程序多的潜力,工程他们在细胞内环境正确折叠和溶解度,同时维持对结合靶抗原的能力是具有挑战性的。的还原性的细胞质环境防止通常所需的全长抗体和抗体片段,包括单链可变片段(scFv)抗体6,7的稳定折叠的二硫键的形成。许多定向进化方法已被用来工程用H抗体对于目标IGH亲和力抗原8-10。这些方法通常使用噬菌体展示,酵母表面展示或细菌表面展示筛选抗体11-13的大型图书馆。这些方法是有力及有效用于鉴定结合靶的抗体,但它们依赖于分泌途径运输将要显示14-16蛋白质。分泌途径易位从还原细胞质折叠蛋白成在酵母或进入细菌的周质中的内质网腔。然后将蛋白质在氧化条件下折叠并显示在细胞表面上或包装入噬菌体颗粒来筛选结合亲和力17,18。其结果是,使用这些技术中分离的抗体不一定会在细胞质折好,和细胞内的溶解度通常必须单独设计,如果抗体将在细胞内的应用中使用。
为了改善那些在细胞质以及折叠工程抗体的效率,我们先前报道的MAD-TRAP(对于基于康达识别相关联蛋白的膜锚定显示器)的成功,用于筛选使用大肠杆菌党内的scFv抗体文库的方法膜显示19。细菌内膜显示依赖于双精氨酸易位(TAT)途径用于传输显示抗体,而相比之下,使用分泌途径其他常见的显示方法。 Tat途径包含质量控制机制,只允许可溶的,正确折叠的蛋白从大肠杆菌运大肠杆菌胞质中,穿过内膜并进入周质20,21。 (定位到具有N末端融合到Tat信号肽ssTorA Tat途径即 ,蛋白质),其在细胞质以及折叠过表达的Tat基板形成长寿命易位与N-末端中间体In中的细胞质和周质19中的C末端。这允许正确折叠的Tat基材,包括抗体片段的显示,在大肠杆菌的周质面大肠杆菌内膜。通过酶消化除去外膜以产生原生质球后,抗体被暴露于细胞外空间( 图1)。这允许对内部隔膜显示达衬底被筛选用于结合到一个特定的目标。重要的是,利用对细胞表面显示Tat途径确保只有在该库被在细胞质以及折叠抗体将被询问的结合,允许结合亲和力和细胞内折叠同步工程。在这个协议中,我们描述了如何显示在E.一个单链抗体库大肠杆菌内膜,平移针对靶抗原的文库,并进行二次筛选,以确定该文库的最有希望的成分。虽然我们关注关于scFv的协议,该方法可以应用于工程的应用要求绑定和细胞内折叠任何蛋白质。
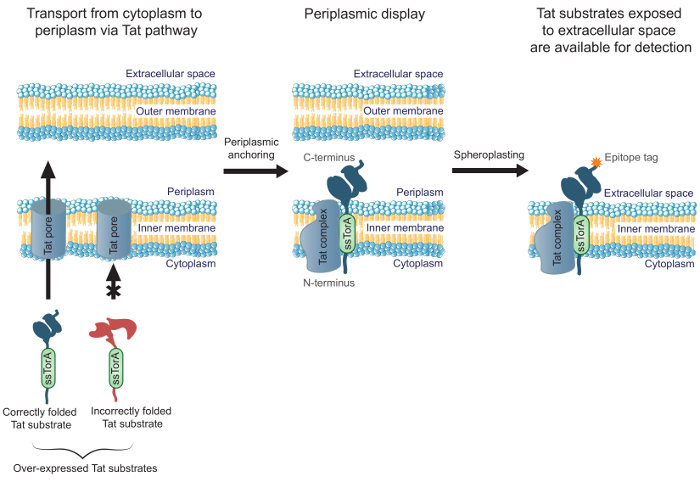
图1.达内膜显示,在E.大肠杆菌,被表达为融合到ssTorA信号序列和正确地在细胞质折叠的scFv抗体穿过内膜运输。 A易位中间形式,其中所述的scFv在与在细胞质N端和在周质的C-末端的内膜锚。 大肠大肠杆菌外膜被酶消化,以形成球状体,从而暴露锚定的抗体与细胞外的空间,使它们可用于检测通过使用结合于所显示的抗体的C-末端融合表位标签的抗体。负载/ 54583 / 54583fig1large.jpg“目标=”_空白“>点击此处查看该图的放大版本。
Protocol
1.准备单链抗体库中的融合到ssTorA信号序列
- 获得含有单链抗体基因的变体脱氧核糖核酸(DNA)的文库。
注:该库,也可以使用任何适当方式来产生在整个scFv基因或靶向域多样性构造22( 例如 ,第三互补决定区,CDR3的)。 - 插入DNA文库成使用标准的分子克隆方法23 PIMD质粒( 图2)。
注意:此质粒表达的scFv作为遗传融合到ssTorA信号序列(N端至抗体)和FLAG表位标签(C-末端到的scFv)。该质粒为内膜显示器的设计已先前19描述。该PIMD质粒是从作者。

图2内膜显示质粒(PIMD)地图(步骤1.2到1.3),该质粒含有lac启动启动子,复制的ColE1起点和氯霉素抗性基因。插入的scFv基因融合至ssTorA信号序列的scFv的靶向Tat途径和一个FLAG表位标签,与在同一阅读框架的所有三个。限制性酶位点表示。对于的XbaI和NotI限制性酶切位点之间插入一个图书馆,质粒的大小为2219 bp的加单链抗体的大小。 请点击此处查看该图的放大版本。
- 变换包含库到MC4100 大肠杆菌的质粒DNA 大肠杆菌细胞23。恢复和发展图书馆的这种细菌形态。离心机在4000×g下在室温下15分钟以收集细胞。去除上清,并悬浮收集的细胞在的Luria-BERTANI(LB)培养基的25%甘油。直到需要在-80°C储存分装,或进行第2步。
注:该协议已被证实与MC4100细胞,但其他E.大肠杆菌菌株也有望成为与该协议兼容。电穿孔是用于转化的优选方法中,由于其高转化效率。库通常应该包含至少10 9个单链抗体在这阶段的变体,并且每个等分试样应包含足够的细胞,例如该库覆盖100倍。
2.快递图书馆和原生质球准备
- 解冻细菌库(来自步骤1.3)在RT中的一个等分试样,并且等分试样加入到含有100毫升LB培养基用20微克/毫升氯霉素(CM)的烧瓶中。生长在37℃,3小时和225 RPM在培养摇床。
- 3小时后,取出从37℃孵育摇瓶。允许抗体库的表达进行O / N在20℃15〜22小时和225 RPM在培养摇床。
注意:使用PIMD质粒时,由于启动子是漏无需诱导。注意,MC4100细胞不过表达lac阻遏(和了LacI上未质粒找到)。

图3. 大肠杆菌 细胞原生质。(A)E. 大肠杆菌细胞在形状上是圆柱形的。 (B) 用EDTA和溶菌酶, 大肠杆菌的外膜原生质球后大肠杆菌细胞破裂,将所得原生质球在形状上是球形的。使用上的倒置显微镜100X的目标,获得微分干涉对比(DIC)显微镜图像。 请Ç舔此处查看该图的放大版本。
- 准备库原生质球。
注:原生质球是由破裂大肠杆菌的外膜形成大肠杆菌和是球形( 图3)。- 准备必要的缓冲区。
注:所有缓冲区应该是无菌的。- 通过溶解8克氯化钠将0.2g氯化钾,1.44克Na 2 HPO 4在蒸馏水中的 H 2 O操作1000 ml的终体积;(pH 7.4的PBS中),及0.24g的KH 2 PO 4制备1×磷酸盐缓冲盐水。置于冰上。
- 用0.1%制备的PBS(重量/体积)牛血清白蛋白(BSA)通过0.2克BSA溶解在200ml 1×PBS中。置于冰上。
- 通过混合7.5无菌过滤的1M蔗糖,1毫升的1M Tris缓冲液(pH 8.0),和1.5毫升蒸馏水H 2 O中的溶液制备的分馏缓冲液(FB)置于冰上。
- 通过加入30微升ö制备1mM的乙二胺四乙酸(EDTA)˚F的0.5MEDTA 14.97毫升蒸馏H 2 O.
- 制备的0.5M的MgCl 2通过在100毫升4.76克的MgCl 2溶于蒸馏水H 2 O置于冰上。
- 从摇床中取出烧瓶,并使用分光光度计,以确定细胞密度测量在600nm的光密度(OD)。计算诱导培养的容积所需的,使得每个样品为原生质球具有1×10 10细胞。
注:指示用于E的10 9细胞/ ml的浓度的OD 1 600的近似大肠杆菌可用于24。 - 离心机诱导培养的在1.5ml微量离心管中的计算量在12,000×g下在室温下5分钟。在这一个问题在样品制备发生的事件至少准备两个样品。
- 从离心培养物除去上清,悬浮于100μl冰冷FB的各细胞沉淀。离心机在12000×g的室温进行1分钟,然后通过移液除去上清液。重悬每个粒料在冰冷的FB的350微升补充有3.5微升的10毫克/毫升的溶菌酶。
- 慢慢涡每管同时加入,滴加700微升1mM EDTA中,然后在室温孵育管20分钟,同时在管旋转体缓慢旋转混合样本。从旋转器中取出试管,添加50微升冰冷的0.5M 的 MgCl 2到每个管中,并孵育他们在冰上10分钟。离心机在4℃,10分钟,在11000×g下的管中。
- 隔离原生质体沉淀。
- 使用微量1毫升尖慢慢拉起颗粒的一部分。在保持管中于直接一个新的1.5毫升管上方的开口的角,慢慢抬起枪头出上清液,沉淀滑入新管中。
- 如果上清液显著体积转移到新管中,通过移液除去它。如果颗粒不坚定ENO啊转移,重新离心机在11000×g下2分钟,企图粒料隔离一次。
- 重悬在每个管原生质球沉淀在1ml冰冷的1×PBS中。吸液和在涡流保持器慢慢涡旋直至沉淀完全重悬之间交替。在一个时间不保持样品落冰超过2分钟,而从冰再次除去之前返回到冰上至少5分钟。保持球状体在4℃下(用于2天),直至用于步骤4平移。
- 准备必要的缓冲区。
3.固定靶标抗原到磁珠
- 生物素化在大肠杆菌重组生产过程中在体内靶抗原大肠杆菌细胞。另外,使用化学结合25或已经购买生物素化靶抗原,并继续执行步骤3.2。
- 816克N-二加至50毫升的水,使10×N-二缓冲器。稀释BUFFER为1×蒸馏水的 H 2 O和热至50℃。添加14.7毫克生物素到12毫升加热1×N-二羟乙基甘氨酸缓冲,以使生物素的解决方案,是在10mM N-二羟乙基甘氨酸缓冲液的5mM生物素。直到需要在-20°C储存。
- 表达和使用pAK400cb-BCCP质粒26,其允许生产靶抗原的作为融合到生物素羧基载体蛋白(BCCP)生物素化靶蛋白。
注:E。大肠杆菌细胞本身生物素化BCCP,无需纯化和化学上链霉抗包被的珠子固定之前生物素化靶蛋白。本机E.大肠杆菌生物素连接酶生物素化酶BirA足以用于生物素化融合蛋白。- E.成长含有生物素化的质粒(插入作为融合到BCCP的N-末端的靶抗原)O- 大肠杆菌 / N为15至18小时,在5在37℃下补充有20微克/毫升厘米,而在摇动LB培养基中的溶液225转。
- 使用分光光度计测量在600nm处的OD值,并计算培养需要(Ⅴ 加法 )传代培养的容积在25 0.05毫升的新鲜LB培养基中,使用20μg/ ml的CM有关的方程式起始外径:V 添加 =(0.05× 25ml)中/(OD 600 - 0.05),其中, 外径 600是O / N培养和V 添加的光密度是O / N培养物体积添加到新鲜LB.继代培养并生长至0.5的OD〜0.8在温育摇床上,在37℃和225转。
- 添加异丙基β-D-1-硫代半乳糖苷至100μM的终浓度和生物素为5微米的最终浓度。诱导表达在15至22小时的温育摇床上在20℃和225转。
- 收获细菌通过在4000×g下离心4℃10分钟。去除上清。储存在-20℃沉淀,直到准备使用。
- 加1毫升每0.2克细胞沉淀的细胞裂解洗涤剂。通过移液重悬并轻轻旋转20分钟以裂解细胞。裂解后,以16,000×g下离心4℃20分钟。吸取可溶性裂解物(上清液)到一个新的1.5毫升管。
- 使用3 kDa的分子量截留柱以除去未结合的生物素。根据制造商的说明在20℃吸取溶解物入列,和离心机。用1×PBS洗涤,直至在裂解物中的生物素已稀释100倍和洗过的溶胞产物的体积等于裂解物的原始体积。转移裂解物至新管。
- 固定生物素化的靶抗原上的链霉包被的磁珠。
- 如步骤2.3.1描述制备1×PBS中1×PBS中的0.1%(重量/体积)BSA中。
- 制备磁珠。
注意:这要求使用磁分离机架。- 悬浮streptav啶包被的磁珠在原小瓶中。任一涡旋至少30秒或旋转5分钟。
- 转移7-10×10 9珠到1.5ml管中。
注意:需要的量将依赖于由制造商提供的珠的浓度。 - 上放置装有磁铁架珠的管2分钟以收集在管侧的珠子。与管仍然在磁铁,小心地通过吸液而不破坏珠除去上清液。
- 来洗,从磁体取出管,和不产生气泡,通过移液重悬珠子在1ml 1×PBS中。返回管到磁体2分钟来收集珠,并小心地通过吹打除去上清液。重复该过程两次,总共洗涤三次。确保没有液体留在管中的最终洗涤之后。
- 含有生物素化抗原的裂解物添加到磁性BEADS。
- 从磁体取出管并在1ml裂解物(来自步骤3.1.5)的重悬珠子。孵育在室温下进行30分钟,同时轻轻地旋转。
- 放置在磁体的管3分钟以收集所述抗原包被的珠子。如在步骤3.2.2.3至3.2.2.4描述的1×PBS洗涤包被的珠五次用0.1%BSA的相同方法。最后一次洗涤后,重悬珠子在1×PBS中的0.1%BSA的最多在步骤3.2.2.2使用的相同的体积。
- 如果固定的靶抗原是稳定的,在4℃下,直到需要用于摇摄存储涂覆的珠粒于4℃。否则,继续执行步骤4。
4.屏幕的单链抗体库通过淘选对目标抗原(图4)

图4.平移(步骤4)。抗原包被的磁珠ARË孵育表达抗体库变体原生质球。从珠结合的球状体的质粒DNA被回收并用于生成一个子库,这是使用基于ELISA的次级筛过筛。相应的协议步骤说明。 请点击此处查看该图的放大版本。
- 孵育原生质涂珠。
- 使用原生质球对珠粒的大约5:1的比例。添加4×10 9球状体和8×10 8珠无菌的15毫升管。
注意:假设无细胞在原生质球过程中丢失,所以浓度仍然为1×10 10原生质球/ ml的。 - 加1×PBS中的0.1%BSA,使总体积至4毫升分装成4节1.5毫升管与每1ml。孵育反应在4℃下进行5小时,同时轻轻地旋转。
- 使用原生质球对珠粒的大约5:1的比例。添加4×10 9球状体和8×10 8珠无菌的15毫升管。
- 镨epare用于聚合酶链式反应(PCR)的珠粒结合的原生质球。
- 放置平移反应管上的磁铁3分钟。通过移液除去上清液,并如步骤3.2.2.3描述3.2.2.4用冰冷洗涤珠结合的球状体的四倍冷1×PBS中的0.1%BSA的相同的方式。重悬在每个管中的珠结合的原生质球在25微升蒸馏水H 2 O的存放珠子-20℃或继续执行步骤4.3。
- 在珠结合的原生质进行全质粒PCR扩增含有珠结合的scFv的基因质粒。
- 得到的引物具有下列序列:5'CCAACTCTTTTTCCGAAGGTAACTG3'(正向引物)和5'TAGCTCTTGATCCGGCAAACAAA3'(反向引物)。
注意:这些将结合端至端上PIMD质粒的相反链( 图2)并设计成退火PIMD的一个共同的特征,所以无论将发生扩增单链抗体变体序列。 - 磷酸化的引物。
注意:如果没有磷酸化,不会发生重新结扎。引物也可以与5'-磷酸化排序,而不是在该协议使用该磷酸化方法。- 在0.5毫升管,设置为如表1所记载的正向PCR引物的磷酸化反应。重复此过程的反向引物。
- 孵育反应在37℃进行1小时。然后在65°C孵育他们20分钟以停用T4多核苷酸激酶(PNK)。储存在-20℃的磷酸化引物。
- 进行PCR。
- 在PCR管中,如表2中所述制备的PCR反应。
注意:多个反应可以是更高的产率制备。未使用的珠粒结合的球状体可存放在-20℃。 - 加热在98℃下进行15分钟的PCR反应在热循环仪,以确保完全裂解的球状体。从热循环仪取出试管,并添加0.5微升高保真聚合酶到每个。返回管,以热循环仪并使用表3中详述的程序运行。
- 池PCR产物为宜。储存在-20°C或继续执行步骤4.4。
- 在PCR管中,如表2中所述制备的PCR反应。
- 得到的引物具有下列序列:5'CCAACTCTTTTTCCGAAGGTAACTG3'(正向引物)和5'TAGCTCTTGATCCGGCAAACAAA3'(反向引物)。
表1. PNK磷酸化反应(步骤4.3.2.1)。
| 试剂 | 体积(微升) |
| 蒸馏水2 O | 15 |
| 10×T4 DNA连接酶反应缓冲液 | 2 |
| 100μM底漆 | 2 |
| T4多核苷酸激酶(PNK) | 1 |
| 试剂 | 体积(微升) |
| 蒸馏水2 O | 28.5 |
| 5X高保真聚合酶缓冲液 | 10 |
| 10μM磷酸化正向引物 | 2.5 |
| 10μM磷酸化反向引物 | 2.5 |
| 的40mM dNTP混合物(10mM的每种dNTP) | 1 |
| 珠结合的原生质 | 五 |
表3. PCR程序(步骤4.3.3.2)。
| 步 | 温度(℃) | 时间(分:秒) | 周期数 |
| 初始变性 | 98 | 0:30 | 1 |
| 变性 | 98 | 0:10 | 35 |
| 退火 | 69 | 0:30 | |
| 延期 | 72 | 0:30每KB | |
| 最终延伸 | 72 | 6:00 | 1 |
| 保持 | 12 | 无穷 | 1 |
- 重新环化整个质粒的PCR产物,并使用连接产物转化MC4100 大肠杆菌大肠杆菌细胞。
- 通过运行在琼脂糖凝胶上23上的PCR反应中,在凝胶中23染色的DNA,并使用纯化的PCR产物凝胶净化试剂盒由以下由制造商提供的说明来纯化线性化的质粒。使用分光光度计在260nm处测量的浓度。在-20℃保存纯化的片段,直到需要,或继续执行步骤4.4.2。
- 再环化从PCR产物的质粒。
- 为了防止在PCR产物的分子间连接,进行连接反应具有低浓度为1ng / PCR产物微升27。计算出所需的制备在该浓度的800微升连接反应的体积。
- 准备冰上连接反应。在一个管中,加入在PCR产物的步骤4.4.2.1计算出的体积,80微升10×连接酶缓冲液中,和蒸馏水2 O至800微升。加入4微升T4 DNA连接酶,立即将管在16℃的水浴或热循环仪。孵育在16℃的CO / N代表14至18小时。存储完成连接反应在-20°C直到需要,或继续执行步骤4.4.3。
- 放置在65℃的加热块上的连接反应15分钟以加热-灭活DNA连接酶。然后用微透析膜或DNA净化试剂盒脱盐连接的DNA。保存在20°C或继续执行步骤4.4.4。
- 采用全热灭活,脱盐连接产物转化大肠杆菌 MC4100 大肠杆菌细胞23。制备甘油原种,如在-80℃下在步骤1.3中描述的含有所得平移子库的细胞,并储存等分试样。
- 利用由步骤4.4.4等分试样上做的子库的第二摇摄其全部重复步骤4。
注:第二次淘洗有助于充实为很好地结合靶抗原19库成分。
5.执行二级屏幕采用酶联免疫法,以确定进一步鉴定有前途的克隆(图5)</ P>

基于ELISA的图5.二次筛选(步骤5)。从淘选过程中富集的子库(A)库变体接种在生长和表达一个培养板的各个孔中。 (B)将ELISA板涂有靶抗原。 (C)的库变体使用在协议中描述的基于ELISA的二次筛过筛。当从二次筛选得到的数据的分析中,感兴趣的变体进行选择和进一步的特征。相应的协议步骤说明。 请点击此处查看该图的放大版本。
- 解冻平移子库(来自步骤4.4.4)和板到LB琼脂平板上的一个管。板数稀释在浓度足够低,以确保单个菌落( 例如 ,10:2 - 〜10 6倍稀释)。孵育15到18小时将板在37℃。存储板在4℃或继续执行步骤5.2。
- 从平移子库的文化和诱发殖民地。执行所有的无菌条件下的步骤。使用涉及96孔板步骤多通道移液器。
- 加入200μl的LB用20微克/毫升厘米到圆底96孔培养板的每个孔中。
- 接从琼脂板的个体菌落用移液管尖,将吸头中的96孔板的第一井,并轻轻搅拌来接种。使用新的提示为每个很好。接种一个菌落到每个孔中。作为对照,将包括至少一个无菌对照孔没有菌落接种。
- 重复步骤5.2.1和5.2.2接种几个96孔板。
- 放置96孔板上在310转微孔板摇床上。在376;下进行20至24小时表达的scFv。
- 在步骤5.2制得的每个培养板,涂布一96孔ELISA板与靶抗原。
- 稀释纯化的靶抗原,以1×PBS中适当的浓度( 例如,1微克/毫升至4微克/毫升),以使该涂层溶液中。使溶于5ml涂敷液为每个96孔板中。
注:将合适的浓度取决于所使用的具体抗原,可能需要进行调整。 - 加入50μl的涂覆溶液,以96孔高结合透明聚苯乙烯ELISA板的各孔中。轻轻拍打台式表面上的板,以确保每个孔的整个表面被涂覆。重复每个板。在4℃CO / N孵育所述板。
- 稀释纯化的靶抗原,以1×PBS中适当的浓度( 例如,1微克/毫升至4微克/毫升),以使该涂层溶液中。使溶于5ml涂敷液为每个96孔板中。
- 复制从96孔培养板中的菌落到琼脂平板上。
- 放置无菌聚苯乙烯复制到培养板的孔中收集理趣的少量ID。小心抬起复制和转移到15厘米的LB琼脂平板上,这样所有的技巧都在触摸板。一旦液体已转移,解除复制直线上升。重复每个培养皿。
- 以正确的方向,使得从在96孔板的二次筛选结果可与在板的正确复制菌落如果进一步表征期望相匹配,标号琼脂板。生长于37℃下进行15至18小时,然后在4℃下储存直到需要。
- 执行ELISA二次屏幕。
- 通过使2%(重量/体积)在1×PBS中干乳制备封闭溶液。清空从ELISA板的涂布溶液。加入100微升的封闭溶液的各孔中。孵育在室温下至少2小时,或嵌段O / N在4℃。
- 通过添加聚山梨醇酯20为0.05%,以1×PBS中的终浓度制备洗涤缓冲液。让每ELISA板250毫升
- 加入20微升的浓缩细胞裂解洗涤剂向圆底培养板的各孔中,并孵育在室温在微孔板振荡器上15至20分钟的培养板。开始的同时,该ELISA板的阻塞是完整,以使裂解和洗涤步骤5.5.4可以同时进行裂解。
- 清空从ELISA板的封闭溶液。用每孔200μl的洗涤缓冲液的每次洗涤洗阻断ELISA板四次。清空从孔的洗涤缓冲液。
- 转移从细胞裂解板的每个孔加入50μl到相应井ELISA板,采用为每一个新的前端井。孵育在室温的ELISA板进行1至2小时。
- 制备抗体溶液检测结合的scFv。
- 使用辣根过氧化物酶(HRP)标记的结合融合到库的scFv FLAG表位标签的主要抗体。
- 稀释抗体与适当稀释在ELISA使用(见商&#39的推荐)的2%(w的1×PBS中的0.05%聚山梨酸酯20 /体积)奶粉。制备5-毫升每个板。
- 如步骤5.5.4所述洗涤ELISA板用洗涤缓冲液四次。
- 加入50μl抗体溶液到ELISA板的各孔中。孵育1至2小时在RT。
- 通过同时避免光蒸馏水溶解Ø苯二胺二盐酸盐(OPD)片剂2 O按照制造商的协议准备HRP底。每次准备酶标板20毫升
- 根据需要稀释浓H 2 SO 4蒸馏水2 O 3准备MH 2 SO 4。每次准备酶标板5毫升
注意:H 2 SO 4是强酸。一定要戴适当的个人防护装备。 - 用洗涤缓冲液洗涤ELISA板四次,如步骤5.5.4说明。
- 孵育ELISA板与HRP substra德。
- 添加200微升HRP底物的每个孔中。为了尽量减少光曝光,衬底一次添加到一个ELISA板,并在继续下一盘前用铝箔包裹。孵育所述板在黑暗中在RT 30至60分钟。
- 第30分钟后,检查板为基材变黑,并在必要时进行可视化显色孵化时间。
- 加入50μl的3 MH 2 SO 4到各孔中以终止反应。使用不同的提示为每个很好,没有起泡,轻轻上下吹打混合在孔中的溶液。一致性和以防止饱和,SO 4快速前仔细对各板的溶液混合添加的 H 2的所有ELISA板的。
- 用酶标仪测定各板的孔中的溶液的吸光度在492nm处。
- 分析的吸光度数据来识别的scFv变体即exhib它有前途的结合信号和刻画这些有前途的scFvs。选择表现出比背景信号比在每个板中的平均信号越来越高吸光度信号的scFv。
注:吸光度水平将取决于所使用的,与分离在筛选的scFv的变体的强度沿抗原和抗FLAG抗体的性质。
Representative Results
在大肠杆菌 Tat途径的细胞内蛋白质折叠质量控制机制大肠杆菌限制穿过内细胞膜转运到在还原细胞质环境良好折叠的蛋白质。通过超表达的scFv的ssTorA信号序列(从TORA蛋白,这是自然的Tat途径20传送的信号序列)的融合,易位被停止,从而导致在内侧膜19 scFv的显示。外膜的酶破坏之后,所显示的抗体用于筛选抗原结合活性可用。取Tat途径为单链抗体显示的优点的能力是由Karlsson 等所示的19( 图6)。所述scFv抗体scFv13和scFv13.R4被融合到缺少由公认的精氨酸 - 精氨酸残基对天然ssTorA序列或修饰的ssTorATat途径。 scFv13.R4通过马丁纽等人通过四轮定向进化工程。从scFv13并且已知在细胞质9折好。这种单链抗体被显示在内膜,但只有当作为融合到天然ssTorA信号序列表达( 图6)。反之,scFv13没有很好地折叠细胞质9,所以它不是在内部膜显示良好,无论信号序列其所稠合。此外,如果的scFv被在于缺乏TATC蛋白,康达机械20,28的一个重要组成部分,显示没有观察到,显示内膜显示和Tat途径之间的重要链接细胞中表达。这些结果表明,仅包含Tat信号肽和蛋白质正确折叠在细胞质中被显示在内膜,允许传输通过Tat途径充当细胞内FOL一个屏幕鼎。

图6.检测上内膜显示scFv的流式细胞仪分析,以检测在其内的膜折叠很差scFv13和良好的折叠scFv13.R4的显示。的scFv进行融合到天然ssTorA或ssTorA(KK),其中精氨酸 - 精氨酸对在ssTorA序列修饰成Lys赖氨酸。用异硫氰酸荧光素(FITC)检测上的scFv的C-末端FLAG表位标签缀合抗FLAG抗体。没有TATC蛋白(ΔtatC)和ssTorA-scFv13没有FLAG标记细胞进行了测试作为对照。 M表示中间荧光值。从权限参考19转载。 请点击此处查看该图的放大版本。
19。为了证明这一点,根据scFv13,其具有对β半乳糖苷酶结合亲和力的低电平的易错PCR文库,是针对靶抗原β半乳糖苷酶使用显示器和平移在协议中所述方法平移。 scFv的1-4被一轮诱变和淘选后分离,并表现出较高的结合亲和力β-半乳糖苷比scFv13( 图7A)和细胞质溶解度更高水平( 图7B)。
一个新的图书馆,以单链抗体1-4,使用易错PCR制成,以及对第二代文库淘β半乳糖苷酶使用所描述的协议的变形完成。第二轮演进抗β半乳糖苷酶的声像是在纯化的,可溶性scFv 14的存在下完成的作为竞争对手,以改善与比的scFv 1-4更高的亲和性分离的克隆的可能性。第二轮诱变和平移后,单链抗体2-1和2-3单链抗体使用基于ELISA的二次筛选分离。这些scFv的不仅表现出对β半乳糖苷酶比scFv13更高的结合亲和力,但也表现出比第一轮克隆的scFv 1-4更好的结合。的scFv 2-1表现出β半乳糖苷酶结合比得上scFv13.R4( 图7A)的。的scFv 2-3还示出了在细胞质溶解度进一步增加相比的scFv 14,突出的溶解性和抗原结合的同步工程。由于scFv的亲和力和可溶性表达筛选,用于同时,可能的是一个选定的scFv已国防部中心提供全方位溶解性,但高结合,反之亦然。例如,单链抗体2-1具有比单链抗体2-3可溶表达低,但它具有较高的结合亲和力β-半乳糖苷。

图7.目标结合和scFv细胞质表达变异体内部采用膜分离出的显示(A)的scFv在大肠杆菌的细胞质中表达大肠杆菌细胞( 例如 ,没有ssTorA信号序列)与六组氨酸(6×-His)标签,并使用镍-氮川三乙酸自旋柱纯化。对β-半乳糖苷酶的结合的纯化的scFv的用的ELISA测定。纯化的scFv被装载到β半乳糖苷酶包被ELISA板,并用抗6×-His抗体检测结合的scFv的。的数据是六个重复的平均,误差棒表示平均值的标准误差。(B)中的细胞裂解物的从表达细胞质通过用抗6×-His抗体探测Western印迹分析的scFv细胞的可溶和不溶组分。总蛋白质浓度用于标准化样品的加载。转载(A)和经许可的参考19改编(B)。 请点击此处查看该图的放大版本。
Discussion
工程抗体的细胞质活动是一个艰巨的任务,由于细胞质,阻碍稳定的二硫键6,7-形成的还原环境。这会导致最抗体,除非它们被设计为在细胞质中的稳定性和溶解度,除了被设计为结合亲和力是细胞质无效。噬菌体展示,细菌表面展示,酵母表面展示方法的现有方法都使用分泌途径14-16工程抗体的显示,但这些方法都没有办法工程师胞内折叠。因为Tat途径的折叠质量控制阻止了不良折叠并在细胞质中不稳定的抗体的转用内膜显示工程化抗体具有改善的细胞质的稳定性和溶解性。此方法简化了工程细胞内抗体的迭代过程对亲和性的第二溶解度,因为这两个性质在一个步骤工程改造。虽然这种方法被设计用于工程抗体与降低细胞内环境的溶解度,它也可以适用于工程抗体在非还原条件下发挥作用,因为蛋白质使用这种方法维持其在周质的氧化环境的折叠设计。
尽管该技术简化了工程以高亲和力和高细胞质溶解度抗体的过程中,一些限制是重要的使用该协议时要考虑的。在分析了二次筛选的ELISA信号,以确定有前途的单链抗体变体,潜在的有趣的变体和那些可能不会出现足够的抗原结合之间挑剔的门槛并不容易,直到经过多次克隆得到了进一步的特点是显而易见的。重要的是要寻找改良过的亲本抗体结合是很重要的;然而,异常高的信号可以是指示亲和力29或聚集效应30,其不是唯一的内膜显示筛选方法的挑战。要记住一个关键限制使用此协议时,是无法平移后的恢复原生质球,因为他们都是非可行的(未公布数据)。这就需要DNA扩增和改造步骤来恢复编码抗体的质粒。
该协议的几个关键步骤,使折叠和抗体结合的同步工程。用于筛选是成功的,所述scFv文库被筛选必须表达为融合到ssTorA信号肽。没有该序列中,抗体将不被引导到Tat途径,因而将不被转运到周质19。另外,至关重要的是,C-末端表位标签融合至抗体以允许在bin显示抗体的检测鼎检测。显然, 大肠杆菌大肠杆菌菌株用于表达的scFv还必须有必要的Tat途径机械,但是这是在通常使用的大肠杆菌的真大肠杆菌菌株。
修改这个协议是可能的,以改善其电势以分离具有所需特性的抗体。减色层叠步骤可以淘洗针对靶抗原耗尽非所需成分的抗体库之前完成。库球状体,可以单独用或涂覆涂覆有BCCP用非所需的蛋白的磁珠孵育,并结合于那些珠的球状体可筛选其余未结合的球状体结合于期望的目标之前被丢弃。如在代表结果所提到的,一个方法来改善的分离的scFv的亲和力是包括在平移反应的可溶性竞争者与对球状体显示的scFv竞争。由于可溶性补偿etitor是纯化的蛋白质,没有DNA从它放大,所以只对球状体显示的scFv的序列将在PCR反应中回收。此外,这种方法可以扩展到工程其他类型的抗体或非抗体结合蛋白。
大肠杆菌内的膜显示为工程抗体以高亲和力和高含量的胞内溶解度的有力的平台。这种方法特别适用于设计成在细胞内的环境中发挥作用的抗体的有效的工程。这些细胞内的抗体已经被探索作为在许多领域的潜在治疗剂,包括神经变性疾病,癌症和病毒感染31。这种技术可以使更广泛地使用胞内抗体作为在这些领域的研究和医学并且其中正在学原位蛋白靶标是所期望的任何其他字段的工具。
Materials
| Name | Company | Catalog Number | Comments |
| scFv library | Varies | A suitable scFv library should be obtained from a commercial or academic source. | |
| MC4100 E. coli cells | Coli Genetic Stock Center | 6152 | Cells need to be chemically competent or electrocompetent, depending on the selected transformation method. |
| Glycerol | Fisher Scientific | BP229-4 | |
| Difco dehydrated culture media LB Broth, Miller (Luria-Bertani) | BD | 244610 | |
| Chloramphenicol (Cm) | Fisher Scientific | BP904-100 | |
| Sodium chloride (NaCl) | Fisher Scientific | BP358-1 | |
| Potassium chloride (KCl) | Fisher Scientific | BP366-500 | |
| Sodium phosphate, dibasic (Na2HPO4) | Fisher Scientific | BP332-500 | |
| Potassium phosphate, monobasic (KH2PO4) | Fisher Scientific | BP362-500 | |
| Bovine serum albumin (BSA) | Fisher Scientific | BP9706-100 | |
| Sucrose | Fisher Scientific | BP220-1 | |
| Tris base | Fisher Scientific | BP1521 | |
| Ethylenediaminetetraacetic acid (EDTA), 0.5 M | Fisher Scientific | BP2482-500 | |
| Magnesium chloride (MgCl2) | Fisher Scientific | BP214-500 | |
| Lysozyme | Sigma Aldrich | L3790-10X1ML | |
| Vortex mixer | VWR | 97043-564 | |
| Bicine | Fisher Scientific | BP2646100 | |
| D-Biotin | Fisher Scientific | BP232-1 | |
| Isopropyl β-D-1-thiogalactopyranoside | Fisher Scientific | BP1755-1 | |
| BugBuster Master Mix (cell lysis detergent) | EMD Millipore | 71456 | |
| Vivaspin 2 MWCO, 3,000 daltons | GE Healthcare Sciences | 28932240 | |
| Target antigen | Varies | N/A | Purified target antigen may be purchased or produced/purified. |
| Dynabeads MyOne Streptavidin T1 | Invitrogen | 65601 | |
| Dynamag-2 magnet | Invitrogen | 12321D | |
| Tube rotator | VWR | 13916-822 | |
| PCR primers | IDT | N/A | Primer sequences are as described in the protocol. |
| 10x T4 DNA ligase reaction buffer | New England BioLabs | B0202S | |
| T4 Polynucelotide kinase (PNK) | New England BioLabs | M0201S | Make sure the T4 ligase buffer used in the primer phosphorylation reaction contains 1 mM ATP. |
| 5x Phusion HF buffer pack | New England BioLabs | B0518S | |
| Deoxynucleotide (dNTP) solution mix, 10 mM each dNTP | New England BioLabs | N0447L | |
| Phusion DNA polymerase | New England BioLabs | M0530S | Other high-fidelity polymerases may be used as an alternative, but the annealing temperature in Table 3 must be adjusted. |
| C1000 Touch thermal cycler with dual 48/48 fast reaction module | Bio-Rad | 185-1148 | |
| Agarose | Promega | V3121 | |
| SYBR Safe DNA gel stain | Invitrogen | S33102 | |
| Wizard SV gel and PCR clean-up system | Promega | A9281 | |
| T4 DNA ligase | New England BioLabs | M0202S | |
| Microdialysis membrane filter | EMD Millipore | VSWP04700 | |
| Agar | BD | 214030 | |
| 96-well polystyrene round-bottom cell culture plates | VWR | 10062-902 | |
| Costar general polystyrene assay plate lids | Corning | 3931 | |
| Microtitre plate shaker | VWR | 12620-926 | |
| Costar 96 well EIA/RIA Easy Wash clear flat bottom polystyrene high bind microplate | Corning | 3369 | |
| Bel-blotter polycarbonate 96-well replicating tool | Bel-Art Products | 378760002 | |
| Instant nonfat dry milk | Quality Biological | A614-1000 | |
| Tween 20 (polysorbate 20) | Fisher Scientific | BP337-500 | |
| PopCulture reagent (concentrated cell lysis detergent) | EMD Millipore | 71092-3 | |
| Monoclonal ANTI-FLAG M2-Peroxidase(HRP) antibody produced in mouse | Sigma Aldrich | A8592 | |
| SigmaFast OPD | Sigma Aldrich | P9187-50SET | |
| Sulfuric acid (H2SO4), 10 N solution | Fisher Scientific | SA200-1 | |
| Reynolds Wrap aluminum foil | VWR | 89079-075 | |
| BioTek Epoch microplate spectrophotometer | Fisher Scientific | 11120570 |
References
- Biocca, S., Pierandrei-Amaldi, P., Campioni, N., Cattaneo, A. Intracellular immunization with cytosolic recombinant antibodies. Biotechnology (NY). 12 (4), 396-399 (1994).
- Chen, S. Y., Bagley, J., Marasco, W. A. Intracellular antibodies as a new class of therapeutic molecules for gene-therapy. Hum. Gene Ther. 5 (5), 595-601 (2008).
- Gargano, N., Biocca, S., Bradbury, A., Cattaneo, A. Human recombinant antibody fragments neutralizing human immunodeficiency virus type 1 reverse transcriptase provide an experimental basis for the structural classification of the DNA polymerase family. J Virol. 70 (11), 7706-7712 (1996).
- Mhashilkar, A. M., et al. Inhibition of HIV-1 Tat-mediated LTR transactivation and HIV-1 infection by anti-Tat single chain intrabodies. Embo J. 14 (7), 1542-1551 (1995).
- Strube, R. W., Chen, S. Y. Characterization of anti-cyclin E single-chain Fv antibodies and intrabodies in breast cancer cells: enhanced intracellular stability of novel sFv-F-c intrabodies. J. Immunol. Meth. 263 (1-2), 149-197 (2002).
- Mössner, E., Koch, H., Plückthun, A. Fast selection of antibodies without antigen purification: adaptation of the protein fragment complementation assay to select antigen-antibody pairs. J. Mol. Biol. 308 (2), 115-122 (2001).
- Wörn, A., et al. Correlation between in vitro stability and in vivo performance of anti-GCN4 intrabodies as cytoplasmic inhibitors. J Biol Chem. 275 (4), 2795-2803 (2000).
- Knappik, A., Plückthun, A. Engineered turns of a recombinant antibody improve its in vivo folding. Protein Eng. 8 (1), 81-89 (1995).
- Martineau, P., Jones, P., Winter, G. Expression of an antibody fragment at high levels in the bacterial cytoplasm. J Mol Biol. 280 (1), 117-127 (1998).
- Steipe, B., Schiller, B., Plückthun, A., Steinbacher, S. Sequence statistics reliably predict stabilizing mutations in a protein domain. J. Mol. Biol. 240 (3), 188-192 (1994).
- Daugherty, P. S. Protein engineering with bacterial display. Curr Opin Struct Biol. 17 (4), 474-480 (2007).
- Lener, M., et al. Diverting a protein from its cellular location by intracellular antibodies. Eur. J. Biochem. 267 (4), 1196-1205 (2000).
- Lynch, S. M., Zhou, C., Messer, A. An scFv intrabody against the nonamyloid component of α-synuclein reduces intracellular aggregation and toxicity. J. Mol. Biol. 377 (1), 136-147 (2008).
- Gai, S. A., Wittrup, K. D. Yeast surface display for protein engineering and characterization. Curr. Opin. Struc. Biol. 17 (4), 467-473 (2007).
- Kieke, M. C., et al. Selection of functional T cell receptor mutants from a yeast surface-display library. Proc. Natl. Acad. Sci. USA. 96 (10), 5651-5656 (1999).
- Steiner, D., Forrer, P., Stumpp, M. T., Pluckthun, A. Signal sequences directing cotranslational translocation expand the range of proteins amenable to phage display. Nat. Biotechnol. 24, 823-831 (2006).
- Pugsley, A. P. The complete general secretory pathway in gram-negative bacteria. Microbiol. Rev. 57 (1), 50-108 (1993).
- Rapoza, M. P., Webster, R. E. The filamentous bacteriophage assembly proteins require the bacterial SecA protein for correct localization to the membrane. J. Bacteriol. 175 (6), 1856-1859 (1993).
- Karlsson, A. J., et al. Engineering antibody fitness and function using membrane-anchored display of correctly folded proteins. J. Molec. Biol. 416 (1), 94-107 (2012).
- DeLisa, M. P., Tullman, D., Georgiou, G. Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc Natl Acad Sci U S A. 100 (10), 6115-6120 (2003).
- Fisher, A. C., Kim, W., DeLisa, M. P. Genetic selection for protein solubility enabled by the folding quality control feature of the twin-arginine translocation pathway. Protein Sci. 15 (3), 449-458 (2006).
- Maynard, J., Georgiou, G. Antibody engineering. Annu Rev Biomed Eng. 2, 339-376 (2000).
- Green, M. R., Sambrook, J. Molecular Cloning: A Laboratory Manual. 1, Fourth, Cold Spring Harbor Laboratory Press. (2012).
- Milo, R., Jorgensen, P., Moran, U., Weber, G., Springer, M. BioNumbers--the database of key numbers in molecular and cell biology. Nucleic Acids Res. 38, D750-D753 (2010).
- Hermanson, G. T. Bioconjugate Techniques. , Third, Elsevier/Academic Press. (2013).
- Tayapiwatana, C., Chotpadiwetkul, R., Kasinrerk, W. A novel approach using streptavidin magnetic bead-sorted in vivo biotinylated survivin for monoclonal antibody production. J Immunol Methods. 317 (1-2), 1-11 (2006).
- Zhu, G., Song, L., Lippard, S. J. Visualizing inhibition of nucleosome mobility and transcription by cisplatin-DNA interstrand crosslinks in live mammalian cells. Cancer Res. 73 (14), 4451-4460 (2013).
- Bogsch, E. G., et al. An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J. Biol. Chem. 273, 18003-18006 (1998).
- Julian, M. C., et al. Co-evolution of affinity and stability of grafted amyloid-motif domain antibodies. Protein Eng. Des. Sel. 28 (10), 339-350 (2015).
- Garber, K. Bispecific antibodies rise again. Nat. Rev. Drug. Discov. 13 (11), 799-801 (2014).
- Marschall, A. L., Dübel, S., Böldicke, T. Specific in vivo knockdown of protein function by intrabodies. MAbs. 7 (6), 1010-1035 (2015).
